

Abstract
A model-based strategy was used to inform the early clinical development of anacetrapib, a novel cholesteryl ester transfer protein inhibitor under development for the treatment of hyperlipidemia. The objectives of this model-based approach were to enable bridging variable pharmacokinetic effects, differences among formulations used in development, and to identify an appropriate dose for the phase III confirmatory program. Nonlinear mixed effects PK/PD models were initially developed based on data obtained from multiple phase I studies and later were updated with data from a phase IIb study. The population pharmacokinetic model described differences between the liquid-filled capsule used in phase I and phase IIb and the hot-melt extruded (HME) tablet formulation introduced in phase III, allowing for bridging of the two formulations, and quantified the complex relationship of apparent anacetrapib bioavailability with subject meal intake. Proportional Emax models quantified the relationships between anacetrapib trough concentration and lipoprotein effects (LDL-C and HDL-C), with covariate effects of study population (normal volunteers vs. patients), and co-administration with HMG-CoA reductase inhibitor (“statin”). The interaction between anacetrapib and atorvastatin suggested pharmacological independence, i.e., that when given together, each agent exerts the same proportional lipid effect observed from monotherapy. Clinical trial simulation was used to examine the robustness of the effects to random dietary indiscretion, and found that the results were robust as long as patients generally adhered to a low-fat diet. These results allowed the selection of the 100 mg dose with the HME formulation for phase III development even though this dose and formulation were not specifically studied in a phase IIb trial.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-011-9254-0) contains supplementary material, which is available to authorized users.
Key words: anacetrapib, cetp inhibition, clinical trial simulation, modeling
INTRODUCTION
Anacetrapib is a potent and selective inhibitor of cholesteryl ester transfer protein (CETP) currently under development for the treatment of primary hypercholesterolaemia and mixed hyperlipidaemia. Anacetrapib has been generally well tolerated in phase I and II studies, and has demonstrated lipid-altering effects greater than any member of its class (1–4). Key development questions for this drug included selection of an optimal dose for phase III development, understanding the effects of formulation or fed state on drug exposure and lipid effects, elucidation of a predictive exposure–response relationship between anacetrapib and high-density lipoprotein (HDL-C) or low-density lipoprotein cholesterol (LDL-C), and the likely extent to which these effects change when patients are treated with anacetrapib in combination with an HMG-CoA reductase inhibitor (statin).
The pharmacokinetics of anacetrapib exhibits significant variation in the fasted and fed states. In controlled phase I studies of a hot-melt extruded tablet (HME), administration of a standard high-fat breakfast with single or multiple doses of 150 mg anacetrapib resulted in ∼7.5-fold increase in area under curve (AUC) and ∼17-fold increase in Cmax relative to the fasted state. A low-fat meal increased AUC ∼2.2-fold (∼3.7-fold increase in Cmax) (2,4). An Imwitor/Tween liquid-filled capsule (LFC) formulation was used in early phase I development while the HME tablet was introduced in phase III following limited phase I evaluation. The pharmacodynamic effects of anacetrapib were reflected by changes in CETP activity and concentrations, by increases in HDL and decreases LDL cholesterol (HDL-C and LDL-C, respectively).
A model-based strategy was applied to anacetrapib development to facilitate the (1) optimal selection of a clinical dose for phase III that is not limited to doses specifically used in phase IIb; and (2) characterization of a formulation with a similar but not bioequivalent pharmacokinetic profile to that used in phase IIb; and (3) assessment of the effects of meal condition on pharmacokinetic (PK) exposures and effects on lipid endpoints. Thus, the objectives of the work described here were: (1) to characterize the population pharmacokinetics and pharmacodynamics of anacetrapib in healthy volunteers and in patients with dyslipidemia for the two formulations of interest, and under different meal conditions and (2) to conduct appropriate simulations predicting the mean and observed effects of anacetrapib as a function of dose when given as monotherapy or coadministered with a statin under different meal conditions.
METHODS
A population PK model was first developed based on data from several completed phase I studies. Initial pharmacodynamic models were then developed, followed by updates to the population PK model and PD models, based on emerging data from the phase IIb study. These are further detailed in the following sections.
PK Model Development
The studies, doses, formulations, and sampling details for the population PK analysis are summarized in Table IA in the Electronic Supplementary Material. Pharmacokinetic model development was performed in NONMEM Version 5.0 using a first-order conditional estimation with interaction (“FOCE INTER”) (5). Diagnostic plots, point, and interval estimates of parameters, residuals, and the minimum value of the NONMEM objective function (“OFV”) were used to guide model building and to assess goodness-of-fit using standard methods (5). Pharmacokinetic model evaluation included various compartment structures with first-order absorption. The model assessed effects of dose, fat content of meals, number of capsules per dose (for LFC only), and of formulation on bioavailability.
