

Abstract
mRNA expression profiling has suggested the existence of multiple glioblastoma subclasses, but their number and characteristics vary among studies and the etiology underlying their development is unclear. In this study, we analyzed 261 microRNA expression profiles from the Cancer Genome Atlas (TCGA), identifying five clinically and genetically distinct subclasses of glioblastoma that each related to a different neural precursor cell type. These microRNA-based glioblastoma subclasses displayed microRNA and mRNA expression signatures resembling those of radial glia, oligoneuronal precursors, neuronal precursors, neuroepithelial/neural crest precursors or astrocyte precursors. Each subclass was determined to be genetically distinct, based on the significant differences they displayed in terms of patient race, age, treatment response and survival. We also identified several microRNAs as potent regulators of subclass-specific gene expression networks in glioblastoma. Foremost among these is miR-9, which suppresses mesenchymal differentiation in glioblastoma by downregulating expression of JAK kinases and inhibiting activation of STAT3. Our findings suggest that microRNAs are important determinants of glioblastoma subclasses through their ability to regulate developmental growth and differentiation programs in several transformed neural precursor cell types. Taken together, our results define developmental microRNA expression signatures that both characterize and contribute to the phenotypic diversity of glioblastoma subclasses, thereby providing an expanded framework for understanding the pathogenesis of glioblastoma in a human neurodevelopmental context.
Keywords: MicroRNA, Microarray, Glioma, Tumor, Genome
Introduction
Glioblastoma is the most common and most malignant intrinsic brain tumor (1, 2). Because of the extremely unfavorable prognosis of glioblastoma (median survival of 14 to 16 months), it is important to develop more effective diagnostic and therapeutic strategies that are based on a biologically and clinically relevant disease subclassification system (3). Recent studies have proposed at least two mRNA-based classification systems for glioblastoma: ‘proneural – proliferative –mesenchymal’ (4) and ‘proneural – neural – classical – mesenchymal’ subtypes (5). These studies differ regarding the number of subclasses, their relationship to neural differentiation, and whether they have prognostic value. Although there is a consensus that the expression signature of one of the subclasses resembles that of a ‘proneural’ precursor cell (4, 5), the relationship of other glioblastoma subclasses to neural differentiation is less clear. Moreover, there is disagreement regarding the relationship of the subclasses to patient survival (4, 5).
MicroRNAs are short, non-coding RNAs that are key regulators of neural development and cancer. Previous studies suggest that expression-based clustering using microRNAs may yield more accurate histological and prognostic sample classification than clustering based on mRNA expression (6, 7). The recently published Cancer Genome Atlas (TCGA) data set for glioblastoma includes the expression profiles of microRNAs as well as data for mRNA expression, somatic mutations and copy number changes for tumors from more than 260 glioblastoma patients (8). In this study, we use microRNA expression-based clustering to identify five clinically and genetically distinct glioblastoma subclasses, each of which corresponds to a specific neural precursor cell type. We describe the genetic and clinical features of each glioblastoma subclass, and we show that microRNAs help to establish these subclasses by regulating neurodevelopmental growth and differentiation programs.
Materials and Methods
Consensus clustering of microRNA expression profiles
MicroRNA expression profiling was performed using Agilent 8 × 15K Human microRNA-specific microarrays in The Cancer Genome Atlas (TCGA) pilot study (8). Processed (level 3) microRNA expression data as well as the clinicopathological annotations for 261 glioblastoma patients were downloaded from TCGA portal (http://cancergenome.nih.gov/dataportal/). We first removed viral origin microRNAs. The expression data for the remaining 470 microRNAs were mean centered, and the standard deviation was normalized to one per array. We then filtered those with low variability in expression level (median absolute deviation or MAD < 0.1). We used three additional criteria to further select a group of highly informative microRNAs (Supplementary Fig. S1 and Table S1). These criteria included (i) microRNAs showing highly variable expression (MAD > 1.0; n = 45), (ii) patient survival-related microRNAs (significance in univariate Cox model < 0.1; n = 58) and (iii) neurodevelopment-related microRNAs (n = 57) which were manually curated from the published literature (Supplementary Table S2). Supplementary Table S1 also lists the regression coefficients and P values for these microRNAs, which were estimated using a univariate Cox regression model. The resultant group of 121 microRNAs was used for sample- and microRNA-clustering using a consensus clustering algorithm (9). Consensus clustering was performed using the hierarchical clustering method with average linkage and 1 minus the Pearson's correlation coefficient as the distance measure. A total of 100 permutation tests were performed with a subsampling ratio of 0.8. Consensus clustering was performed both for glioblastoma (n = 261 patients) and microRNA (n = 121 microRNAs) subgrouping. The optimal number of glioblastoma and microRNA subgroups was determined using a consensus clustering cumulative distribution function (CDF) and consensus matrices (Supplementary Fig. S2).
Genomic alterations in GBM subclasses
The mutation profiles as well as the genomic alteration profiles for glioblastoma were downloaded from TCGA portal. For mutation analyses, we first collected 1320 validated somatic mutations for 147 TCGA glioblastoma samples (8). We further selected 1065 non-silent mutations for 140 TCGA glioblastomas that were available for microRNA-based cluster membership. The significance of mutation frequency across the five microRNA-subclasses was calculated using a two-sided Fisher's exact test. For each gene, the significance was calculated as the probability of observing the mutation frequency in each microRNA subclass given the total number of mutations for that gene and the number of samples in each subclass. In calculating the significance, we ignored the mutations present in the seven hypermutation samples (annotated in a previous publication) (8). The mutation frequency of the top 23 most frequently-mutated genes is available in Supplementary Table S3.
For copy number profiles, we used the circular binary segmentation (CBS) smoothed copy number profiles of Agilent 244K array comparative genomic hybridization profiles (10). Significant copy number changes for the five microRNA subclasses were identified using GISTIC software (11). The significantly recurrent amplifications and deletions (false discovery rate or FDR < 0.25) were considered as microRNA-based glioblastoma subclass-specific alterations. Subclass-unique alterations were further identified by filtering significant alterations in one glioblastoma subclass with those in the remaining 4 glioblastoma subclasses. Genes belonging to unique alterations occurring in each microRNA-based glioblastoma subclass are provided in Supplementary Table S4.
For gene expression data analysis, we used unified expression profiles across three different gene expression microarray platforms (Affymetrix U133A, Affymetrix Exon, and Agilent custom 244K expression) as available in the published literature (4). The unified expression level of 11861 genes was used for expression analysis across 197 TCGA glioblastoma samples that were available for the microRNA-based five cluster membership. Annotations for the four mRNA-based glioblastoma subclasses (proneural, neural, classical and mesenchymal) were also downloaded from the published literature (4) and correlated with the microRNA-based glioblastoma subclasses.
