

Abstract
In this study, a pharmacodynamic model is developed, based on calcium–parathyroid hormone (PTH) homeostasis, which describes the concentration–effect relationship of a negative allosteric modulator of the calcium-sensing receptor (CaR) in rats. Plasma concentrations of drug and PTH were determined from plasma samples obtained via serial jugular vein sampling following single subcutaneous doses of 1, 5, 45, and 150 mg/kg to male Sprague–Dawley rats (n = 5/dose). Drug pharmacokinetics was described by a one-compartment model with first-order absorption and linear elimination. Concentration-time profiles of PTH were characterized using a model in which the compound allosterically modulates Ca+2 binding to the CaR that, in turn, modulates PTH through a precursor-pool indirect response model. Additionally, negative feedback was incorporated to account for tolerance observed at higher dose levels. Model fitting and parameter estimation were conducted using the maximum likelihood algorithm. The proposed model well characterized the data and provided compound specific estimates of the Ki and cooperativity constant (α) of 1.47 ng/mL and 0.406, respectively. In addition, the estimated model parameters for PTH turnover were comparable to that previously reported. The final generalized model is capable of characterizing both PTH–Ca+2 homeostasis and the pharmacokinetics and pharmacodynamics associated with the negative allosteric CaR modulator. As such, the model provides a simple platform for analysis of drugs targeting the PTH–Ca+2 system.
KEY WORDS: allosteric, bone, calcium sensing receptor, ionized calcium, osteoporosis, parathyroid hormone, pharmacodynamics, pharmacokinetics
INTRODUCTION
It has been estimated that over 200 million people suffer from osteoporosis worldwide (1). Fragility fractures associated with this disease incur significant cost, morbidity, and mortality (2,3). The majority of osteoporotic patients are treated with antiresorptive agents, which slow the resorption of bone by several mechanisms. Currently, there are relatively few anabolic treatment options, namely full or partial length forms of recombinant parathyroid hormone (PTH 1–84, Preotact™ or PTH 1–34 Forteo™, respectively). PTH is known to maintain extracellular Ca+2 concentrations (1.2 mM) through multiple sites of action, which involve the kidney (increased reabsorption of Ca+2), intestines (activation of vitamin D), and bone (4). Although the underlying mechanism of recombinant PTH action is still under investigation, these agents produce distinct anabolic effects in patients with osteoporosis (5–9). However, issues related to cost, label restrictions, and the necessity for daily injection limit the clinical utility of these agents. As a result, their use is primarily restricted to a subset of patients with preexisting fractures, very low bone mineral density, or an unsatisfactory response to antiresorptive therapy.
The calcium sensing receptor (CaR) represents an attractive alternative to the development of effective anabolic therapies for osteoporosis. This receptor is a class C, G-protein-coupled receptor located on the parathyroid cell surface where it functions as one of the key regulators of bone homeostasis (4,10). Secretion of PTH in response to signaling from the CaR is one of the key endocrine regulators of Ca+2 (11). The relationship between Ca+2 and PTH is characterized by a steep inverse concentration–response curve, whereby slight decreases in extracellular Ca+2 result in disproportionate increases in PTH concentrations (12). This has led to efforts to identify orally available, small molecule antagonists, or calcilytics, of the CaR in hopes of achieving anabolic effects through elevation of endogenous PTH. The amino alcohol-based compound, NPS 2143, is the first calcilytic that emerged from these efforts (13,14). Several classes of amino alcohol-based CaR antagonists have now been described in the literature (15–19), with SB-423557 and its biologically active component SB-423562 showing transient elevations of endogenous PTH in a manner similar to that produced by subcutaneous PTH formulations (19). These transient elevations have been associated with increased bone formation and strength in an ovariectomized rat model of bone loss. However, concerns still exist due to the paradoxical effects of pulsatile versus sustained elevations in PTH that have been associated with bone formation and loss (20–22). Chronic CaR antagonism may, in theory, lead to parathyroid hyperplasia, hyperparathyroidism, and hypocalcemia.
Thus, understanding the quantitative relationship between CaR antagonist concentration and PTH elevation is critically important in the discovery and development of safe and effective calcilytics. However, to date, no mathematical model has been proposed for the pharmacokinetic–pharmacodynamic (PK/PD) behavior of calcilytics. Models have been proposed to characterize the underlying PTH–Ca+2 system, which include a four-parameter logistic model (23), a simple biexponential model to describe induced PTH secretion by rapid lowering of blood Ca+2 (24), and a minimal model of calcium homeostasis involving Ca+2, PTH, and calcitriol (25). In addition, we have proposed an integrated mechanism-based PK/PD model (26), which simultaneously describes the interplay of PTH and Ca+2 through occupancy of the CaR. This model captures the time course of PTH concentrations in response to lowering of serum Ca+2 by a chelating agent, and is applicable to data obtained from multiple studies and across species (rats and humans). As such, it represents an ideal base model upon which to develop a PK/PD model for calcilytics. In this study, we develop a model to understand the PK/PD properties of an investigational amino alcohol-based CaR antagonist, by incorporating a model for allosteric receptor modulation.
