

Abstract
Histones, the fundamental packaging elements of eukaryotic DNA, are highly decorated with a diverse set of post-translational modifications (PTMs) that are recognized to govern the structure and function of chromatin. Ten years ago, we put forward the histone code hypothesis, which provided a model to explain how single and/or combinatorial PTMs on histones regulate the diverse activities associated with chromatin (e.g. gene transcription). At that time, there was a limited understanding of both the number of PTMs that occur on histones as well as the proteins that place, remove and interpret them. Since the conception of this hypothesis, the field has witnessed an unprecedented advance in our understanding of the enzymes that contribute to the establishment of histone PTMs, as well as the diverse effector proteins that bind them. While debate continues as to whether histone PTMs truly constitute a strict “code”, it is becoming clear that PTMs on histone proteins function in elaborate combinations to regulate the many activities associated with chromatin. In this special issue, we celebrate the 50th anniversary of the landmark publication of the lac operon with a review that provides a current view of the histone code hypothesis, the lessons we have learned over the last decade, and the technologies that will drive our understanding of histone PTMs forward in the future.
“Small changes modifying the distribution in time and space of the same structures are sufficient to affect deeply the form, the functioning, and the behavior of the final product…. It is always a matter of using the same elements, of adjusting them, of altering here or there, of arranging various combinations to produce new objects of increasing complexity. It is always a matter of tinkering.”
– François Jacob, “Evolution and Tinkering” (Science 1977)
The adult animal was in actuality the final product that François Jacob was referring to in this eloquent statement taken from his article “Evolution and Tinkering”.1 Yet, as chromatin biologists, we delight in the applicability of Jacob’s quote regarding the plasticity of a single template to the chromatin landscape. However, François Jacob is not best known for his theories on how patterns of gene expression affect evolution, but rather for his seminal work with Jacques Monod establishing the basis of the lac operon. In celebration of the 50th anniversary of François Jacob and Jacques Monod’s landmark publication on the lac operon2, we are honored to contribute this piece in which we reflect on how several of the scientific themes put forward by Jacob and Monod in their historic work are widely applicable to topics as diverse as chromatin biology and the histone code hypothesis.
In simplistic terms, an operon is a functional genomic unit comprised of a cluster of genes that is controlled by a single regulatory element or promoter.3 Complementary genetic and biochemical studies revealed that the basic principle underlying the lac operon is that the coordinated expression of the genes necessary to metabolize lactose is under the control of the lac repressor protein and activator protein CAP, which negatively and positively control transcription of the lac operon, respectively.2 From the pioneering studies on the lac operon completed by Jacob and Monod, we now know that there are three major types of regulatory DNA sequences that function in the control of gene expression: (1) promoter sequences to which RNA polymerase binds; (2) operator sequences to which transcriptional repressors bind; and (3) positive control elements to which transcriptional activator proteins bind.4 While the lac operon provides a simple yet elegant mechanism by which gene expression is controlled in prokaryotes, it is unreasonable to think that such a system would adequately provide a means by which efficient regulation of gene expression could occur in eukaryotes, where DNA must be highly compacted to fit within the confines of the nuclear space. The need for differential patterns of gene expression to specify diverse types of tissues from a single genome in multicellular organisms also calls for the existence of additional regulatory mechanisms. For example, cellular identity must be faithfully maintained through cell divisions for a lifetime, despite differentiation occurring earlier during embryonic development. The plasticity of cellular differentiation and the stability of cellular memory are thought to represent epigenetic phenomena wherein inherited changes in phenotype occur independently of changes in the underlying DNA sequence and without the need for trans-factors that establish the initial programs of coordinated gene regulation. Hence, while the historic work of Jacob and Monod reveals an elegant mechanism for prokaryotic gene regulation, it is clear that more sophisticated means of gene regulation involving components that do more than engage the DNA template alone are necessary for processes such as cellular memory in multicellular eukaryotes.
Based on many insightful studies on chromosome structure, we know that in eukaryotes, DNA is assembled on a histone scaffold to form chromatin.5 The nucleosome core particle, or fundamental repeating unit of chromatin, consists of approximately 147 base pairs of DNA wrapped around an octamer containing one tetramer of histones H3 and H4 (two copies each) and two histone H2A-H2B dimers.5; 6; 7; 8 Nucleosomes are packaged into progressively higher-order structures to ultimately form chromosomes. Chromatin structure largely affects DNA-templated processes such as transcription, thus necessitating that access to DNA be tightly controlled to allow factors that function in such processes to make appropriate contacts with the DNA template itself.5; 9 Post-translational modifications (PTMs) to the histone proteins themselves can significantly affect the levels of chromatin compaction by creating generally condensed “heterochromatic” or more open “euchromatic” regions, and therefore provide a means by which rapid and localized access to DNA can be accomplished.10; 11 Additionally, other well-studied mechanisms, such as ATP-dependent chromatin remodeling and the exchange of primary sequence histone variants, introduce meaningful variation into the chromatin polymer, “tinkering” in such a way that one relatively stable genome can give rise to the demands of multicellular development.12; 13; 14
The ‘histone code hypothesis’: the first ten years
In 2000, we proposed what has commonly come to be referred to as the ‘histone code hypothesis’, which, in its original form, posits that “multiple histone modifications, acting in a combinatorial or sequential fashion on one or multiple histone tails, specify unique downstream functions”.15 Parallels to François Jacob’s quote from “Evolution and Tinkering” are readily apparent. The same fixed set of amino acids that make up the histone proteins have the potential of being post-translationally modified within the chromatin template, where distinct spatiotemporal patterns of modifications ultimately shape functional outcome. One of the more striking phenomena predicted by such a code is that subtle variations to the same template can result in vastly different outcomes, especially in the context of regulation of gene expression.