We obtained the promoter methylation status of the MGMT gene, which was profiled using the Illumina DNA Methylation Cancer Panel I (8). The calculated methylation values (β value) of two CpG dinucleotides located at 281bp and 271bp upstream of the MGMT transcription start site were rescaled as previously described (8). The 59 glioblastoma cases with MGMT promoter methylation (rescaled β > 0.25) were selected and compared with the remaining cases for survival differences in each microRNA-based glioblastoma subclass.
Combined analysis with mRNA expression profiles
To examine the expression-level association of microRNA and mRNA signatures, we used module and gene set matrix analysis (12-14). First, we collected 7 gene sets (mRNA signatures) whose gene members show higher expression in different types of differentiated neural cells (4 gene sets) or stem cells (3 gene sets). For the differentiated neural cell types, we obtained expression profiles representing mouse neurons, oligodendrocytes or (cultured) astrocytes (15). Four neural cell types were distinguished according to the original tissue descriptions in the dataset (GEO accession, GSE9566), and these distinctions were further confirmed by hierarchical clustering. We next used a t-statistic to select the top 500 up-regulated genes for each of the four neural cell types, and subsequently used these genes as representative gene sets for the four differentiated neural cell types. We also obtained 3 stemness-related gene signatures from MSigDB database (16) representing up-regulated genes in human embryonic, hematopoietic and neural stem cells (STEMCELL_EMBRYONIC_UP, _HEMATOPOIETIC_UP and NEURAL_UP, respectively). The correlation (Pearson's correlation coefficient) between individual genes in each gene set and the expression of individual microRNAs was determined. The mean correlation level in each gene set was then converted into a Z-score and shown in a heatmap (Fig. 1). In the heatmap, a higher Z-score indicates that the expression level of the microRNA in question is highly correlated with gene expression in the gene set across the glioblastoma samples. Lower Z-scores are indicative of negative correlations between the expression of the microRNA and that of the gene set in the context of the glioblastoma samples. For Supplementary Fig. S4, the 7 gene sets were measured for the extent of enrichment in individual glioblastoma samples by calculating the Z-score as the normalized mean expression intensity of genes in the corresponding glioblastoma sample.
Figure 1. MicroRNAs identify five neural precursor-related glioblastoma subclasses.
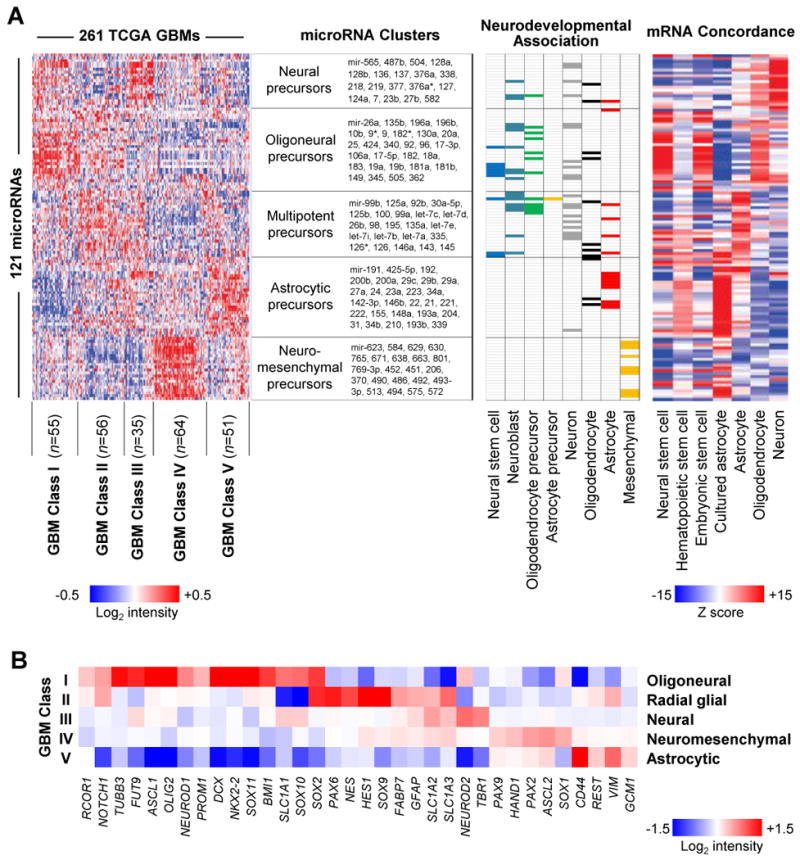
(A) (left) Consensus clustering of 121 highly variable, survival-related or neurodevelopmentally-related microRNAs from 261 glioblastomas revealed five glioblastoma subclasses and five microRNA clusters (see also Supplementary Fig. S3). The neurodevelopmental annotations of the five microRNA clusters are shown along with the microRNAs contained within each cluster. Chart (middle) illustrates the association of selected microRNAs with eight different stages of neural cell differentiation (see also Supplementary Table S2). Heatmap (right) illustrates the extent of correlation between microRNAs and the mRNA signatures of neurons, oligodendrocytes, astrocytes and hematopoietic, embryonic or neural stem cells. Red and blue represent positive and negative correlation, respectively. The order of 121 microRNAs in the chart (middle) and the heatmap (left) is identical to that in the correlation heatmap (right). (B) The mRNA expression level of 34 neural differentiation markers among the five microRNA-based glioblastoma subclasses is illustrated in a heatmap.
For extended functional correlation analysis, we used gene ontology (GO) categories. Gene sets representing GO categories were downloaded from MSigDB (http://www.broadinstitute.org/gsea/msigdb/; c5 GO categories). We further selected 1268 GO terms with ≥ 5 genes and ≤ 500 genes. We first performed parametric gene set enrichment analysis (PAGE) for 1268 GO sets across 121 microRNAs (17). The correlations between 121 microRNAs and 11861 mRNAs were then measured and used to calculate a Z-score. Then, the significance of the degree of enrichment (the Z-score) was calculated by PAGE algorithm for individual microRNA and GO category pairs (17). We further selected 1190 GO categories that showed significant enrichment (FDR < 0.05) with at least one microRNA. A matrix was constructed across 1190 GO categories and 121 microRNAs by placing the Z-score only for GO category/microRNA pairs with significant (FDR < 0.05) enrichment. In Fig. 5, hierarchical clustering was performed on the Z-score matrix, and the Dynamic Tree Cut software program was used to group the 1190 GO categories into 15 functional categories according to the correlation similarities (18).
Figure 5. MicroRNAs regulate subclass-specific pathways.