MATERIALS AND METHODS
Materials
An amino alcohol CaR antagonist, (R)-2-chloro-6-([3-((1,1-dimethyl-2-[4-(methyloxy)phenyl]ethyl)-amino)-2-hydroxypropyl]oxy)benzonitrile (abbreviated as AA for this paper; Fig. 1), was prepared as described previously (27). HEK293 cells expressing the human calcium receptor (NP_000379) were scaled up, and purified membranes were obtained utilizing a sucrose gradient purification scheme. Briefly, cells were collected and lysed using a hypotonic lysis buffer containing 0.5 mM sodium phosphate pH 7.0, 0.1 mM EDTA containing Roche Complete Protease Inhibitor Cocktail Tablets. Following centrifugation at 100,000×g for 40 min at 4°C, the pellet was resuspended in the hypotonic lysis buffer and dounce homogenized at which time an equal volume of 0.5 M sucrose was added and the homogenate was recentrifuged at 100,000×g for 40 min at 4°C. The pellet was resuspended in incubation buffer (0.25 M sucrose, 10 mM Tris–HCl, pH 7.4 with Roche Complete Protease Inhibitor Cocktail Tablets) and dounce homogenized. The crude membrane fraction was then layered onto a 38% sucrose gradient in 5 mM HEPES/KOH, pH 7.4, and centrifuged at 280,000×g for 2 h at 4°C. The interface containing the membrane fraction was collected, rehomogenized, washed, recentrifuged, and resuspended in incubation buffer for use in a binding assay.
Fig. 1.

Molecular structure of (R)-2-chloro-6-([3-((1,1-dimethyl-2-[4-(methyloxy)phenyl]ethyl)-amino)-2-hydroxypropyl]oxy)benzonitrile
Radioligand Binding Assay
Membranes were diluted in binding buffer containing 50 mM Tris pH 7.5, 1 mM EDTA, 3 mM MgCl2, and 0.01% P104 containing Roche Complete Protease Inhibitor Cocktail Tablets, and 120 μL was aliquoted into a 96-well Costar 3357 polypropylene plate on ice. A 100-fold stock of the compounds which had been serially diluted in DMSO was added to the membranes (for a final top dose of 10 μM and a final concentration of 1% DMSO) followed by 120 μL of the 3H AA at 4 nM (for a final concentration of 2 nM). The reaction was incubated at 4°C for 1 h and then filtered onto a Millipore 96-well glass filter plate MAHFC pretreated with 0.3% polyethyleneimine. Wells were washed five times with 200 μL of binding buffer containing 1% DMSO and then allowed to dry. Plates were sealed before adding scintillant and counted on a Microbeta Trilux to determine the amount of radioactivity. Nonspecific binding was determined by the addition of a 1,000-fold excess of the nonlabeled ligand. Radioactivity as a percent of control was plotted against nonlabeled AA concentrations, and a sigmoid Imax model was used to estimate the IC50 for AA. This assay was performed in triplicate on 13 different occasions in order to estimate the 95% confidence interval for IC50. The equilibrium dissociation constant for AA (Ki) was calculated according to the Cheng–Prusoff equation:
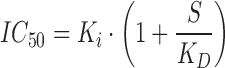 |
1 |
where S is the concentration of radiolabeled AA in the incubation (2 nM). In this case, because radiolabeled AA was used to assess binding of cold AA, Ki also equals KD. This enables Ki to be calculated as:
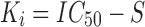 |
2 |
such that it could be compared to the estimate obtained by modeling the PK/PD data obtained in rats.
Drug Assay
Concentrations of AA in plasma were determined using a Sciex API model 3000 LC-MS/MS triple quadrupole mass spectrometer. Plasma samples were treated with equal volumes of acetonitrile containing an internal standard (midazolam), centrifuged (4,000×g) and the supernatant injected into the LC-MS/MS system. Analytes were chromatographically separated using a Hewlett Packard Series 1100 HPLC system. An autosampler was programmed to inject 20 μL on a Phenomenex Primesphere 5 μ C18-HC 30 × 2.0-mm column using a mobile phase consisting of 10 mM ammonium acetate buffer–acetonitrile (60:40 v/v) containing 0.2% (v/v) triethylamine, and 0.1% (v/v) acetic acid at a flow rate varying from 1 to 1.5 mL/min. Ionization was conducted in the positive ion mode at the ionspray interface temperature of 400°C, using nitrogen as the nebulizing and heating gas. The ion spray voltage was 5.0 kV, and the orifice voltage was optimized at 30 eV. AA and midazolam were analyzed in the MRM mode using the transitions m/z 389 → 227 and 326 → 291, respectively. Calibration curves were prepared by plotting the appropriate peak area ratios against the concentrations of AA in plasma using 1/× weighting of AA/internal standard peak height ratios. The concentration of AA in the plasma samples was determined by interpolation from the standard curve, and the dynamic range of the assay was 1–1,000 ng/mL. As measured by quality control samples, precision (% coefficient of variation) and accuracy (% deviation from expected) of the assay were acceptable (CV% less than 15%).
PTH Assay
Plasma PTH concentrations were analyzed by use of a commercially available ELISA kit (Rat Intact PTH ELISA Cat. no. 60-2500; Immutopics, San Clemente California) according to kit instructions. Intact rat PTH 1–84 was detected with a sensitivity of 1.6 pg/mL and a detection range from 0 to 3,000 pg/mL. The intra- and interassay coefficient of variation was less than 2.5% and 6.0%, respectively.