At the time that we proposed the histone code hypothesis, we had a limited understanding of the true breadth of the number and type of PTMs that exist on histone residues either on the unstructured N-terminal tails that protrude from the nucleosomal surface or within the structured globular domains. Acetylation and phosphorylation were the best-characterized modifications at that time, with multiple sites and several of the enzymes responsible for their placement and removal having been identified. However, investigations on the dynamics of histone methylation were in their infancy. Only a handful of sites modified by methylation were known at the time, and the function of histone methylation was largely unclear, primarily because the enzyme systems responsible for the steady-state balance of methyl marks (histone methyltransferases and demethylases) were not yet identified and the intricacies associated with a modification that could exist in multiple states (mono-, di-, or trimethyl) complicated studies. Insight into other modifications was even more rudimentary. Today, we know that a number of PTMs exist, including acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP-ribosylation, proline isomerization, citrullination, butyrylation, propionylation, and glycosylation (Table 1).11; 16; 17 Numerous studies using both biochemical and genetic approaches have revealed many of the enzymes that are responsible for placement or removal of these modifications on specific amino acid residues on histones as well as non-histone proteins. While the functional significance of some of these modifications remains to be determined, the collective field of chromatin biologists has made great strides toward identifying the biological consequence of others. For example, modifications can disturb contacts between histones in contiguous nucleosomes or histones with DNA, resulting in alteration of higher-order chromatin structure. Specifically, acetylation of lysine residues on histone tails neutralizes the basic charge of the residue on which it occurs, thereby disrupting histone contacts with other histones and/or DNA and in turn chromatin compaction.9 While it had been known that histone modifications such as methylation did not disrupt nucleosomal contacts by altering the charge of the modified residue, we now know that specialized domains within effector proteins facilitate recognition and binding to methyl marks in a defined state on specific residues to mediate downstream effects. Domains characterized thus far as being able to bind to methylated residues include chromodomains, tudor domains, PHD fingers, MBT domains, Ankyrin repeats, PWWP domains, HEAT domains and WD40 repeats (Table 1).18; 19; 20; 21; 22 Other domains that recognize and bind to specifically modified histone forms have also been characterized. For instance, where bromodomains can bind to acetylated lysine residues, 14-3-3, BRCT, and BIR domains can bind to phosphorylated threonine and serine residues (Table 1).19; 23
Table 1.
Histone modification types and the interacting domains that “read” them
| Modification types | Residue(s) modified | Reader domain(s) |
|---|---|---|
| Unmodified lysine | Lysine | PHD |
| Acetylation | Lysine | Bromo |
| Methylation | Lysine/Arginine | Ankyrin, Chromo, HEAT, MBT, PHD, Tudor, PWWP, WD40 |
| Phosphorylation | Serine/Threonine | 14-3-3, BIR, BRCT |
| Ubiquitylation | Lysine | ? |
| Sumoylation | Lysine | ? |
| ADP-ribosylation | Lysine | ? |
| Citrullination | Arginine | ? |
| Butyrylation | Lysine | ? |
| Propionylation | Lysine | ? |
| Glycosylation | Serine/Threonine | ? |
The chromatin-modifying enzymes that facilitate alterations to the chromatin landscape by placing, removing, or interpreting modifications to establish variable states have been more recently come to be generally referred to as writers, erasers, and readers, respectively, of the histone code (Figure 1). Returning to the idea of tinkering with chromatin, we are now in a position to appreciate the true potential of a “toolkit”24 of writers, erasers, and readers of the histone code in the establishment of proper spatiotemporal patterns of modifications necessary for cellular identity and function. At defined points, writers place marks on defined histone residues, which are in turn interpreted by readers harboring specialized domains that facilitate recognition and binding to the specific mark of interest to drive the progression of a specific biological phenomenon. At a time when such signaling needs to be terminated, erasers are recruited to their defined target(s) to remove the mark, thereby ending the associated functional outcome of the previously defined reader. Admittedly, the situation is made vastly more complicated by the fact that particular amino acid residues can house more than one type of modification (this is largely true for lysine residues, which can be methylated, acetylated, ubiquitylated, or sumoylated), and that some enzymes can write, erase, or read more than one modification. Moreover, one mark can often recruit multiple effector proteins.25; 26 Such complications, however, support the general notion of tinkering with combinatorial pattern of PTMs to control proper recruitment of effector proteins or complexes in which they reside.
Figure 1. Toolkit for modifying the chromatin template.
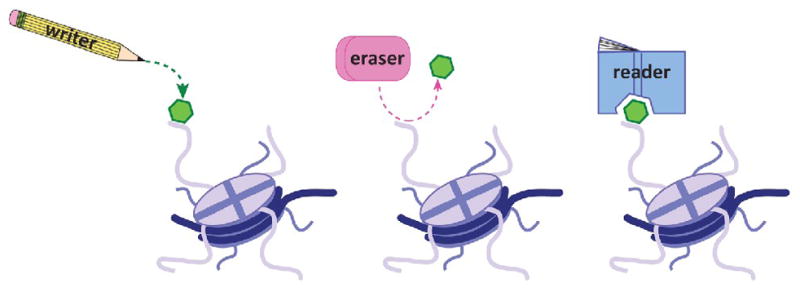
Schematic illustrating the concept that writers place post-translational modifications on histone proteins (left), erasers remove such modifications from histone proteins (middle), and readers function to interpret these covalent modifications (right) to mediate diverse downstream processes.