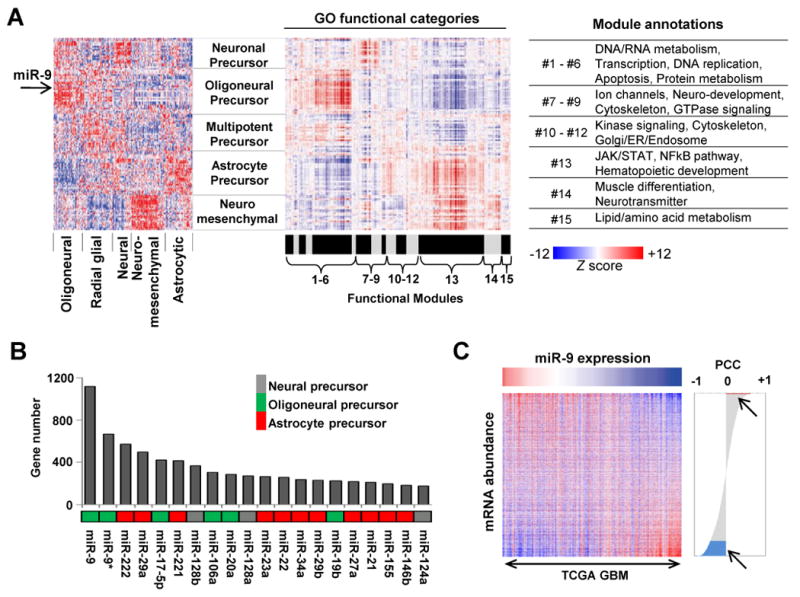
(A) The extent of correlation is calculated between 121 microRNAs and 1190 GO categories and rendered as a heatmap (middle panel). Red and blue represent positive or negative correlation between the corresponding microRNA and genes in the GO category, respectively. The correlation heatmap (middle panel) is shown along with the microRNA expression heatmap generated by consensus clustering (left panel, from Fig. 1A) for comparison. The location of miR-9 is indicated by an arrow. The 1190 GO categories are further separated into 15 modules with their respective functional annotations listed to the right. (B) The number of significantly correlated genes is plotted for the top 20 most highly connected microRNAs. The subclass-specific grouping of individual microRNAs is indicated using different colors. (C) The total number of genes (n = 11,861) is sorted in order of correlation with miR-9, placing the positively and negatively correlated genes at the top and bottom of the list, respectively. The heatmap shows the differences in mRNA expression of the sorted genes relative to the expression of miR-9. In the correlation plot, the red and blue areas (arrows) contain 32 positively- and 1184 negatively-correlated genes, respectively.
Co-expression network and microRNA regulon analysis
The expression profiles of 121 microRNAs and 11861 genes were separately prepared and merged across 197 glioblastoma samples for which both microRNA and mRNA expression data was available. Pearson's correlation coefficients were measured for all possible combinations of entries in a pairwise manner, excluding self-to-self comparisons. The distribution of correlations for three different combinations (microRNA-vs-microRNA, microRNA-vs-gene and gene-vs-gene) is shown in Supplementary Fig. S9. To identify the cutoff of significant correlation for microRNA-vs-gene pairs, we performed 100 permutation tests in which the Pearson correlation was measured for label-permuted expression profiles. The distribution of Min and Max correlation levels in each permutation test is shown in Supplementary Fig. S9. The correlation cutoff for positive and negative correlations between microRNA-vs-gene pairs was set to be the lowest value of upper 5 percentile of Max correlation (+0.60) and the highest value of lower 5 percentile of Min correlation (-0.40), respectively. Applying these cutoffs, we obtained 8752 significant correlations between 74 microRNAs and 2822 mRNAs. We measured the connectivity (i.e., the number of directly connected mRNAs) for individual microRNAs. In the case of miR-9, we collected the predicted miR-9 target genes from 3 different algorithms (miRanda, PicTar and TargetScan). The miRanda miR-9 targets (n = 1283) and TargetScan miR-9 targets (n = 629) were obtained from their websites (http://www.microrna.org and http://www.targetscan.org, respectively) (19, 20). The PicTar (21) miR-9 targets (5-way conserved n = 191 and 4-way conserved n = 491) were obtained from the UCSC Genome Browser (http://genome.ucsc.edu/). Significant enrichment of miR-9 predicted target genes among the total number of genes correlated with miR-9 expression was determined by Fisher's exact test.
Cell culture and reagents
The human U251 glioblastoma cell line was authenticated by and obtained from the American Tissue Type Culture Collection (ATCC) within the last ten years. The cells were expanded by culturing for less than two passages, and were subsequently frozen in liquid nitrogen for storage. For experiments, low passage cells were thawed and used within four months. Primary human CD133+ glioblastoma cancer stem cells were isolated from surgical specimens and expanded as tumorspheres in serum-free medium for less than three passages as described (22), and were subsequently frozen in liquid nitrogen for storage. For experiments, low passage cells were thawed and used within four months. Methods for lentivirus construction, immunoblotting, BrdU proliferation assays and the use of microRNA mimics, inhibitors and controls (100 μM) were as described (22). Antibodies used were STAT3, phospho-STAT3 (Cell Signaling); JAK1, JAK2, JAK3, CD44, GCM1, CEBP-β and β-actin (Abcam).
Results
Consensus clustering using microRNA expression profiles
We obtained microRNA expression profiles for 261 glioblastomas from The Cancer Genome Atlas portal (8). We selected for analysis 121 microRNAs (Supplementary Fig. S1 and Table S1) that demonstrated highly variable expression, were related to patient survival or were previously associated with neural development (23-28) (see Materials and Methods). Consensus clustering of 261 glioblastomas using these 121 microRNAs identified five microRNA clusters and five distinct glioblastoma subclasses (Fig. 1A; Supplementary Fig. S2 and Fig. S3).
A review of published literature (Supplementary Table S2) revealed that four of the five microRNA clusters were enriched for microRNAs expressed in oligoneuronal precursors, multipotent neural precursors, neuronal precursors or astrocytes, respectively (Fig. 1A; middle panel). The remaining cluster contained microRNAs that regulate differentiation and metabolism in both neural and mesenchymal tissues. For example, miR-206, a muscle-enriched microRNA that is also highly expressed in the cerebellum and dorsal root ganglion, inhibits oligodendrocyte and osteoblast differentiation and promotes muscle differentiation (25, 29, 30). MiR-451 is expressed in the cortex and in dorsal root ganglion, regulates invasion and metabolism in glioblastoma and promotes erythroid differentiation (25, 31, 32).
A highly orchestrated and unique progression of microRNA expression accompanies each stage of development. We therefore examined the correlation between the expression of individual microRNAs and several differentiation-related mRNA signatures across the glioblastoma samples, and subsequently rendered these correlations as a heatmap (Fig. 1A; right panel). mRNA signatures representing four murine neural cell types and three human stem cell types were used (see Materials and Methods). MicroRNA expression in the oligoneuronal precursor microRNA cluster correlated with mRNA signatures from oligodendrocytes, embryonic stem cells and neural stem cells. The multipotent precursor cluster was associated with the mRNA expression patterns of astrocytes as well as hematopoietic, embryonic and neural stem cells. The neuronal precursor microRNA cluster was associated with neuronal and oligodendrocyte mRNA expression patterns. The neuromesenchymal microRNA cluster was associated with the mRNA signature of cultured astrocytes and, to a lesser degree, embryonic and neural stem cells. The astrocytic microRNA cluster was accompanied by mRNA patterns characteristic of cultured astrocytes and neural/hematopoietic stem cells. Thus, both the microRNA and mRNA signatures associated with each microRNA cluster corresponded to a specific stage of neural precursor differentiation.