Animal Studies
All animal care and in vivo procedures were approved and conducted in accordance with guidelines of the Pfizer Animal Care and Use Committee. AA was administered subcutaneously (s.c.) at doses of 1, 5, 45, and 150 mg/kg (n = 5/dose) to fasted male Sprague–Dawley rats (11–13 weeks old; Taconic Farms, Spafas, NY). Animals were fed following collection of the 4-h blood samples. AA was formulated in a 20% aqueous solution of 2-hydroxypropyl β-cyclodextrin (Sigma/RBI, Natick, MA). After dosing, serial plasma samples were collected via tail vein sampling at appropriate times and kept frozen at −20°C until analysis of AA and PTH concentrations.
PK/PD Model
Experimental data were characterized using a mechanistic PK/PD model, the schematic of which is shown in Fig. 2. The concentration-time profiles of AA were described using a standard two-compartment linear disposition model with first-order absorption:
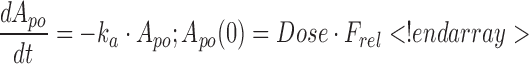 |
3 |
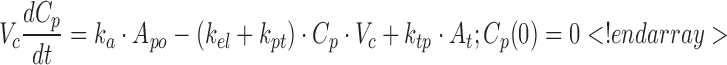 |
4 |
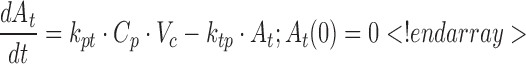 |
5 |
where ka is the first-order absorption rate constant after subcutaneous dosing, kpt and ktp are the first-order rate constants for nonspecific distribution of the drug to/from a peripheral tissue compartment, and kel is the first-order elimination rate constant. Equation 3 describes the amount of drug (A) at the site of absorption after subcutaneous dosing. The central compartment is denoted as Cp, and its volume of distribution is Vc. The amount of drug in the peripheral compartment (At) is described by Eq. 5. Preliminary noncompartmental analysis of the PK data suggested incomplete absorption for the 150-mg/kg dose level (not shown). A multiplicative term, krel, was estimated for the highest dose level, and is operational on ka, where the product allowed for simultaneous characterization of all PK data.
Fig. 2.

Integrated pharmacokinetic/pharmacodynamic (PK/PD) model for quantitative assessment of a calcium-sensing receptor (CaR) antagonist on the parathyroid hormone–extracellular calcium in vivo regulatory system. Turnover parameters (k in_Ca and k out_Ca) were fixed to give stationary and time-invariant ionized calcium (Ca+2) concentrations. Model information and abbreviations for rate constants have been defined under PK/PD model in “MATERIALS AND METHODS” section
Our model of PTH–Ca+2 dynamics has been described elsewhere (26). Briefly, a precursor-dependent indirect response model (28) was implemented to characterize PTH turnover. The model was modified and is described by the following differential equations:
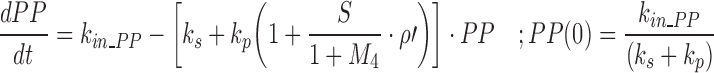 |
6 |
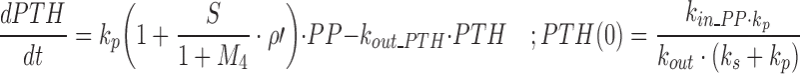 |
7 |
where PP and PTH are the PTH concentrations in the precursor pool and plasma, and the initial conditions for each equation are given as PP(0) and PTH(0). The precursor compartment is representative of a composite measure of preproparathyroid hormone, proparathyroid hormone, and stored intact PTH. A zero-order production rate constant, kin_PP, is assumed to account for the constant production of these precursors within the gland. The first-order rate constants ks, kp, and kout_PTH represent degradation of stored PTH, secretion of intact PTH from the gland, and loss of PTH from plasma. Loss of PTH from circulation is primarily mediated by the liver and kidneys (29,30). A basic assumption for the model is that ks, kp, kout_PTH, and PP fully account for the production and loss of PTH. All of these system turnover parameters for PTH were estimated in the modeling process. The S parameter linearly relates Ca+2/CaR occupancy to the modulation of PTH release from the precursor pool. This is consistent with the known modulation of PTH exocytosis associated with binding of extracellular Ca+2 to the CaR (31). The value of S was fixed to 20.4, which was obtained from initial model runs. The PK/PD model links Ca+2 and PTH interaction through changes in Ca+2/CaR occupancy relative to baseline (ρ′). In the absence of antagonist, fractional Ca+2/CaR occupancy is defined as:
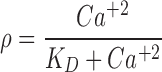 |
8 |
with a KD, the equilibrium dissociation constant of Ca+2 with CaR, of 1.2 mM (32). The plasma concentration of Ca+2 was assumed to be time-invariant and not expected to change as a result of the CaR antagonist over the short term (fixed to 1.2 mM). Parameter estimates for the Ca+2 turnover model were fixed to values obtained from a previous analysis (26). Amino alcohol-based compounds, like AA, are known to antagonize the CaR receptor via an allosteric modulation mechanism (14), the basic principles of which have been presented elsewhere in detail (33,34). Briefly, negative allosteric modulators bind to the receptor at a different site other than the endogenous ligand. In the presence of a negative allosteric modulator, the affinity of the endogenous ligand is altered, and this effect is saturable after the modulator occupies all allosteric binding sites. Any excess concentration of the negative allosteric modulator will only prolong the duration of antagonism, whereas the magnitude of the response is not further altered. Thus, Eq. 8 was modified to a closed form solution of the Ehlert model for receptor allosterism (33,35):
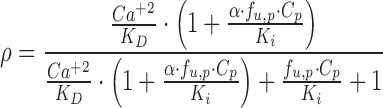 |
9 |
which includes the equilibrium dissociation constant (Ki) and cooperativity constant (α) for AA. If α is greater than 1, then the interaction is termed as “positively cooperative,” and if less than 1, then the interaction is “antagonistic.” Both Ki and α were estimated in the modeling process. The unbound fraction of AA in plasma (fu,p) was fixed to the mean estimate determined from standard in vitro equilibrium dialysis experiments (fu,p = 0.0065 ± 0.0014; data not shown) such that only unbound concentrations of the antagonist are assumed to interact with the CaR. The final model (Eqs. 6 and 7) includes a fractional change in Ca+2/CaR occupancy from baseline (ρ′) as the driving force for PTH release from the precursor pool:
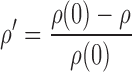 |
10 |
where ρ(0) is the Ca+2/CaR occupancy at baseline conditions.