We appreciate that the ‘histone code hypothesis’, as originally articulated by us in 2000, evolved into an influential review on the function(s) of covalent histone modifications. We acknowledge that this hypothesis, and extensions of it, rest heavily on the foundation of many biologists and biochemists who were dedicated to the general view that chromatin was going to be much more than a passive way to package the genome. However, because of the rapid pace of research in chromatin biology and the complexity associated with chromatin modifications such as those mentioned above, we must continually refine how we define the histone code. In fact, the mere existence of a code in the first place has been a point of contention.27 Beyond discussions in the field as to whether a strict histone code truly exists, there is also debate over whether it is most appropriate to define it as “code” in which definite combinations lead to an absolute outcome (as exemplified by the genetic code). Some see it more in terms of a “language”, where complex combinatorial patterns of modifications form words that ultimately give rise to a vocabulary of histone crosstalk.28 Others yet prefer to think of it more specifically in terms of an “epigenetic code” that is defined by combinations of histone PTMs which are predictive of, and necessary for, expression patterns of differentiation and developmental-specific genes.29 On the other hand, it has been argued that histone modifications are not truly “epigenetic”, as the nature of their heritability (a requisite condition to be defined in the classical sense of epigenetic) is questionable30, thereby disputing the appropriateness of an “epigenetic code”. At some point, the question of how exactly to define the histone code becomes somewhat rhetorical, as at their very essence, all definitions ultimately seem to convey the same fundamental principle that histone PTMs act in concert to elicit downstream biological outcomes. Here we reflect on the many forms the ‘histone code hypothesis’ has come to take since the time of its inception a decade ago, and suggest that individual definitions may not be mutually exclusive of one another, but are perhaps instead complementary.
Transcribing the ‘histone code’: chicken or egg?
Although applicable to a diverse set of cellular processes, the histone code is most commonly considered in the context of transcription regulation. Within this realm, there has been much debate as to whether a putative code formed by combinatorial modifications can formally regulate transcription itself or rather, if patterns of modifications are generally associated with a particular transcriptional state. On one side is the argument that genes are not necessarily regulated by chromatin modifications per se, but rather are regulated by specific DNA-binding proteins that recruit activating and repressive complexes to genomic loci to modulate transcriptional activity. According to this line of reasoning, the histone-modifying machinery is recruited by canonical transcriptional activators and repressors (as would be defined in the classical sense by Jacob and Monod), and the placement of modifications by these enzymes then contributes to transcription by creating a more or less permissive chromatin environment for the further recruitment of downstream factors that regulate transcription. In support of this idea, it has long been known that histone acetylation is associated with active genes31, and functions to facilitate the disruption of higher-order chromatin structure prior to gene activation.9 Thus, one would argue that it is the action of the activators that directly determine transcriptional output, and that the targeting of acetylation to histones via activators that bind to specific upstream activating sequences functions to make the chromatin environment more permissive for transcriptional regulation.9; 32 In an analogous fashion, binding of transcriptional repressors to upstream repressive sequences facilitates recruitment of histone deacetylase (HDAC) enzymes to chromatin, which in turn remove acetyl marks to contribute to transcriptional repression through chromatin compaction.32; 33; 34 By this argument, modifications are thereby associated with gene activation and/or silencing (much like RNA polymerase II (RNAPII) is associated with active genes), but do not formally regulate transcription itself. An extension of this position would be that chromatin modifications themselves do not intrinsically regulate gene expression alone because an element of targeting or recruitment is necessary (in other words, how do the enzymes know where to place the marks?). Once set, PTMs putatively function in transcriptional regulation by promoting or excluding the binding of elements that directly function in regulation (i.e., activators and/or repressor) to such regions.
One counterargument that could be made in response to the aforementioned view of the histone code whereby chromatin-modifying machinery is recruited by transcriptional activators or repressors would be that histone modifications are a prerequisite for recruitment of certain elements of the transcriptional machinery. For example, two TBP-associated factor (TAF) subunits of the transcription factor complex TFIID have been shown to bind directly to histone PTMs, which would suggest that modification of histone proteins is necessary for binding of the transcriptional machinery. The double bromodomain of Taf1, the largest subunit of TFIID, binds preferentially to diacetylated histone H4.35 Taf3 harbors a PHD finger that is selective for binding to trimethylated H3K4, and loss of this chromatin mark results in reduced TFIID association with and transcriptional activity from certain promoters 36, providing support for the role of histone PTMs as a requisite component in the recruitment of transcription factors.