These five differentiation-related microRNA clusters contributed in various combinations to define five subclasses of glioblastoma, suggesting a relationship between each subclass and a distinct stage of neural differentiation. We therefore examined the expression of mRNAs for stage-specific markers of neural differentiation in each microRNA-based glioblastoma subclass (Fig. 1B). Subclass I, which was dominated by expression of the oligoneuronal precursor microRNA cluster, showed increased oligodendrocyte/neuronal precursor markers such as NKX2-2, OLIG2, SOX2, SOX10, SLC1A1 and ASCL1. Subclass II showed increased expression of the astrocytic, oligoneural and multipotent precursor microRNA clusters and expressed numerous radial glia markers, including PAX6, SOX2, NES, FABP7, SLC1A3, GFAP and others. Subclass III displayed increased expression of the neuronal precursor microRNA cluster and expressed neuronal markers such as TBR1, SLCA1 and NEUROD2. Subclass IV showed increased expression of the neuromesenchymal microRNA cluster, was enriched for SOX1, PAX2, PAX9 and HAND1, and displayed modest upregulation of radial glia markers. SOX1 inhibits the differentiation of neuroectoderm into radial glia (33). HAND1, PAX2 and PAX9 are expressed in cephalic neural crest precursors (34-37) and regulate their neuromesenchymal differentiation repertoire (i.e. neurons, glia, melanocytes, osteocytes, chondrocytes and myofibroblasts). Subclass V showed increased expression of the astrocytic microRNA cluster and astrocyte markers such as GCM1 and REST (38, 39). Furthermore, sample-wise gene expression in the five glioblastoma subclasses was closely related to the expression signatures of the differentiated progeny of the parent neural precursors (Supplementary Fig. S4). Based upon these findings, we have named each glioblastoma subclass according to its associated stage of neural precursor cell differentiation, i.e. ‘oligoneural’, ‘radial glial’, ‘neural’, ‘neuromesenchymal’, and ‘astrocytic’.
Clinical characteristics of GBM subclasses
When compared to the glioblastoma subclasses identified using mRNA consensus clustering (4), the microRNA-based oligoneural, radial glial and astrocytic subclasses were enriched in tumors from the mRNA-based proneural, classical and mesenchymal subgroups, respectively (Fig. 2A). However, 20 to 50% of the tumors in the mRNA-based subclasses were reclassified into other groups using microRNA expression. The neural and neuromesenchymal subclasses contained a mixture of tumors from all four mRNA-based glioblastoma subclasses.
Figure 2. Distinct clinical characteristics define glioblastoma subclasses.
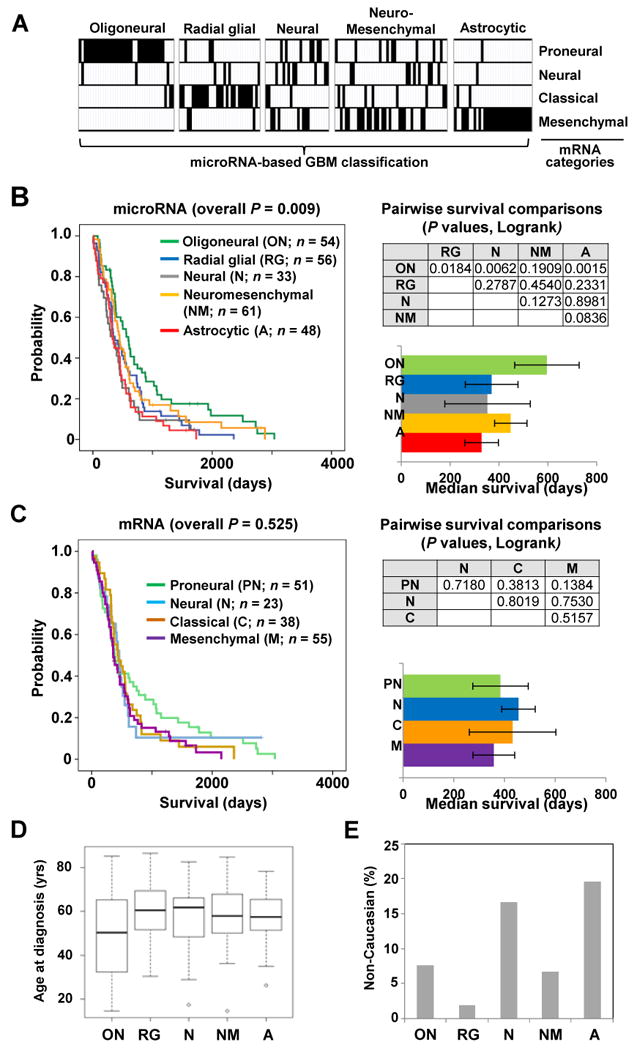
(A) The relationship between the microRNA-based glioblastoma subclassification scheme (vertical columns) and the previously published mRNA-based subclassification (horizontal rows) is shown using the same tumors for analysis. (B) (left) Kaplan-Meier survival plots for the five microRNA-based glioblastoma subclasses. (upper right) Table listing P values for survival differences calculated in a pairwise manner between microRNA-based subclasses. (lower right) The median survival of individual microRNA-based subclasses (error bar represents standard error of the mean). (C) Kaplan-Meier survival plots are shown for four mRNA-based glioblastoma subclasses (left) with the significance of survival differences between subclasses (upper right) and median survival (lower right). (D) Box plot illustrates the mean age at diagnosis for five microRNA-based glioblastoma subclasses (P < 0.001, ANOVA). (E) Graph illustrates percentage of Non-Caucasian (Asian and Black) patients in each glioblastoma subclass (P = 0.021, Fisher's exact).