A negative feedback mechanism was required to characterize the decrease in PTH concentrations at later times in the presence of sustained drug exposure at higher doses. Serum PTH concentrations were used to initiate a negative feedback through a series of transit compartments (n = 4):
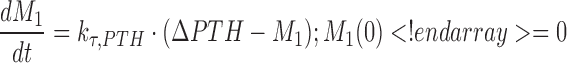 |
11 |
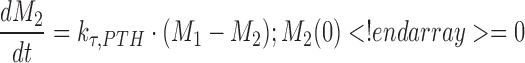 |
12 |
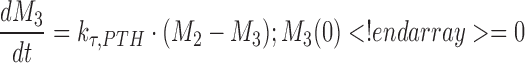 |
13 |
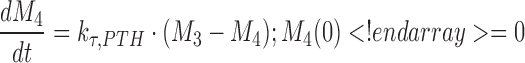 |
14 |
where Mn is the nth delay compartment, and kτ,PTH is a first-order rate constant for signal propagation. The signal in the last compartment (M4) was used to attenuate the drug effect as shown in Eqs. 6 and 7.
Data Analysis
A naïve-pooled approach was used, and the proposed model (Fig. 2) was fitted to dose-ranging and placebo data. Parameter estimation was conducted in sequential stages. First, concentration-time profiles of AA were characterized, and PK parameters were estimated. In the second stage, PK parameters were fixed and used to drive the PD model describing allosteric binding of the compound to the CaR, PTH turnover, and delayed feedback regulation. In the final run, all dose-ranging PK/PD data were characterized simultaneously using the integrated model and initial estimates obtained from the first two stages.
Parameter estimation was conducted using nonlinear regression analysis with ADAPT II (36) by the maximum likelihood method. The variance model was defined as:
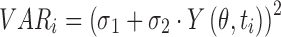 |
9 |
where σ1 and σ2 are the variance model parameters, and Y(θ, ti) is the ith predicted value from the PK/PD model. Distinct variance parameters were used for the CaR antagonist and PTH state variables. Model selection was based on goodness-of-fit criteria, which included model convergence, Akaike Information Criterion, estimation criterion value for the maximum likelihood method, and visual inspection of predicted versus observed values and residual plots.
RESULTS
Binding Assay
The binding IC50 of AA for the CaR was determined to be 4.9 nM with a 95% CI of 1.7–8.1 nM (Fig. 3). For purposes of comparison to the Ki derived from PK/PD modeling, Eq. 2 was applied to these results to provide a calculated mean Ki estimate of 2.9 nM (1.0 ng/mL).
Fig. 3.

Representative plot of binding assay results used in the determination of the K i of AA. Assay was run 13 times in triplicate in order to obtain a mean K i estimate of 2.9 nM (1.0 ng/ml)
Pharmacokinetics
The model fitted concentration-time profiles of AA are shown in Fig. 4. The linear two-compartment model with first-order absorption well characterized the naïve-pooled data for all four dose levels, and uncertainty (CV%) associated with the parameter estimates was low. Final PK parameter estimates for the compound are reported in Table I. The estimated value for the central volume of distribution (Vc/F = 0.688 L/kg) was associated with a large CV% value and was therefore fixed to an estimate obtained from earlier model runs. Preliminary analysis of the data suggested differences in the extent of absorption for the highest dose. For example, the peak plasma concentration was 2,110 ng/mL for the 45-mg/kg dose level and 2,020 ng/mL for the highest dose. Inclusion of the multiplicative term for the 150-mg/kg dose (krel = 0.126) stabilized the structural model and the precision of estimated parameter values. The low estimate allows for a slower rate of absorption that may result in the relatively low peak plasma concentration. The model-fitted profiles for the 1- and 5-mg/kg doses showed slight overestimation in the terminal phase. The rate of absorption (ka = 0.0119 min−1) is slow relative to the elimination rate (kel = 0.138 min−1).