Despite the seemingly opposite lines of reasoning regarding the role of histone modifications in transcriptional regulation, we maintain that the nature of the histone code may not necessarily be as clear-cut as histone PTMs functioning solely as a consequence of or prerequisite for recruitment of the canonical transcriptional machinery. It is likely that both arguments hold true in their own rights with respect to transcription (as well as other DNA-templated processes), and that possibly no absolute rule exists favoring either position over the other, thereby necessitating examination of such codes on an individual basis. It is, therefore, perhaps more judicious to focus our discussion on the histone code in the context of how it more generally contributes to the physical organization of eukaryotic genomes. Three major principles have developed during the evolution of the histone code hypothesis over the past ten years: (1) interactions between histone modifications are not limited to a single tail; (2) a single mark can recruit more than one protein; and (3) proteins acting alone or in the context of a macromolecular complex can contain multiple domains to facilitate binding to chromatin (Figure 2). At the time when the histone code hypothesis was put forward, we had a relatively limited scope of the existent histone PTMs, the combinations in which they exist, and how they affect downstream functionality. That marks located in close proximity to one another often times exhibit functional interplay was demonstrated by examples such as phosphorylation of serine 10 of histone H3 (H3S10ph) reducing the affinity of the chromodomain of heterochromatin protein 1 (HP1) for di- and trimethylated lysine 9 of histone H3.37 At present, the chromatin field continually refines our understanding of how individual modifications affect placement of another, especially in the context of how modifications on one histone tail affects placement of marks and recruitment of effector proteins on other tails. A clear example of this idea is provided by studies that have demonstrated that a signal cascade in which 14-3-3 is recruited to the enhancer of FOSL1 by binding to H3S10ph and itself subsequently recruits the HAT MOF, which acetylates histone H4 on lysine 16 (H4K16ac) to create a doubly-modified H3S10ph/H4K16ac nucleosome38. These PTMs then function as a platform for the bromodomain-containing protein BRD4 (which in turn recruits the positive transcription factor b (P-TEFb)) to activate transcription elongation, thus providing an elegant example of the numerous intricacies associated with interactions between multiple histone PTMs across multiple tails.38 It is becoming increasingly clear that modifications that work together to form a putative code are not limited to a single histone tail, but are likely to span multiple tails within one nucleosome, between adjacent nucleosomes, or between non-adjacent nucleosomes that are physically located in close proximity to one another due to higher-order chromatin structure. Examples of histone crosstalk continue to evolve, and many more are likely to surface from future work, thereby shedding light on the growing complexity associated with the many permutations of a histone code.
Figure 2. Mechanisms of histone-recognition modules binding their target modification.
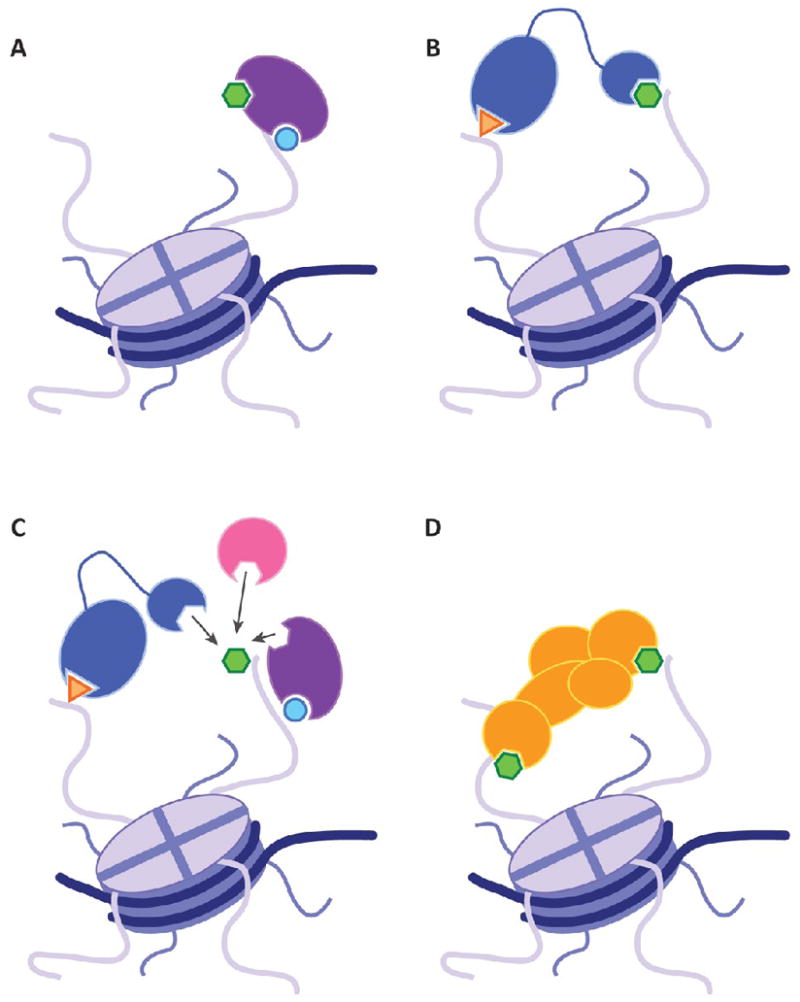
Binding of specialized domains to histone post-translational modifications can occur in cis, where contact is made to a series of modifications on the same histone tail (A), or in trans, where contacts are made to distinct modifications across histone tails (B). Often, a single modification can serve as a docking site for more than one protein, in which secondary signals (e.g. other PTMs) may serve to dictate which protein is recruited to the specific mark (C). Proteins acting alone (A–B), or in the context of a macromolecular complex (D) can harbor multiple domains capable of facilitating chromatin recognition and binding. For clarity, no attempts have been made to depict histone recogntion between nucleosomes in either the same or distinct polynucleosome fibers, but these modes of binding recognition are also likely (reviewed in Ruthenburg et al. 2007).