We observed that microRNA-based consensus clustering yielded robust survival differences among glioblastoma subclasses (Fig. 2B, P = 0.009, Logrank). Patients with oligoneural glioblastomas lived significantly longer than patients with radial glial (P = 0.018, Logrank), neural (P = 0.006, Logrank) or astrocytic tumors (P = 0.002, Logrank), and those with neuromesenchymal glioblastomas showed a trend toward longer survival (P = 0.084, Logrank) when compared to patients with astrocytic tumors (Fig. 2B). In contrast to our findings, previous reports using mRNA-based consensus clustering failed to identify significant survival differences among glioblastoma subclasses (4, 40). Consistent with these reports, we also do not observe significant survival differences among mRNA-based glioblastoma subclasses (Fig. 2C). Detailed analysis revealed that the difference derives primarily from a 50% increase in median survival for the microRNA-based oligoneural subclass when compared to the median survival for the mRNA-based proneural subclass (see Fig. 2B and Fig. 2C). When the proneural subclass was further divided into those samples that were also categorized as oligoneural and those that were not, we observed a significant survival advantage for proneural tumors that were also designated as oligoneural (P = 0.039, Logrank; Supplementary Fig. S5). Furthermore, we continued to observe significant survival differences between glioblastoma subclasses when clustered using only the highly variable (n = 45; Logrank P = 0.001), survival-related (n = 58; Logrank P = 0.002) or neurodevelopment-related (n = 57; Logrank P = 0.043) microRNA subsets as independent data sets (Supplementary Fig. S6).
The age at diagnosis was significantly different (P = 0.011, one-way ANOVA) among glioblastoma subclasses. On average, patients with oligoneural glioblastomas developed disease at a younger age (Fig. 2D; Supplementary Fig. S7) (4, 5). Additionally, we observed significant racial differences across microRNA-based glioblastoma subclasses (P = 0.021, Fisher's exact) (Fig. 2E; Supplementary Fig. S7). An increased percentage of non-Caucasian patients was observed among the neural and astrocytic subclasses when compared to the radial glial subclass (P = 0.03, Proportion test).
For each subclass, we compared the clinical response of patients treated with radiation (at least 54 Gy) and temozolamide (2 or more cycles administered separately or concurrent with radiation) to that of patients treated with all other regimens (Fig. 3A). A significant survival benefit of radiation and temozolamide was observed for patients with tumors in the astrocytic subclass, but not for those with tumors in the oligoneural, neural or neuromesenchymal subclasses. A trend toward improved survival was observed in the radial glial subclass (P < 0.085, Logrank), and this trend became significant (P < 0.014, Logrank) after exclusion of a single long-surviving outlier.
Figure 3. Subclass-specific treatment response and the effect of MGMT methylation on survival.
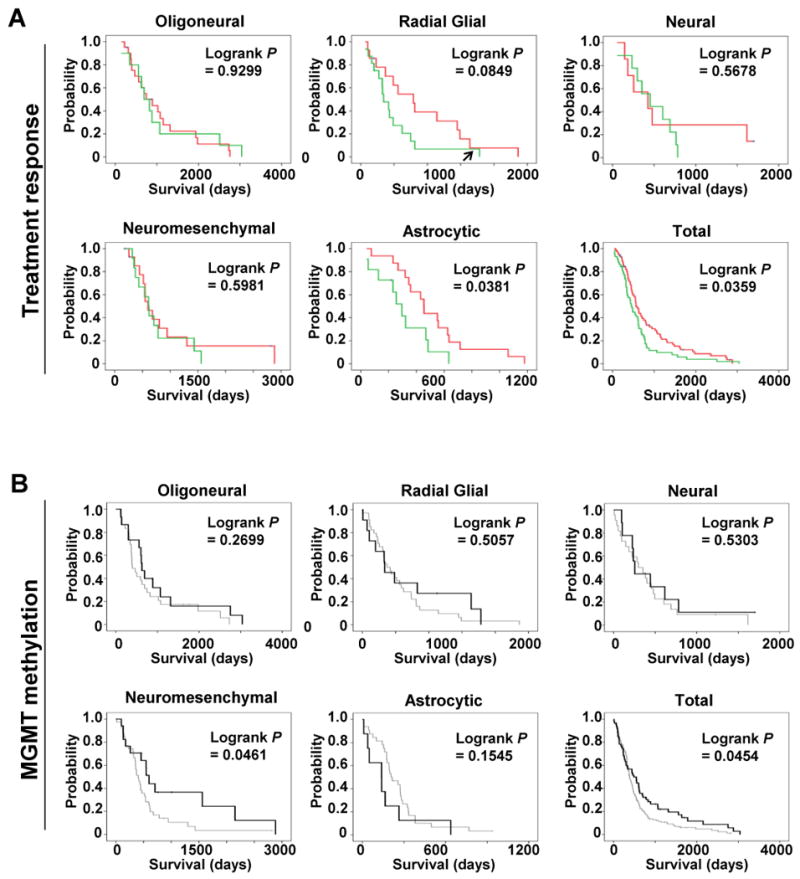
(A) Kaplan-Meier survival analyses for glioblastoma patients in each subclass or for the entire cohort are shown. The survival of patients treated with radiation (at least 54 Gy) and two or more cycles of temozolamide (red) versus all other regimens (green) are distinguished. Survival was calculated from the date of diagnosis. The arrow in the radial glial plot identifies a long-surviving outlier which, if excluded, P < 0.014, Logrank. (B) Kaplan-Meier survival analyses for glioblastoma patients in each subclass or for the entire cohort are shown. The survival of patients with tumors harboring MGMT promoter methylation was compared to those with tumors lacking MGMT methylation in each glioblastoma subclass. Black and grey lines represent the survival of patients with tumors with or without MGMT methylation, respectively.
We also analyzed the relationship between MGMT promoter methylation and patient survival in each of the microRNA subclasses (Fig. 3B). Overall, we observed that tumors harboring MGMT promoter methylation have a more favorable prognosis (P = 0.045, Logrank). However, individual subclasses showed differential patterns of survival with respect to MGMT promoter methylation. In particular, a significant association between MGMT promoter methylation and longer survival was observed in the neuromesenchymal glioblastoma subclass (P = 0.046, Logrank).
Mutation and copy number profiles of GBM subtypes
Each microRNA-based glioblastoma subclass displayed a distinct pattern of somatic mutations, some of which were observed in studies using mRNA for glioblastoma subclassification (Fig. 4A; Supplementary Table S3) (4). The oligoneural subclass was enriched for IDH1 and PIK3R1 mutations, but lacked NF1 mutations. Interestingly, six of seven “hypermutator” tumors (8) were grouped into the oligoneural subclass. EGFR mutations were more common in the radial glial subclass. The neuromesenchymal subclass was enriched for PTEN and FKBP9 mutations but lacked RB1 mutations, while the astrocytic subclass was enriched for PTEN and RB1 mutations.
Figure 4. Distinct genetic alterations characterize glioblastoma subclasses.