Fig. 4.

Predicted and observed concentration-time profiles of AA after subcutaneous dosing at four dose levels (1 (triangles), 5 (circles), 45 (inverted triangles), and 150 mg/kg (squares)). A standard linear two-compartment model was used to characterize the PK
Table I.
Estimated Model Parameters for the Integrated PK/PD Model Applied to AA
| Parameter (units) | Definition | Final estimate | %SEM |
|---|---|---|---|
| Pharmacokinetic parameters | |||
| k el (1/min) | First-order elimination rate constant | 0.138 | 4.62 |
| k a (1/min) | First-order absorption rate constant | 0.0119 | 9.22 |
| k pt (1/min) | First-order rate constant for distribution of drug from plasma to tissues | 0.221 | 16.9 |
| k tp (1/min) | First-order rate constant for distribution of drug from tissues to plasma | 0.00605 | 15.3 |
| k rel | Multiplicative term on k a for 150 mg/kg dose level | 0.126 | 10.1 |
| V c /F (mL/kg) | Central volume of distribution | 688 | –a |
| Allosteric binding and PTH model parameters | |||
| α | Allosteric binding cooperativity constant | 0.406 | 21.9 |
| K D (mmol/L) | Equilibrium dissociation rate constant of Ca+2 with CaR | 1.20 | –a |
| K i (ng/mL) | Equilibrium dissociation rate constant of AA with CaR | 1.47 | 29.5 |
| S | Linear stimulation coefficient for PTH secretion | 20.4 | –a |
| k s (1/min) | First-order degradation rate constant of PTH precursor pool | 0.0397 | 30.7 |
| k p (1/min) | Steady-state first-order basal PTH production rate constant | 0.0132 | 29.5 |
| k out_PTH (1/min) | First-order loss rate constant of PTH from plasma | 0.544 | 7.86 |
| k τ_PTH (1/min) | First-order feedback delay rate constant | 8.67 E−04 | –a |
aFixed value
Pharmacodynamics
The model-fitted PTH concentration-time profiles are shown in Fig. 5. Transient dose-dependent stimulation of PTH from baseline in response to AA administration was observed for all doses. For the 1- and 5-mg/kg dose levels, elevated PTH concentrations returned to baseline values within 6 h. In contrast, sustained elevation of PTH was seen for the 45- and 150-mg/kg doses with a delayed return to baseline after 8 h. The integrated PK/PD model well characterized the PTH release in response to CaR antagonism. System parameters for the precursor-dependent PTH model are listed in Table I. All turnover parameters were estimated, and their values were similar to previously reported estimates (26). The kin_PP values were estimated and values ranged between 208 and 499 pg/mL/min. This range is indicative of the variability associated with the baseline data of PTH expression in different groups. The model predicted half-life for PTH (1.09 min) is similar to the previously reported half-life estimate (1 min) in rats (26).
Fig. 5.

Predicted and observed parathyroid hormone concentrations in normal adult rats for placebo and four dosing arms of AA. Lines represent model-fitted profiles
The Ki of AA for the CaR was estimated to be 1.47 ng/mL (4.3 nM). This estimate is similar to the experimental estimate for binding affinity to the human receptor of 1.0 ng/mL (2.9 nM) determined in vitro. The model estimate for α (0.406) indicates antagonistic activity of AA toward the CaR receptor.
Although there is a dose-dependent increase in PTH secretion, an apparent disconnect between the elevated exposure of the drug and resultant PTH concentrations exists at later times for the two higher dose levels. PTH concentrations were seen to approach baseline despite the relatively high drug concentrations detected in plasma at the 24-h time point. This was also evident from early attempts to characterize the PTH profiles using a model without negative feedback regulation (data not shown). The feedback model uses persistent PTH concentrations as a signal that has been delayed through transit compartments (n = 4). The delayed signal (M4) modulates the maximal stimulation of PTH to inhibit further secretion of PTH. The calculated transit time (τ) of 19.2 h is suggestive of a significant temporal delay in PTH feedback.
DISCUSSION
Current anabolic therapy approved for use in osteoporosis involves the use of full-length recombinant PTH or a fragment of the PTH hormone. To avoid the limitations associated with protein-based therapeutics, a new class of orally active small molecule compounds, CaR antagonists, or calcilytics are currently under development. To date, calcilytics are primarily amino alcohol-based compounds that are structurally related to the type II calcimimetics (37). In order to ensure efficacy and safety, CaR antagonists must elicit PTH secretion in a manner that is consistent with its pulsatile pharmacokinetic profile observed following daily subcutaneous dosing. The extent of PTH stimulation following administration of a CaR antagonist is dependent upon the rate and extent of decrease in Ca+2 binding to the CaR. Although such an understanding is critically important to research efforts aimed at developing negative allosteric CaR modulators, a formal mathematical analysis of these important kinetic properties has not been described to date.