As alluded to above, modifications of histone residues in defined states can serve as platforms for binding of more than one effector protein. For example, multiple proteins (including JMJD2A, Rag2, BPTF, Ing2 and Taf3) have all been demonstrated to bind to trimethylated lysine 4 of histone H3 (H3K4me3). 36; 39; 40; 41; 42; 43; 44 Such promiscuity by a defined mark for multiple readers indicates that secondary levels of specification must exist. One possible explanation is that one protein can harbor multiple domains that cooperatively facilitate recognition and binding to chromatin.45 For example, Tsai et al have recently shown that the tandem PHD finger and bromodomain of the protein TRIM24, a co-activator of oestrogen receptor α (ERα), bind combinatorially to unmodified H3K4 and acetylated H3K23 to facilitate chromatin recognition and contribute to ERα-mediated transcription activation.46 Alternatively, more than one histone PTM (or the recognition of unmodified histone residues with modified ones) can function in concert to form a recognition code for a single protein with multiple chromatin-binding domains or multiple proteins within a chromatin-associated complex.47 One example of this type of nucleosomal interaction is provided by the Rpd3S histone deacetylase complex, which stably interacts with H3K36 methylated nucleosomes via recognition of H3K36 methylation by the chromodomain-containing subunit Eaf3 and H3 recognition by a PHD finger within in Rco1 subunit of this same complex.48 Thus, it is becoming increasingly clear that the one mark:one reader (or writer or eraser in certain instances) ratio does not allow for generation of enough physically distinct relationships to sufficiently impart the degree of information necessary to mediate diverse outcomes, supporting the existence of numerous levels of complexity built into the histone code. Such complexity would allow multiple ways to tinker with the same chromatin landscape to promote diverse biological outcomes.
Tinkering the ‘histone code hypothesis’ in years to come
The key question that remains then, is perhaps not one of mulling over how to best define the histone code, but rather, what form will the histone code hypothesis take over the years to come? Given the rapidity of chromatin-based research and the prominent role of chromatin in numerous DNA-based processes, research in the years to come is likely to continue along the same fruitful path of discovery that it has witnessed in the past ten years, demonstrating additional levels of complexity by which intrinsic cellular machines tinker with the chromatin template. While studies aimed at identifying additional writers and erasers of the histone code as well as novel marks remain ever important, investigations elucidating how chromatin marks act in concert to recruit readers are of equal significance. Technological advancements and new methodologies have significantly progressed our efforts in both areas of study, and are expected to continue to do so well into the future.49; 50 Histone PTMs have traditionally been identified by metabolic labeling, microsequencing, the generation of immunological reagents, and more recently, mass spectrometry (MS).51 Advancements in MS technology include the recently developed top-down methodology, which analyzes intact proteins samples (as opposed to the more canonical bottom-up approach where proteins are fragmented prior to analysis). Because proteins are analyzed at the whole-molecule level, top-down MS allows for identification of combinatorial patterns of modifications that exist within one histone protein.52 For example, top-down MS analysis has now been completed on all three human histone H3 variants (H3.1, H3.2, and H3.3), revealing complex patterns of modified H3 forms.53; 54 Additionally, analysis of asynchronously grown HeLa cells treated with the HDAC inhibitor sodium butyrate has revealed a surprising and complex number of combinatorially-modified species of histone H3.2 and H4.53; 55 Though still in its infancy, studies such as these have made it readily apparent that top-down MS analysis will be a highly utilized technique in future studies to decipher how combinatorial patterns of histone modifications contribute to the regulation of diverse biological processes.49
Identification of the histone marks themselves and the combinatorial patterns in which they exist is not enough to understand functional consequences of their placement. The availability of modification-specific antibodies has allowed for immunoprecipitation of DNA fragments associated with a particular mark by chromatin immunoprecipitation (ChIP). It should be formally noted that one major limitation to be kept in mind when designing and/or interpreting experiments involving ChIP is the requirement for a high-quality antibody that can specifically recognize a defined modification state (e.g., a dimethylated but not trimethylated lysine residue). Moreover, as neighboring modifications may unpredictably impact antibody specificity, it is becoming increasingly clear that rigorous validation of antibody quality is essential for any ChIP-based analysis to effectively provide insight into the location of a particular modification in a defined state.56; 57; 58
Early approaches for studying chromatin-modifications on a genome-wide level utilized ChIP combined with DNA microarray analysis (ChIP-chip). More recently, ChIP coupled with next-generation sequencing technology (ChIP-seq) has provided considerable insight into the function of histone PTMs, allowing for the identification of genome-wide patterns of specific modifications as well as transcription factors and the machinery responsible for modifying the chromatin landscape under defined biological conditions.59 Early ChIP-seq analyses mapping histone modifications in CD4+ T cells or mouse embryonic stem (mES) cells revealed a number of findings.60; 61; 62 For instance, a comparative ChIP-seq analysis of mES, neural progenitor and embryonic fibroblasts confirmed the existence of bivalent domains characterized by the co-localization of H3K4 and H3K27 trimethylation that function in cellular plasticity and commitment to a defined lineage.61; 63 However, how widespread bivalent domains occur in various developmental contexts remains unclear and is under active investigation. Genome-wide association studies derived from ChIP-seq analyses completed to date have led many to see the histone code less in terms of as sets of definite combinations that produce an absolute outcome, but rather, more as patterns of modifications that when in combination tend to favor a specified outcome. In that vein, the ramifications of the histone code are correlative rather than causal in that combinatorial patterns provide a bias for a specific outcome rather than serve as an absolute mark of one. ChIP-seq analyses has, for example, revealed that in general, higher levels of H3K9me1 and H2BK5me1 in the 5′ end, H3K27me1 distributed throughout, and H3K36me3 in the 3′ end of a transcribed region mark actively transcribed regions.60 Furthermore, another study found that there is a combinatorial pattern of methylation and acetylation events on histone tails that are co-associated with each other on a significant fraction of genes within the human genome.62 Such studies provide important insight, in that they demonstrate that actively transcribed regions of the genome, as well as functional elements in general, bear distinct histone PTM signatures.64 Additional studies will surely expand upon whether the histone code is characterized by a fixed set of combinatorial patterns that establish defined chromatin states (also referred to frequently as chromatin ‘signatures’) or rather, if certain combinations tend to tip the balance in favor of a certain state. For example, recent work published by the modENCODE Consortium has provided great insight into the genome-wide chromatin organization in the model organisms Caenorhabditis elegans and Drosophila melanogaster, which together have vastly advanced our understanding how various histone PTMs are associated with genomic regulatory elements in defined developmental states.65; 66; 67 Newcomers to this field should refer to these studies to become oriented not only to some of the principal PTMs that mark chromatin domains, but also to the staggering complexities underlying the combinatorial nature with which gene bodies and regulatory elements are specified and defined in a chromatin context. Indeed the language is colorful and must be interpreted in context, especially in a developmental setting.