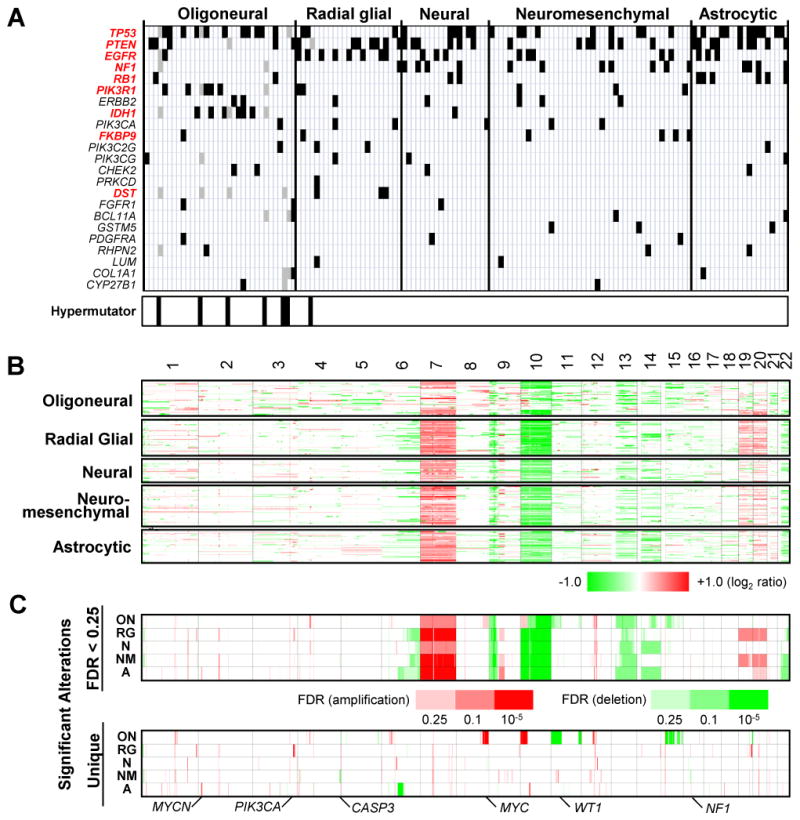
(A) The distribution of somatic mutations is shown for 23 frequently mutated genes across five glioblastoma subclasses (black bars). Genes with significant differences in mutation frequency (P < 0.1, Fisher Exact test) are indicated in red. Each vertical column contains data from a single tumor. The location of seven hypermutator tumors is indicated at the bottom of the figure. Hypermutator mutations (gray bars) were excluded from significance calculations. (B) Genome-wide copy number alterations for 261 glioblastomas across five subclasses are illustrated in a heatmap. Red and green represent copy number gains and losses, respectively. (C) (upper panel) Significant alterations (false discovery rate or FDR < 0.25) are shown for five glioblastoma subclasses. The significance of recurrent alterations were measured using the GISTIC algorithm. (lower panel) Significant subclass-specific DNA alterations exclusive to each glioblastoma subclass are shown.
Each subclass also displayed a unique pattern of copy number alterations (Fig. 4B and 4C; Supplementary Fig. S8 and Table S4). For example, MYC, PIK3CA, WT1, and MYCN were amplified in the oligoneural, radial glial, neuromesenchymal, and astrocytic subclasses, respectively, while loci containing CASP3 and NF1 were deleted primarily in the neuromesenchymal subclass.
Integrative functional analysis using microRNA and mRNA profiles
To identify pathways activated in each glioblastoma subclass, we measured the extent of correlation between the expression of individual microRNAs and that of mRNAs in 1190 curated GO categories. The GO categories were further grouped into 15 modules based upon hierarchical clustering of the correlation matrix linking the individual microRNAs with their putative functions (Fig. 5A). From this analysis, we inferred upregulation of proliferative and neurodevelopmental pathways in the oligoneural, radial glial and neural subclasses, and downregulation of RNA and DNA metabolism in the neuromesenchymal and astrocytic subclasses. Importantly, the neuromesenchymal and astrocytic subclasses displayed upregulation of the NFkB and JAK/STAT pathways.
MicroRNA contribution to subclass phenotype
Analysis of microRNA-mRNA correlations (41, 42) revealed significant correlations between 74 microRNAs and 2822 genes (Supplementary Fig. S9). Some microRNAs (such as miR-9, miR-9* and miR-222) displayed disproportionately high connectivity (Fig. 5B and 5C), suggesting that they might serve as core regulators of subclass-specific gene expression in glioblastoma. To investigate this possibility, we focused on miR-9, which was upregulated in the oligoneural subclass and identified as the microRNA with the largest correlated gene expression network. Genes correlated with miR-9 showed significant enrichment for predicted miR-9 target genes (Supplementary Fig. S9). Importantly, miR-9 promotes neural stem cell differentiation and cooperates with miR-124a to inhibit STAT3 phosphorylation in neural stem cells by an unknown mechanism (24). We used microRNA target prediction algorithms (http://www.targetscan.org; http://www.microrna.org) to identify putative binding sites for miR-9 in the 3′-UTR of JAK1, JAK2 and JAK3, all of which phosphorylate STAT3. MiR-9 was anti-correlated with JAK2 and JAK3 mRNA expression in glioblastoma (Pearson's correlation coefficient or PCC of -0.34 and -0.56, respectively), and a miR-9 mimic decreased expression of luciferase mRNA fused to the JAK1 or JAK2 3′-UTR (Supplementary Fig. S10). In human U251 glioblastoma cells or in CD133+ glioblastoma cancer stem cells (GCSCs) obtained from surgical specimens, exposure to a miR-9 mimic (100 μM) or lentiviral-mediated overexpression of miR-9 decreased JAK1, JAK2 and JAK3 protein expression (Fig. 6A). Furthermore, miR-9 alone (but not miR-124a) (25) decreased STAT3 phosphorylation in U251 glioblastoma cells and in primary human CD133+ glioblastoma stem-like cells, and it also decreased expression of the STAT3 transcriptional target, CEBP-β, in CD133+ glioblastoma cells (Fig. 6B). An essential role for STAT3 and CEBP-β in maintaining the mesenchymal phenotype in glioblastoma has been reported (43). Accordingly, the miR-9 mimic decreased expression of astrocytic/mesenchymal markers (GCM1, CD44 and GFAP), increased expression of the neuronal marker, TuJ1 (Fig. 6C) and inhibited GCSC proliferation (Fig. 6D).
Figure 6. miR-9 regulates subclass differentiation in glioblastoma stem cells.