In this study, a model-based analysis of preclinical PK/PD data was implemented for an investigational amino CaR antagonist, AA. A structural model describing the underlying biology of the PTH–Ca+2 system (26) was modified to include the allosteric interaction of the antagonist with its target receptor to elicit the desired pharmacological response. The integrated PK/PD model (Fig. 2) was able to successfully characterize the PK and the dose-dependent increase of circulating PTH driven by the interaction of the antagonist with the CaR receptor.
The PK profiles of AA were well characterized by a standard linear two-compartment model with first-order absorption (Fig. 4). The mean data for the 150-mg/kg dose reveal a shallower terminal phase slope (λZ = 0.001 min−1) as compared to the 1- and 5-mg/kg dose levels (λZ = 0.01 min−1). At the highest dose, complete washout of the drug is not achieved by 24 h, and as a result, the true terminal phase of the drug is not likely observed. The relatively shallow terminal phase at the higher dose might suggest a decreased systemic clearance of the drug at such dose levels. Attempts to capture these differences using a nonlinear elimination pathway were met with partial success only (data not shown). The parameter estimates for such a model were not precise and resulted in model instability. The overestimation in PK at the low doses is modest, and hence, a linear model was preferred as a parsimonious approach. Moreover, systematic deviations in terminal phases do not seem to significantly affect PTH profiles, especially as the PD response had returned to baseline values. To account for the apparent discrepancy in the observed peak plasma concentration for the highest dose level, the multiplicative krel parameter was estimated, and the low estimate (0.126) suggests that the rate constant of absorption is smaller at this dose level. The lower Cmax might also indicate differences in the extent of drug absorbed at the highest dose compared to lower dose levels.
The proposed pharmacodynamic model is biologically plausible and is based on the underlying biology of the Ca+2/PTH system. The secretion of PTH by the parathyroid glands is primarily under the control of the CaR and is governed by multiple factors (10). The principle mechanism is release of PTH from stored reserves in secretory vesicles. Enough PTH is stored such that secretion can last up to 60–90 min for a low sustained hypocalcemic stimulus (38). The structural model accounts for continuous synthesis of PTH, stimulatory PTH release from stores, and degradation of PTH to fragments (predominantly 7–84 amino acid long) within the storage vesicles (11). PTH concentrations in rats exhibit a circadian rhythm (39). However, a circadian rhythm for longitudinal PTH concentrations in rats was not included in the present model, as PTH data from our animal studies did not indicate a trend or influence of daily rhythms. Moreover, the placebo data are associated with significant variability. With the exception of the last time point, PTH concentrations are reasonably described by the baseline model profiles. Also, the last time points were not significantly lower than the initial time points. Overall, a trend or baseline shift is not well supported in the current dataset. The model well characterized PTH profiles that result from CaR antagonist administration (Fig. 5). The estimated PD parameter values for PTH (i.e. ks, kp, kout_PTH) are similar to previous estimates obtained from modeling the PTH–Ca+2 regulatory system in rats (26). The identical structural model has also been applied to describe the PTH–Ca+2 regulatory system in humans (26). These aspects of the model confer confidence in parameter estimates and indicate that the model may be useful in evaluating the translational pharmacology of agents affecting this system across species.
A simple model of receptor allosterism was used to codify the interactions of both Ca+2 and AA with the CaR (Eq. 9). In addition to providing a good characterization of PTH secretion and disposition, this model provided an estimate of Ki (1.47 ng/mL; 4.3 nM) that is generally consistent with the Ki value determined in vitro (1 ng/mL; 2.9 nM). The model estimate for the cooperativity constant (α = 0.406) is lower than 1, which indicates antagonism of the CaR. An α of 0.406 implies that AA decreases CaR affinity for Ca+2 by a factor of 2.46 (Kobs = KD/α) at AA concentrations in excess of its Ki. In contrast, AA would have an apparent behavior consistent with a simple competitive antagonist at concentrations below its Ki.
Finally, the proposed PK/PD model includes a functional feedback component to characterize the decrease in PTH concentrations associated with the sustained exposure to AA at later time points following the administration of high dose levels. At the two lowest doses (1 and 5 mg/kg), where linear pharmacokinetics is observed, the return of plasma PTH concentrations to baseline values was commensurate with the concentration of AA relative to its Ki for the CaR. However, at the two highest doses, plasma PTH concentrations returned to baseline values 24-h post-dose despite the maintenance of AA concentrations in excess of its Ki. The exact molecular mechanism of this feedback is not clear, and further research is needed to implement a more mechanistic approach to model this functional adaptation. However, the proposed empirical feedback component of the model was effective in capturing the observed decrease in PTH at these later times. The derived empirical transit-time parameter (τ) of 19.2 h is indicative of a relatively slow feedback regulation mechanism, which may not be important under therapeutic conditions in which the goal is to produce pulsatile elevations in PTH. Nevertheless, this aspect of the model highlights additional complexity in how the pharmacokinetics of these agents relate to associated changes in plasma PTH concentration and reinforces the utility of model-based analysis for the development of CaR antagonists.