Novel methods are also being developed to characterize combinatorial patterns that facilitate binding of effector proteins as well as identify novel proteins that can bind to modified histone tails. Use of combinatorial peptide libraries based on the N-terminal histone tails has become a widely used practice to identify how the presence of additional marks enhances or weakens the affinity of an effector protein for its target binding module. Peptide libraries have been synthesized as various types of platforms, including resin-bound PTM-containing histone tail libraries and custom peptide microarrays.56; 58; 68; 69 Such platforms have recently begun to be used to identify synergistic and antagonistic combinations of histone modifications that ultimately affect the binding of effectors. For example, the H3K9me2 demethylase PHF8 binds to H3K4me3/2, and hybridization of a recombinant GST-PHD(PHF8) fusion protein to a synthetic peptide array containing combinatorial modifications patterns revealed that binding to H3K4me3/2 was also achieved when peptides were acetylated at the H3K9/K14 positions.70 While peptide libraries are advantageous at looking at how effector proteins respond to various combinatorial patterns of modifications, alternative functional technologies are being employed to screen for proteins that bind to a particular modification in an unbiased manner. Recently, a histone peptide pulldown approach paired with SILAC proteomics technology was used to define a large-scale methyl lysine interactome.71 Extending this concept further, designer synthetic nucleosomes in which nucleosomes are reconstituted using recombinant histones harboring specific modifications states have allowed for unbiased identification of cellular proteins that bind to a specific state on a nucleosomal substrate in a technique called SNAP (SILAC nucleosome affinity purification).72 Because the DNA sequence and modifications of interest are user-defined, one could theoretically begin to make oligonucleosomes in which crosstalk both within and across nucleosomes can be addressed. This latter technology holds great potential for future studies in which peptides harboring several modifications are fused to multiple histone tails via native chemical ligation to reconstitute multiply-modified nucleosomes to give a more complete picture of how combinatorial patterns affect binding by chromatin readers in the more-physiologically relevant nucleosomal context.
Strict code versus rich language: exciting either way
At the time of inception, it is always difficult to discern how influential a hypothesis will truly be. We have been privileged to witness that François Jacob and Jacques Monod’s report on the lac operon in the Journal of Molecular Biology in 1961 has revolutionized our understanding of the basic mechanisms underlying gene regulation. We are also beginning to understand the richness of the histone code hypothesis. When we posited this hypothesis, now ten years ago, we had what in retrospect would be described as a quite limited scope of histone post-translational modifications. One decade later, we stand in awe at how the chromatin field, and scientific research community at large, has come together to expand this code to a scope beyond what was imaginable at the time of its conception. For example, never in our wildest dreams had we envisioned a Keystone meeting being dedicated to the singular topic of the ‘Histone Code’: Fact or Fiction (January 10–15, 2011 in Midway, Utah). However, it is with a sense of realism that we recognize that many obstacles remain to be overcome before we can officially declare that this code has been deciphered to its fullest potential. For example, it will be difficult to discern when saturation has been reached and all modifications have been identified, a reality complicated by the fact that organismal differences exist within the chromatin landscape. The staggering complexity of this proposed ‘epigenetic code’ promises to keep many talented scientists busy for the next decade with many more welcomed surprises along the way. Moreover, we are coming to realize that such a code may not pertain specifically to histones, but could potentially be extended to proteins in general. That proteins are modified post-translationally is by no means a novel concept, but the idea that modifications working in concert are predictive of defined downstream biological events has received more thought recently. The tumor suppressor p53 is highly regarded as the model for the existence of a more general protein code, as this protein is subject to a number of PTMs, including methylation, acetylation, phosphorylation, and ubiquitylation.73 The observation that modifications, such as acetylation, correlate with stabilization and activation of p5374 in concert with the idea that one modification can enhance or preclude the placement of another supports a more general mechanism in which modifications are tightly linked to p53 function in an analogous fashion as to how histone PTMs work together to form a functional code. Also worthy of noting is that many of the enzymes responsible for writing, erasing, and reading histone methylation and acetylation on histone proteins are also responsible for modifying the C-terminus of p53 and certainly other non-histone proteins75; 76, echoing Jacob’s visionary sentiment that the same elements are often used to create new products of increasing complexity.