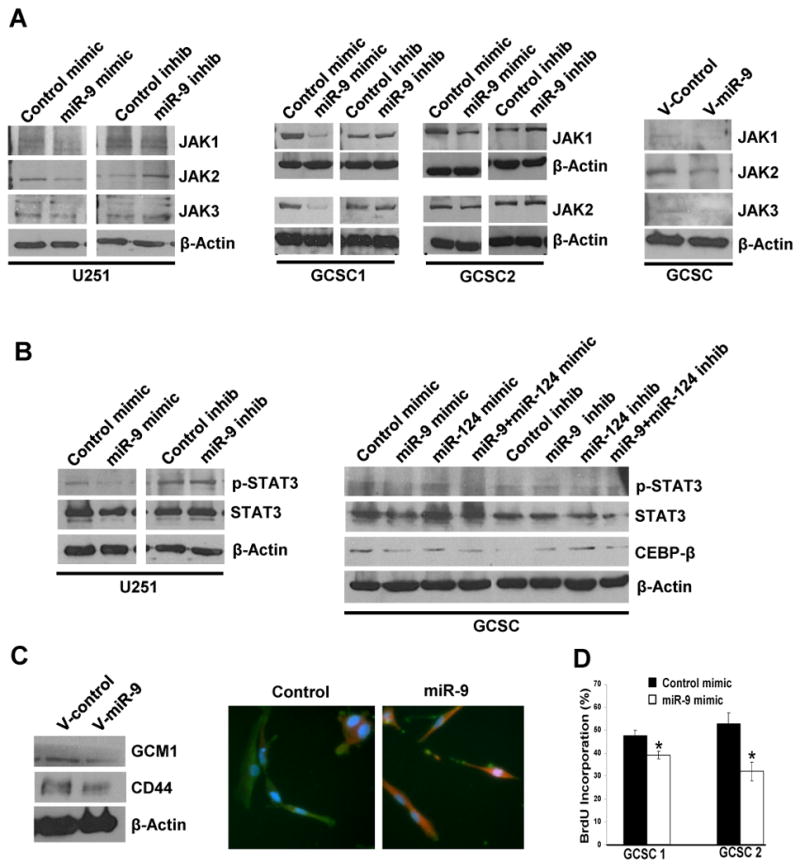
(A) (left and middle panels) Western blot demonstrating JAK1, JAK2 and JAK3 protein expression in established U251 human glioblastoma cells or in primary human glioblastoma cancer stem cells (GCSCs) after exposure to the miR-9 mimic, miR-9 inhibitor or appropriate control oligonucleotides (100 μM). (right panel) JAK1, JAK2 or JAK3 protein expression in GCSCs after transduction using control (V-Control) or pri-miR-9 (V-miR-9) lentivirus. (B) (left panel) STAT3 and phosphorylated STAT3 in established U251 human glioblastoma cells after exposure to miR-9 mimic or inhibitor. (right panel) STAT3, phosphorylated STAT3 and CEBP-β protein expression in GCSCs after exposure to miR-9 or miR-124a mimic, inhibitor or oligonucleotide controls. (C) (left panel) Western blot showing Gcm1 and CD44 protein expression in GCSCs transduced with a control (V-Control) or pri-miR-9 (V-miR-9) lentivirus. (right panel) GFAP (green) and TuJ1 (red) immunoreactivity in GCSCs after exposure to miR-9 mimic (100μM) or an oligonucleotide control. (D) BrdU proliferation assay illustrating GCSC proliferation after exposure to miR-9 mimic (100μM) or an oligonucleotide control (P = 0.04, unpaired t-test).
Other developmentally-regulated microRNAs also contribute to glioblastoma subclass maintenance. For example, we identified miR-124a as a hub microRNA in the neural glioblastoma subclass (see Fig. 5B). This microRNA has been reported to play an instructive role during neuronal differentiation of neural precursors (25), and we and others find that it induces neuronal differentiation and inhibits growth in GCSCs (44).
Discussion
MicroRNAs reveal a greater diversity of glioblastoma subclasses than previously recognized. We identified five glioblastoma subclasses with concordant microRNA and mRNA expression signatures corresponding to each major stage of neural stem cell differentiation. This marked degree of correspondence provides some of the strongest evidence yet in humans that glioblastomas arise from the transformation of neural precursors, as suggested by animal studies (45). Importantly, the signatures correspond to neural precursors at multiple stages of differentiation, suggesting that glioblastomas can arise from cells at each of these stages. Our finding that the largest glioblastoma subclass displays a neuromesenchymal signature resembling that of early neuroepithelial or cephalic neural crest precursors is supported by reports of neuromesenchymal differentiation in CD133+ GCSCs from recurrent glioblastomas (46). The latter result raises the possibility that this signature results from oncogenic reprogramming to a neuromesenchymal-like state (5, 44, 46).
These observations place previously reported effects of microRNAs on glioblastoma growth (23, 32, 44, 47) into a neurodevelopmental context, and reveal that microRNA-dependent regulation of growth and differentiation programs contributes significantly to glioblastoma diversification and patient outcome. The importance of this phenomenon is underscored by the fact that microRNA-defined glioblastoma subclasses display robust differences in genetic alterations, patient demographics, response to treatment and patient survival (Supplementary Fig. S11).
Consistent with previous reports (4, 40), we observed that mRNA-based glioblastoma subclasses do not exhibit significant survival differences. In contrast, microRNA-based glioblastoma subclasses showed robust survival differences among them. Although the mRNA-based proneural subclass has been associated with longer survival (5), our data shows that patients with proneural tumors can be further segregated into two subgroups with significant survival differences using microRNA-based consensus clustering. These findings indicate that the mRNA-based proneural subclass represents a heterogeneous population in terms of survival. This observation is supported by a recent study examining DNA methylation in glioblastoma, which identified a subpopulation of proneural tumors with a hypermethylation phenotype and prolonged survival (40). Such heterogeneity may be partially responsible for the previous inability to build a prognostic model using mRNA expression data (4, 40).
We observed that all of the initial microRNA subsets used in this study (i.e. highly variable (n = 45), neurodevelopmental (n = 57) and survival-related (n = 58) microRNA) yield significant survival differences among glioblastoma subclasses when used independently to cluster the glioblastoma samples (Supplementary Fig. S6). This analysis also revealed that each of the five microRNA-based glioblastoma subclasses was distinguished in at least one of the three alternative clustering paradigms, and four of the five subclasses were identified in at least two of the clustering paradigms, despite the fact that roughly one half to one third the original number of microRNAs was used each time (Supplementary Fig. S6). The neural subclass was only identified clearly using the neurodevelopmental microRNA subset. Importantly, clear evidence for the newest subclass of glioblastoma (neuromesenchymal) was obtained using either the survival-related or the neurodevelopment-related microRNA subsets alone for clustering glioblastoma samples. Thus, the survival and development-related differences observed in our study are not due solely to the inclusion of one (i.e. survival-related or development-related) of the microRNA subsets.
The difference in racial composition observed among glioblastoma subclasses is of particular interest, given the existence of racial differences in the overall incidence of gliomas. Similarly, the finding that only a subset of glioblastoma subclasses responds to standard treatment (i.e. radiation and temozolamide) agrees with a previous report (4) and has significant clinical implications for treatment. We also observed that, overall, MGMT promoter methylation is associated with a more favorable prognosis. This effect is most pronounced in the neuromesenchymal subclass of glioblastoma, and may contribute to the relatively prolonged median survival observed in this glioblastoma subclass. Surprisingly, however, this subclass did not demonstrate responsiveness to temozolamide and radiation. The reasons for this discrepancy are unclear. One possibility is that, in addition to increasing the response to standard therapy, MGMT promoter methylation may be associated with the presence of other molecular changes that contribute to improved survival. Indeed, MGMT methylation is known to promote the acquisition of specific types of mutations that regulate growth in glioblastoma (8).