Pharmacokinetic/Pharmacodynamic (HDL-C and LDL-C) Model Development
Exposure–response models for HDL-C and LDL-C developed for the phase I dataset were used as a basis for further model optimization in the merged dataset that included data from the phase IIb study. Exploratory graphical analysis and initial model development evaluated various metrics for biomarkers of drug effect and of drug exposure as predictors of changes in HDL-C or LDL-C levels, including trough % CETP inhibition, 24 h average % CETP inhibition, trough CETP activity, 24 h average CETP activity, anacetrapib trough concentration, and anacetrapib daily average concentration (both individual and population-predicted trough concentrations were examined).
For the phase I dataset, several model structures were attempted including a linear model, power model, proportional Emax (Eq. 1), and additive Emax (Eq. (2).
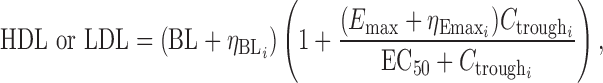 |
1 |
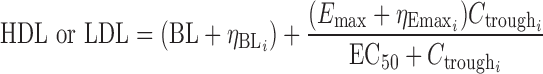 |
2 |
where BL is the estimate of the baseline value of HDL-C or LDL-C, 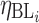 is the intersubject error on the BL for subject i, Emax is the maximum response,
is the intersubject error on the BL for subject i, Emax is the maximum response, 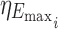 is the intersubject error on Emax,
is the intersubject error on Emax, 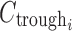 is the anacetrapib trough concentration, and EC50 is the trough concentration producing 50% of the maximum response. Exploratory graphical analysis and preliminary model development were conducted to narrow the range of metrics for model optimization. Concentrations were predicted from individual Bayesian post hoc or population-predicted PK parameter estimates to determine trough and average daily concentrations.
is the anacetrapib trough concentration, and EC50 is the trough concentration producing 50% of the maximum response. Exploratory graphical analysis and preliminary model development were conducted to narrow the range of metrics for model optimization. Concentrations were predicted from individual Bayesian post hoc or population-predicted PK parameter estimates to determine trough and average daily concentrations.
Similar to the development of models based on the initial phase I data, different pharmacodynamics model structures were explored to assess the best fit to the HDL-C and LDL-C data for the merged dataset combining phase I and II studies. Exploratory graphical analysis of the data was performed to suggest preliminary model structures and to identify potential covariate effects. Additional patient covariates included subject health status, i.e. normal healthy volunteers (NHV) versus patients with dyslipidemia, and the effect of co-administration of atorvastatin. The NLME package as implemented in SPLUS version 8.0 (Insightful, Seattle, USA) was used for model development.
Assessment of Model Fit
Point and 95% prediction interval estimates of parameters, likelihood ratio comparisons, diagnostic plots, and plausibility of parameter estimates were used to guide model building and to assess the goodness-of-fit. Nontransformed model parameter estimates were deemed significantly different from zero if their 95% prediction interval excluded zero. Models were compared by likelihood ratio to determine which model provided the better fit to the data, with increased likelihood (reduction in −2log-likelihood) indicating the better fit. When comparing nested models, a p value <0.05 was required for statistically significant improvement in the model likelihood and selection of the more complex model. Diagnostic plots included predicted versus observed response plots, exposure–response plots overlaying the observed and population-predicted response, and residual plots. To identify model lack of fit, weighted residual plots were examined, including residuals or inter-individual random effects versus time and residuals versus covariates of interest. Visual-predictive checks of the pharmacodynamic models were used to verify that the models adequately described the data used to construct them.
Simulations
Both population simulation and clinical trial simulation approaches were used. The purpose of a population simulation was to predict the expected response in a particular population subgroup under a given treatment scenario, irrespective of trial design constraints. In contrast, clinical trial simulations imposed the limitations of a particular clinical trial design, including among other characteristics, a particular sample size, mixture of enrolled patient populations, and observation schedule, to predict the distribution of expected outcomes from a given clinical trial.