CONCLUSION
A biologically plausible model has been developed, which was useful in characterizing the PK/PD data of a negative allosteric modulator of CaR, AA, administered to rats. Additional confidence in the model is derived from the fact that parameter estimates are consistent with those obtained from independent in vitro (i.e., binding assays) and in vivo (i.e., hypocalcemic clamp) studies. These findings suggest that the model may offer a useful framework for predicting rat and human PTH responses to CaR antagonists from preclinical data obtained at the earliest stages of research and development. Pulsatile PTH release is associated with enhanced bone formation, and the clinical implications of the temporal PTH profiles predicted by the final model will require further research. Although the current model does not include end-point measures such as bone mineral density, it may also provide the initial framework upon which additional end points could be added.
Acknowledgments
This study was funded, in part, by the University at Buffalo–Pfizer Strategic Alliance.
References
- 1.Tarantino U, Cannata G, Lecce D, Celi M, Cerocchi I, Iundusi R. Incidence of fragility fractures. Aging Clin Exp Res. 2007;19(4 Suppl):7–11. [PubMed] [Google Scholar]
- 2.Cummings SR, Melton LJ. Epidemiology and outcomes of osteoporotic fractures. Lancet. 2002;359(9319):1761–1767. doi: 10.1016/S0140-6736(02)08657-9. [DOI] [PubMed] [Google Scholar]
- 3.Department of Health and Human Services US. Bone health and osteoporosis: a report of the surgeon general: Rockville, MD: U.S. Department of Health and Human Services, Office of the Surgeon General. 2004.
- 4.Friedman PA. Agents affecting mineral ion homeostasis and bone turnover. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. 11th edn. New York McGraw-Hill Medical Publishing Division; 2006.
- 5.Lindsay R, Nieves J, Formica C, Henneman E, Woelfert L, Shen V, et al. Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350(9077):550–555. doi: 10.1016/S0140-6736(97)02342-8. [DOI] [PubMed] [Google Scholar]
- 6.Reeve J, Meunier PJ, Parsons JA, Bernat M, Bijvoet OL, Courpron P, et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: a multicentre trial. Br Med J. 1980;280(6228):1340–1344. doi: 10.1136/bmj.280.6228.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reeve J, Tregear GW, Parsons JA. Preliminary trial of low doses of human parathyroid hormone 1–34 peptide in treatment of osteoporosis. Calcif Tissue Res. 1976;21(Suppl):469–477. [PubMed] [Google Scholar]
- 8.Black DM, Bouxsein ML, Palermo L, McGowan JA, Newitt DC, Rosen E, et al. Randomized trial of once-weekly parathyroid hormone (1–84) on bone mineral density and remodeling. J Clin Endocrinol Metab. 2008;93(6):2166–2172. doi: 10.1210/jc.2007-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 10.Juppner HW, Gardella TJ, Brown EM, Kronenberg HM, Potts JT., Jr . Parathyroid hormone and parathyroid hormone-related peptide in the regulation of calcium homeostasis and bone development. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Philadelphia: Saunders; 2001. pp. 969–998. [Google Scholar]
- 11.Friedman PA, Goodman WG. PTH(1–84)/PTH(7–84): a balance of power. Am J Physiol Ren Physiol. 2006;290(5):F975–F984. doi: 10.1152/ajprenal.00336.2005. [DOI] [PubMed] [Google Scholar]
- 12.Chen RA, Goodman WG. Role of the calcium-sensing receptor in parathyroid gland physiology. Am J Physiol Ren Physiol. 2004;286(6):F1005–F1011. doi: 10.1152/ajprenal.00013.2004. [DOI] [PubMed] [Google Scholar]
- 13.Gowen M, Stroup GB, Dodds RA, James IE, Votta BJ, Smith BR, et al. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest. 2000;105(11):1595–1604. doi: 10.1172/JCI9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, et al. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther. 2001;299(1):323–331. [PubMed] [Google Scholar]
- 15.Marquis RW, Lago AM, Callahan JF, Rahman A, Dong X, Stroup GB, et al. Antagonists of the calcium receptor. 2. Amino alcohol-based parathyroid hormone secretagogues. J Med Chem. 2009;52(21):6599–6605. doi: 10.1021/jm900563e. [DOI] [PubMed] [Google Scholar]
- 16.Marquis RW, Lago AM, Callahan JF, Trout RE, Gowen M, DelMar EG, et al. Antagonists of the calcium receptor I. Amino alcohol-based parathyroid hormone secretagogues. J Med Chem. 2009;52(13):3982–3993. doi: 10.1021/jm900364m. [DOI] [PubMed] [Google Scholar]
- 17.Arey BJ, Seethala R, Ma Z, Fura A, Morin J, Swartz J, et al. A novel calcium-sensing receptor antagonist transiently stimulates parathyroid hormone secretion in vivo. Endocrinology. 2005;146(4):2015–2022. doi: 10.1210/en.2004-1318. [DOI] [PubMed] [Google Scholar]