Our piece in 2000 was framed as a hypothesis with the hope that it would stimulate discussion and lead to subsequent tests of its central tenets. Much of this has happened, and we look forward to much more along these lines. While contention over use of the word “code” may eventually lead to an alternative designation in future years, we are confident that debates over diction will not hinder the elegant work that the chromatin community has collectively produced at a remarkable pace. We close with a prediction -- we will indeed witness a period of further enlightenment with regard to how cellular enzymes tinker with both histone and non-histone proteins alike to create increasingly complex patterns of regulatory mechanisms in the years to come. Coloring the chromatin code with even more shades will be part of the fun.77; 78
Acknowledgments
We thank the many researchers whose studies have help to expand our understanding of both the lac operon and the histone code, and apologize to those whose work could not be cited here due to space constraints. We also thank Nara Lee, Scott Rothbart, and the members of the Allis lab for insightful conversations and comments on this manuscript, and Nara Lee and Stephen Fuchs for assistance with the illustrations contained in this piece.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jacob F. Evolution and tinkering. Science. 1977;196:1161–6. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- 2.Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–56. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 3.Jacob F, Perrin D, Sanchez C, Monod J. Operon: a group of genes with the expression coordinated by an operator. C R Hebd Seances Acad Sci. 1960;250:1727–9. [PubMed] [Google Scholar]
- 4.Struhl K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell. 1999;98:1–4. doi: 10.1016/S0092-8674(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–94. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 6.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 7.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–71. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 8.Oudet P, Gross-Bellard M, Chambon P. Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell. 1975;4:281–300. doi: 10.1016/0092-8674(75)90149-x. [DOI] [PubMed] [Google Scholar]
- 9.Wolffe AP, Hayes JJ. Chromatin disruption and modification. Nucleic Acids Res. 1999;27:711–20. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 11.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 13.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–84. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–75. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 15.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–9. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A. 2010;107:19915–20. doi: 10.1073/pnas.1009023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15:245–50. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vezzoli A, Bonadies N, Allen MD, Freund SM, Santiveri CM, Kvinlaug BT, Huntly BJ, Gottgens B, Bycroft M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–9. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Reddy B, Thompson J, Wang H, Noma K, Yates JR, 3rd, Jia S. Regulation of Set9-mediated H4K20 methylation by a PWWP domain protein. Mol Cell. 2009;33:428–37. doi: 10.1016/j.molcel.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, Desai A, Dorrestein PC, Glass CK, Rosenfeld MG. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–12. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330:235–9. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–7. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Sims RJ, 3rd, Reinberg D. Histone H3 Lys 4 methylation: caught in a bind? Genes Dev. 2006;20:2779–86. doi: 10.1101/gad.1468206. [DOI] [PubMed] [Google Scholar]
- 27.Smith E, Shilatifard A. The chromatin signaling pathway: Diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell. 2010:40. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–5. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 30.Ptashne M. On the use of the word ‘epigenetic’. Curr Biol. 2007;17:R233–6. doi: 10.1016/j.cub.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 31.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 33.Pazin MJ, Kadonaga JT. What’s up and down with histone deacetylation and transcription? Cell. 1997;89:325–8. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 34.Katan-Khaykovich Y, Struhl K. Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002;16:743–52. doi: 10.1101/gad.967302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–5. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 36.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–22. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 38.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–36. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–51. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–5. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, Walter KL, Utz PJ, Shi Y, Kutateladze TG, Yang W, Gozani O, Oettinger MA. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–10. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, Lacoste N, Cayrou C, Davrazou F, Saha A, Cairns BR, Ayer DE, Kutateladze TG, Shi Y, Cote J, Chua KF, Gozani O. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 45.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–94. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, Aronow B, Gozani O, Fischle W, Hung MC, Patel DJ, Barton MC. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–32. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliver SS, Denu JM. Dynamic Interplay between Histone H3 Modifications and Protein Interpreters: Emerging Evidence for a “Histone Language”. Chembiochem. 2010 doi: 10.1002/cbic.201000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, Gogol M, Carey M, Lee D, Seidel C, Workman JL. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–4. doi: 10.1126/science.1139004. [DOI] [PubMed] [Google Scholar]