Population Simulation
Here, population simulation was used to predict the response to anacetrapib under various dose and/or formulation scenarios possibly differing from the treatment regimens under which the response data were generated. Population simulations also helped to understand the effects of covariates (e.g., formulation and food) and parameter uncertainty on the expected exposure–response. The following algorithm was applied for each simulated treatment condition (i.e., dose, fed state, formulation, etc.): for each replicate (1,000 replicates per condition), (1) a sample of population fixed and random effects was drawn from the final approximate variance–covariance matrix of parameter estimates, (2) the population-predicted trough concentration was calculated, and (3) the expected lipid response was calculated based on the population-predicted trough concentration, the specified treatment condition, and the population covariate conditions of interest. The population simulations assumed that PK parameters are distributed independently from the PD model parameters. Simulation results were uploaded to Drug Model Explorer® (DMX®, Pharsight Corporation, Cary, NC) to provide a quantitative framework for exploring the efficacy profile of anacetrapib under various scenarios of interest.
Clinical Trial Simulations
Because the PK of anacetrapib was sensitive to meals, clinical trial simulation was used here to evaluate the impact of prescribed diet and dietary compliance, in conjunction with dose, on trial outcome. Three types of meal conditions were examined in the anacetrapib clinical development program. The standard low-fat and high-fat breakfast employed in phase I studies were as follows: the standard low-fat meal containing 373 kcal with 20% fat content and contained two slices of toasted white bread, one teaspoon low-fat margarine, one tablespoon jelly, 5 oz skim milk, and 5 oz orange juice; a standard high-fat meal containing 827 kcal with 57% fat content and contained two fried or scrambled eggs, two strips of bacon, two slices of toast with two pats of butter, 4-oz (113 g) hash browns (fried potato), and 240 mL of whole milk. A third meal condition was employed in the phase Ib and phase IIb trials, and consisted of a patient-selected meal based on protocol instructions. The meal selections conformed to the American Heart Association’s TLC diet, and were similar in fat and caloric content to a low-fat meal. A mix of patient-selected meal and low-fat meals was employed in the phase Ib study (see Table IA in the Electronic Supplementary Material) (1).
As part of the strategy to guide dose selection for the phase IIb study, clinical trial simulations were performed to also evaluate the effect of dietary indiscretion on pharmacodynamic endpoints. The steps undertaken for the clinical trial simulations were roughly analogous to those for the population simulations: specifically, for each simulated trial: (1) a sample of population fixed and random effects from parameter uncertainty variance–covariance matrix was drawn; (2) a sample of inter-patient random effects of appropriate sample size was drawn from the random-effects variance–covariance matrix; (3) the population expected trough concentrations were calculated; (4) individual lipid response to protocol-specified treatment specified time points was then simulated; (5) residual error was added to each patient’s expected lipid response at each observation time point; (6) the mean lipid response across patients was calculated; and (7) the protocol-specified test for statistical significance was then conducted. Each simulation was comprised of 1,000 simulated trials. Three different dietary compliance conditions were evaluated in the simulations. These included low-fat diet (100% of patients), high-fat diet (100% of patients), and noncompliant diet (75% of patients with low-fat diet and 25% of patients fasting). The simulated clinical trial protocol included of the following design elements: parallel design, with treatment arms of placebo, 10, 50, 100, 150, 250, and 300 mg, with the assumption of 45 subjects per arm (315 subjects total), allocated by block randomization.
RESULTS AND INTERPRETATION
Data from the eight studies shown in Table IA in the Electronic Supplementary Material were merged to form the final combined dataset for development of the population PK model. The total dataset included information from 576 subjects, approximately 60% of whom were patients. The five studies shown in Table IB in the Electronic Supplementary Material comprise the final dataset used for development of the HDL-C and LDL-C exposure–response models. The total dataset included 546 subjects, including 474 patients and 72 NHVs.
Population PK Model
The model development process initiated with a two-compartment model with 1st-order absorption fit to the phase I data alone, with evaluation of covariate effects on bioavailability. The final merged dataset included data from both phase I and phase II studies. The initial, phase I model development explored effects of meal fat content, number of capsules (for LFC formulation), and formulation type (HME tablets vs. LFC) on bioavailability. The number of capsules was considered as a surrogate for the amount of liquid surfactant contained in the capsule unit making up a given dose (4). The most meaningful improvements to the model upon addition of the phase IIb data were additional effects on bioavailability related to food (patient-selected meals in phase IIb), formulation effects, capsule effects, and inclusion of covariance between apparent clearance and bioavailability.