Although genomic alterations involving microRNAs and their upstream regulators occur in glioblastoma (22), our findings indicate that developmental programs underlie the overall framework of microRNA expression in this cancer. Numerous reports have described important roles for microRNAs in regulating the growth of glioblastoma and other cancers (22, 31, 44, 47). Until now, these reports were interpreted independently. However, our data reveal that many of these “aberrant” microRNA expression patterns and microRNA-target interactions derive from highly coordinated, differentiation-related microRNA expression programs. Importantly, we find that miR-9 downregulates the JAK/STAT pathway and serves as a switch that regulates oligoneural versus mesenchymal decisions in glioblastoma. In addition, we and others have found a role for miR-124 (which is highly expressed in the neural glioblastoma subclass) in promoting neuronal differentiation and decreasing growth in glioblastoma. Thus, our findings reveal a new role for microRNAs in organizing and maintaining the phenotypic and molecular architecture of glioblastoma subclasses.
MicroRNAs are thus useful for subclassifying glioblastomas in a manner that allows for more accurate prognosis and for the development of molecular-based treatment decisions. Taken together, these findings support the adoption of a microRNA-based, neurodevelopmental taxonomy for glioblastoma. The use of such a classification system may aid in prognosis and in the selection of subclass-specific therapies that will improve outcome for glioblastoma patients.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by an NIH Director's New Innovator Award (DP2OD002319), a Brain Science Foundation Research Grant and a Hagerty Fund Research Award to MDJ, and by NIH 1U24 CA126554 (TCGA) to PJP.
References
- 1.DeAngelis LM. Brain tumors. N Engl J Med. 2001;344:114–23. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 2.Behin A, Hoang-Xuan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet. 2003;361:323–31. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- 3.Chin L, Gray JW. Translating insights from the cancer genome into clinical practice. Nature. 2008;452:553–63. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 7.Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–9. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monti S, Tamayo P, Mesirov J, Golub TR. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52:91–118. [Google Scholar]
- 10.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 11.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–8. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 13.Kim TM, Yim SH, Jeong YB, Jung YC, Chung YJ. PathCluster: a framework for gene set-based hierarchical clustering. Bioinformatics. 2008;24:1957–8. doi: 10.1093/bioinformatics/btn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheadle C, Watkins T, Fan J, et al. GSMA: Gene Set Matrix Analysis, An Automated Method for Rapid Hypothesis Testing of Gene Expression Data. Bioinform Biol Insights. 2009;1:49–62. [PMC free article] [PubMed] [Google Scholar]
- 15.Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24:719–20. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 19.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 22.Kim H, Huang W, Jiang X, Pennicooke B, Park PJ, Johnson MD. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci U S A. 2010;107:2183–8. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci. 2008;28:11720–30. doi: 10.1523/JNEUROSCI.1932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006;24:857–64. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua YJ, Tang ZY, Tu K, et al. Identification and target prediction of miRNAs specifically expressed in rat neural tissue. BMC Genomics. 2009;10:214. doi: 10.1186/1471-2164-10-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapsimali M, Kloosterman WP, de Bruijn E, Rosa F, Plasterk RH, Wilson SW. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007;8:R173. doi: 10.1186/gb-2007-8-8-r173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen JA, Lau P, Maric D, Barker JL, Hudson LD. Integrating microRNA and mRNA expression profiles of neuronal progenitors to identify regulatory networks underlying the onset of cortical neurogenesis. BMC Neurosci. 2009;10:98. doi: 10.1186/1471-2202-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006;174:677–87. doi: 10.1083/jcb.200603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehotzky A, Lau P, Tokesi N, Muja N, Hudson LD, Ovadi J. Tubulin polymerization-promoting protein (TPPP/p25) is critical for oligodendrocyte differentiation. Glia. 2010;58:157–68. doi: 10.1002/glia.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godlewski J, Nowicki MO, Bronisz A, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37:620–32. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dore LC, Amigo JD, Dos Santos CO, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci U S A. 2008;105:3333–8. doi: 10.1073/pnas.0712312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suter DM, Tirefort D, Julien S, Krause KH. A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells. 2009;27:49–58. doi: 10.1634/stemcells.2008-0319. [DOI] [PubMed] [Google Scholar]
- 34.Calloni GW, Le Douarin NM, Dupin E. High frequency of cephalic neural crest cells shows coexistence of neurogenic, melanogenic, and osteogenic differentiation capacities. Proc Natl Acad Sci U S A. 2009;106:8947–52. doi: 10.1073/pnas.0903780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makori N, Peterson PE, Wei X, Hummler H, Hendrickx AG. 13-cis-Retinoic acid alters neural crest cells expressing Krox-20 and Pax-2 in macaque embryos. Anat Rec. 1999;255:142–54. doi: 10.1002/(SICI)1097-0185(19990601)255:2<142::AID-AR4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 36.Firulli AB. A HANDful of questions: the molecular biology of the heart and neural crest derivatives (HAND)-subclass of basic helix-loop-helix transcription factors. Gene. 2003;312:27–40. doi: 10.1016/s0378-1119(03)00669-3. [DOI] [PubMed] [Google Scholar]
- 37.Kist R, Greally E, Peters H. Derivation of a mouse model for conditional inactivation of Pax9. Genesis. 2007;45:460–4. doi: 10.1002/dvg.20295. [DOI] [PubMed] [Google Scholar]
- 38.Kohyama J, Sanosaka T, Tokunaga A, et al. BMP-induced REST regulates the establishment and maintenance of astrocytic identity. J Cell Biol. 2010;189:159–70. doi: 10.1083/jcb.200908048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwasaki Y, Hosoya T, Takebayashi H, Ogawa Y, Hotta Y, Ikenaka K. The potential to induce glial differentiation is conserved between Drosophila and mammalian glial cells missing genes. Development. 2003;130:6027–35. doi: 10.1242/dev.00822. [DOI] [PubMed] [Google Scholar]
- 40.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volinia S, Visone R, Galasso M, Rossi E, Croce CM. Identification of microRNA activity by Targets' Reverse EXpression. Bioinformatics. 2010;26:91–7. doi: 10.1093/bioinformatics/btp598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006;1:662–71. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 43.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silber J, Lim DA, Petritsch C, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacques TS, Swales A, Brzozowski MJ, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010;29:222–35. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, Nguyen DH, Dong Q, et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J Neurooncol. 2009;94:1–19. doi: 10.1007/s11060-009-9919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silber J, James CD, Hodgson JG. microRNAs in gliomas: small regulators of a big problem. Neuromolecular Med. 2009;11:208–22. doi: 10.1007/s12017-009-8087-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.