- 49.Young NL, Dimaggio PA, Garcia BA. The significance, development and progress of high-throughput combinatorial histone code analysis. Cell Mol Life Sci. 2010;67:3983–4000. doi: 10.1007/s00018-010-0475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voigt P, Reinberg D. Histone Tails: Ideal Motifs for Probing Epigenetics through Chemical Biology Approaches. Chembiochem. 2010 doi: 10.1002/cbic.201000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia BA, Shabanowitz J, Hunt DF. Characterization of histones and their post-translational modifications by mass spectrometry. Curr Opin Chem Biol. 2007;11:66–73. doi: 10.1016/j.cbpa.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 52.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nature Methods. 2007;4:817–21. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nature Methods. 2007;4:487–9. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 54.Thomas CE, Kelleher NL, Mizzen CA. Mass spectrometric characterization of human histone H3: A bird’s eye view. Journal of Proteome Research. 2006;5:240–247. doi: 10.1021/pr050266a. [DOI] [PubMed] [Google Scholar]
- 55.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266–84. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bock I, Dhayalan A, Kudithipudi S, Brandt O, Rathert P, Jeltsch A. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics. 2011:6. doi: 10.4161/epi.6.2.13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, Gu T, Kharchenko PV, Kuan S, Latorre I, Linder-Basso D, Luu Y, Ngo Q, Perry M, Rechtsteiner A, Riddle NC, Schwartz YB, Shanower GA, Vielle A, Ahringer J, Elgin SC, Kuroda MI, Pirrotta V, Ren B, Strome S, Park PJ, Karpen GH, Hawkins RD, Lieb JD. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2010 doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. Influence of Combinatorial Histone Modifications on Antibody and Effector Protein Recognition. Curr Biol. 2010 doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9:179–91. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 61.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 64.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 65.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K, Agarwal A, Alexander RP, Barber G, Brdlik CM, Brennan J, Brouillet JJ, Carr A, Cheung MS, Clawson H, Contrino S, Dannenberg LO, Dernburg AF, Desai A, Dick L, Dose AC, Du J, Egelhofer T, Ercan S, Euskirchen G, Ewing B, Feingold EA, Gassmann R, Good PJ, Green P, Gullier F, Gutwein M, Guyer MS, Habegger L, Han T, Henikoff JG, Henz SR, Hinrichs A, Holster H, Hyman T, Iniguez AL, Janette J, Jensen M, Kato M, Kent WJ, Kephart E, Khivansara V, Khurana E, Kim JK, Kolasinska-Zwierz P, Lai EC, Latorre I, Leahey A, Lewis S, Lloyd P, Lochovsky L, Lowdon RF, Lubling Y, Lyne R, Maccoss M, Mackowiak SD, Mangone M, McKay S, Mecenas D, Merrihew G, Miller DM, 3rd, Muroyama A, Murray JI, Ooi SL, Pham H, Phippen T, Preston EA, Rajewsky N, Ratsch G, Rosenbaum H, Rozowsky J, Rutherford K, Ruzanov P, Sarov M, Sasidharan R, Sboner A, Scheid P, Segal E, Shin H, Shou C, Slack FJ, et al. Integrative Analysis of the Caenorhabditis elegans Genome by the modENCODE Project. Science. 2010 doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, Linder-Basso D, Plachetka A, Shanower G, Tolstorukov MY, Luquette LJ, Xi R, Jung YL, Park RW, Bishop EP, Canfield TP, Sandstrom R, Thurman RE, Macalpine DM, Stamatoyannopoulos JA, Kellis M, Elgin SC, Kuroda MI, Pirrotta V, Karpen GH, Park PJ. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2010 doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, Washietl S, Arshinoff BI, Ay F, Meyer PE, Robine N, Washington NL, Di Stefano L, Berezikov E, Brown CD, Candeias R, Carlson JW, Carr A, Jungreis I, Marbach D, Sealfon R, Tolstorukov MY, Will S, Alekseyenko AA, Artieri C, Booth BW, Brooks AN, Dai Q, Davis CA, Duff MO, Feng X, Gorchakov AA, Gu T, Henikoff JG, Kapranov P, Li R, Macalpine HK, Malone J, Minoda A, Nordman J, Okamura K, Perry M, Powell SK, Riddle NC, Sakai A, Samsonova A, Sandler JE, Schwartz YB, Sher N, Spokony R, Sturgill D, van Baren M, Wan KH, Yang L, Yu C, Feingold E, Good P, Guyer M, Lowdon R, Ahmad K, Andrews J, Berger B, Brenner SE, Brent MR, Cherbas L, Elgin SC, Gingeras TR, Grossman R, Hoskins RA, Kaufman TC, Kent W, Kuroda MI, Orr-Weaver T, Perrimon N, Pirrotta V, Posakony JW, Ren B, Russell S, Cherbas P, Graveley BR, Lewis S, Micklem G, Oliver B, Park PJ, Celniker SE, Henikoff S, Karpen GH, Lai EC, Macalpine DM, Stein LD, White KP, Kellis M. Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE. Science. 2010 doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bua DJ, Kuo AJ, Cheung P, Liu CL, Migliori V, Espejo A, Casadio F, Bassi C, Amati B, Bedford MT, Guccione E, Gozani O. Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS One. 2009;4:e6789. doi: 10.1371/journal.pone.0006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garske AL, Oliver SS, Wagner EK, Musselman CA, LeRoy G, Garcia BA, Kutateladze TG, Denu JM. Combinatorial profiling of chromatin binding modules reveals multisite discrimination. Nat Chem Biol. 2010;6:283–90. doi: 10.1038/nchembio.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kleine-Kohlbrecher D, Christensen J, Vandamme J, Abarrategui I, Bak M, Tommerup N, Shi X, Gozani O, Rappsilber J, Salcini AE, Helin K. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. 2010;38:165–78. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–80. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 72.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–84. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sims RJ, 3rd, Reinberg D. Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol. 2008;9:815–20. doi: 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- 74.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 75.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 76.Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins. Curr Opin Genet Dev. 2008;18:152–8. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 77.Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–24. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schubeler D. Chromatin in multicolor. Cell. 2010;143:183–4. doi: 10.1016/j.cell.2010.09.045. [DOI] [PubMed] [Google Scholar]