Final Model Description
The final population PK model parameters are provided in Table I. A two-compartment model with first-order absorption described anacetrapib disposition in NHVs and patients. The final model was parameterized in terms of systemic clearance (CL), intercompartmental clearance, central and peripheral compartment volumes (V2 and V3), and bioavailability (F1). Intersubject variability (IIV) could only be identified on CL and F1. The population PK model for anacetrapib is described by Eqs. 3–7.
Table I.
Population PK Model Parameter Estimates
| Parameter | Description | Estimate | SE | % CV |
|---|---|---|---|---|
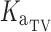 (1/h) (1/h) |
Typical absorption rate constant | 0.48a | Fixed | – |
| CLTV (L/h) | Typical clearance from central compartment | 7.6 | 1.0 | 13.4 |
| V 2TV (L) | Typical central volume of distribution | 55 | 7 | 13.4 |
| Q TV (L/h) | Typical intercompartmental clearance | 5.3 | 0.7 | 13.9 |
| V 3TV (L) | Typical volume of peripheral compartment | 244 | 33 | 13.6 |
| D 50 | Dose of half-maximal effect on bioavailability | 55 | 5 | 10.0 |
| D max | Maximal effect of dose on bioavailability | 1 | Fixed | – |
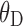
|
Interaction of high-fat meal on bioavailability | 274 | 99.5 | 36.3 |
| T lag | Lag time for appearance of drug in plasma | 0.918 | 0.006 | 0.67 |
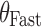 (—) (—) |
Effect of capsule number on bioavailability in fasted subjects | 0.07 | 0.02 | 21.1 |
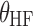 (—) (—) |
Effect of high-fat meal on bioavailability | 2.7 | 0.3 | 11.7 |
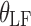 (—) (—) |
Effect of low-fat meal on bioavailability | 2.4 | 0.4 | 14.3 |
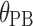 (—) (—) |
Effect of patient-selected meal on bioavailability | 3.3 | 0.4 | 13.1 |
| C 50 | Capsule number for half-maximal effect on bioavailability | 0.67 | 0.14 | 20.4 |
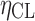
|
Intersubject variability on CL | 0.098 | 9.4e−3 | 9.6 |
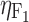
|
Intersubject variability on F 1 | 0.21 | 0.02 | 8.9 |
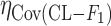
|
Covariance between 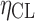 and and 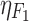
|
0.04 | 8.2e−3 | 20.5 |
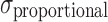
|
Proportional error | 0.184 | 7.4e−3 | 4.0 |
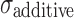
|
Additive error | 32.4 | 9.16 | 28.3 |
Q intercompartmental clearance, SE standard error of parameter estimate
aParameter was fixed at previously estimated value to achieve convergence (rounding errors were preventing convergence)
Clearance is given by:
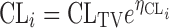 |
3 |
where CLi is the systemic clearance in subject i and CLTV is the population estimate of systemic clearance.
Equation 4 describes apparent bioavailability
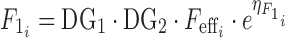 |
4 |
where 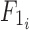 is the bioavailability in subject i,
is the bioavailability in subject i, 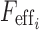 is the effect of the number of capsules or tablets and meal type on bioavailability (Eq. 5), and DG1 and DG2 describe dose and meal effects (Eq. 6) and the effect of capsule number on bioavialibility is given in Eq. 7.
is the effect of the number of capsules or tablets and meal type on bioavailability (Eq. 5), and DG1 and DG2 describe dose and meal effects (Eq. 6) and the effect of capsule number on bioavialibility is given in Eq. 7.