- 18.Balan G, Bauman J, Bhattacharya S, Castrodad M, Healy DR, Herr M, et al. The discovery of novel calcium sensing receptor negative allosteric modulators. Bioorg Med Chem Lett. 2009;19(12):3328–3332. doi: 10.1016/j.bmcl.2009.04.044. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Matheny CJ, Hoffman SJ, Marquis RW, Schultz M, Liang X, et al. An orally active calcium-sensing receptor antagonist that transiently increases plasma concentrations of PTH and stimulates bone formation. Bone. 2010;46(2):534–542. doi: 10.1016/j.bone.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 20.Lotinun S, Sibonga JD, Turner RT. Differential effects of intermittent and continuous administration of parathyroid hormone on bone histomorphometry and gene expression. Endocrine. 2002;17(1):29–36. doi: 10.1385/ENDO:17:1:29. [DOI] [PubMed] [Google Scholar]
- 21.Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138(11):4607–4612. doi: 10.1210/en.138.11.4607. [DOI] [PubMed] [Google Scholar]
- 22.Frolik CA, Black EC, Cain RL, Satterwhite JH, Brown-Augsburger PL, Sato M, et al. Anabolic and catabolic bone effects of human parathyroid hormone (1–34) are predicted by duration of hormone exposure. Bone. 2003;33(3):372–379. doi: 10.1016/S8756-3282(03)00202-3. [DOI] [PubMed] [Google Scholar]
- 23.Brown EM. Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. J Clin Endocrinol Metab. 1983;56(3):572–581. doi: 10.1210/jcem-56-3-572. [DOI] [PubMed] [Google Scholar]
- 24.Momsen G, Schwarz P. A mathematical/physiological model of parathyroid hormone secretion in response to blood-ionized calcium lowering in vivo. Scand J Clin Lab Investig. 1997;57(5):381–394. doi: 10.3109/00365519709084585. [DOI] [PubMed] [Google Scholar]
- 25.Raposo JF, Sobrinho LG, Ferreira HG. A minimal mathematical model of calcium homeostasis. J Clin Endocrinol Metab. 2002;87(9):4330–4340. doi: 10.1210/jc.2002-011870. [DOI] [PubMed] [Google Scholar]
- 26.Abraham AK, Mager DE, Gao X, Li M, Healy DR, Maurer TS. Mechanism-based pharmacokinetic/pharmacodynamic model of parathyroid hormone-calcium homeostasis in rats and humans. J Pharmacol Exp Ther. 2009;330(1):169–178. doi: 10.1124/jpet.109.152033. [DOI] [PubMed] [Google Scholar]
- 27.Van Wagenen BC, Del Mar EG, Sheehan D, Barmore RM, Keenan RM, Kotecha NR, et al., inventors; Calcilytic Compounds patent WO 97/37967 A1. 1997.
- 28.Sharma A, Ebling WF, Jusko WJ. Precursor-dependent indirect pharmacodynamic response model for tolerance and rebound phenomena. J Pharm Sci. 1998;87(12):1577–1584. doi: 10.1021/js980171q. [DOI] [PubMed] [Google Scholar]
- 29.Martin KJ, Hruska KA, Lewis J, Anderson C, Slatopolsky E. The renal handling of parathyroid hormone. Role of peritubular uptake and glomerular filtration. J Clin Invest. 1977;60(4):808–814. doi: 10.1172/JCI108834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daugaard H, Egfjord M, Olgaard K. Metabolism of parathyroid hormone in isolated perfused rat kidney and liver combined. Kidney Int. 1990;38(1):55–62. doi: 10.1038/ki.1990.166. [DOI] [PubMed] [Google Scholar]
- 31.Nemeth EF. Pharmacological regulation of parathyroid hormone secretion. Curr Pharm Des. 2002;8(23):2077–2087. doi: 10.2174/1381612023393387. [DOI] [PubMed] [Google Scholar]
- 32.Chattopadhyay N. Biochemistry, physiology and pathophysiology of the extracellular calcium-sensing receptor. Int J Biochem Cell Biol. 2000;32(8):789–804. doi: 10.1016/S1357-2725(00)00031-5. [DOI] [PubMed] [Google Scholar]
- 33.Kenakin T. Allosteric modulators: the new generation of receptor antagonist. Mol Interv. 2004;4(4):222–229. doi: 10.1124/mi.4.4.6. [DOI] [PubMed] [Google Scholar]
- 34.Kenakin T. Allosteric drug antagonism. A pharmacology primer: theory, application, and method 2nd edn: Academic Press; 2006. p. 127–46.
- 35.Ehlert FJ. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol. 1988;33(2):187–194. [PubMed] [Google Scholar]
- 36.D’Argenio D, Schumitzky A. ADAPT II user’s guide. Los Angeles: Biomedical Simulations Resource; 1997. [Google Scholar]
- 37.Hu J, Reyes-Cruz G, Goldsmith PK, Spiegel AM. The Venus’s-flytrap and cysteine-rich domains of the human Ca2+ receptor are not linked by disulfide bonds. J Biol Chem. 2001;276(10):6901–6904. doi: 10.1074/jbc.C000865200. [DOI] [PubMed] [Google Scholar]
- 38.Brown EM. Calcium receptor and regulation of parathyroid hormone secretion. Rev Endocr Metab Disord. 2000;1(4):307–315. doi: 10.1023/A:1026570518919. [DOI] [PubMed] [Google Scholar]
- 39.Ostrowska Z, Kos-Kudla B, Marek B, Kajdaniuk D, Ciesielska-Kopacz N. The relationship between the daily profile of chosen biochemical markers of bone metabolism and melatonin and other hormone secretion in rats under physiological conditions. Neuro Endocrinol Lett. 2002;23(5–6):417–425. [PubMed] [Google Scholar]