The effect of the number of capsules in the fasted state, or meal type for fed subjects is described in Eq. 5:
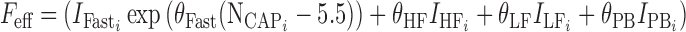 |
5 |
where 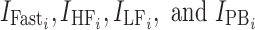 are indicator variables for subject i that equal 1 for fasted dosing, high-fat meal, low-fat meal, and patient-selected meal, respectively, and 0 otherwise. The effect of
are indicator variables for subject i that equal 1 for fasted dosing, high-fat meal, low-fat meal, and patient-selected meal, respectively, and 0 otherwise. The effect of 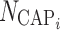 , the number of capsules, is centered on the median number of capsules for fasted subjects. Thus, θFAST, θHF, θLF, and θPB describe the change in bioavailability for fasted subjects or high-fat, low-fat, or patient-selected meals, respectively. Food and formulation effects on bioavailability were dose-dependent. For a 100 mg tablet, the typical value of bioavailability for fasted subjects is 0.35. For subjects on a low-fat, patient-selected, or high-fat diet, relative bioavailability is 0.85, 1.17, or 2.07, respectively. The dose effect is given by Eq. 6:
, the number of capsules, is centered on the median number of capsules for fasted subjects. Thus, θFAST, θHF, θLF, and θPB describe the change in bioavailability for fasted subjects or high-fat, low-fat, or patient-selected meals, respectively. Food and formulation effects on bioavailability were dose-dependent. For a 100 mg tablet, the typical value of bioavailability for fasted subjects is 0.35. For subjects on a low-fat, patient-selected, or high-fat diet, relative bioavailability is 0.85, 1.17, or 2.07, respectively. The dose effect is given by Eq. 6:
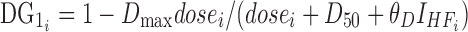 |
6 |
where Dmax, the maximum effect of dose on bioavailability, was fixed at 1, dosei is the anacetrapib dose for the ith subject, D50 is the dose of half the maximal inhibition, θD is the influence of high-fat meal on the half-maximal effect, and 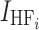 is an indicator variable for consumption of a high-fat meal prior to dosing. Higher doses of anacetrapib exhibited decreased bioavailability, and there was significant interaction of this effect with subject fed state. The half-maximal dose effect was achieved at a 55 mg (44 to 66 mg) dose for the fasted state with a low-fat or patient-selected diet. This inhibition was mitigated by a high-fat diet, which increased the dose required for half-maximal inhibition from 55 to 329 mg (123 to 535 mg).
is an indicator variable for consumption of a high-fat meal prior to dosing. Higher doses of anacetrapib exhibited decreased bioavailability, and there was significant interaction of this effect with subject fed state. The half-maximal dose effect was achieved at a 55 mg (44 to 66 mg) dose for the fasted state with a low-fat or patient-selected diet. This inhibition was mitigated by a high-fat diet, which increased the dose required for half-maximal inhibition from 55 to 329 mg (123 to 535 mg).
The effect of capsule number on bioavailability is given by Eq. 7:
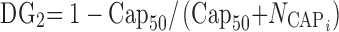 |
7 |
where Cap50 is the number of capsules for a half-maximal effect on bioavailability and 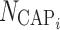 is the number of capsules dosed for subject i. An increase in the number of capsules increases bioavailability by an Emax relationship for fed subjects (high-fat, low-fat, or patient-selected diet), and increases bioavailability exponentially for fasted subjects (when NCAP > 5.5 capsules). A two-capsule dose increases bioavailability by 25% in fed subjects and 35% in fasted subjects. For a six-capsule dose, the increase in fasted subjects is 113% compared with a 50% increase in fed subjects.
is the number of capsules dosed for subject i. An increase in the number of capsules increases bioavailability by an Emax relationship for fed subjects (high-fat, low-fat, or patient-selected diet), and increases bioavailability exponentially for fasted subjects (when NCAP > 5.5 capsules). A two-capsule dose increases bioavailability by 25% in fed subjects and 35% in fasted subjects. For a six-capsule dose, the increase in fasted subjects is 113% compared with a 50% increase in fed subjects.
Model Assessment
Model parameters were generally well estimated. The final PK model parameters are given in Table I. Figure 1 shows the model diagnostic plots for the final model. The lower panels show that residual errors are distributed well over the range of predicted concentration and that the residuals are approximately normally distributed around a mean of zero with a standard deviation ∼1. Weighted residuals show no unexplained variability by trial, diet, number of capsules, dose, formulation, or subject health status (data not shown).
Fig. 1.

Model diagnostic plots for the population PK model. The dashed line is smooth through the data. The solid line is the line of unity in the upper panels and a horizontal line with intercept zero in the lower left panel
PK/PD Model Development
Exploratory data analyses were performed before modeling exposure–response relationships with the pharmacodynamic parameters of interest of HDL-C and LDL-C. Several metrics were evaluated including trough % CETP inhibition, 24 h % CETP inhibition, trough CETP activity, 24 h average CETP activity, anacetrapib trough concentration, and anacetrapib daily average concentration, to determine the metric that was the best predictor of lipid-altering effects.
Exploratory plots of the relationship between HDL-C and trough CETP activity and 24-h average CETP activity showed somewhat paradoxical behavior. Initially, HDL-C levels increased with declining CETP activity, as expected; however, at later time points HDL-C appears to increase with increasing CETP activity. These characteristics are due to the fact that CETP activity declines sharply on the first day of dosing, but the effect is not as pronounced after subsequent doses, even though the lipid effects do not change appreciably over time with continuous daily dosing (2). Trough CETP inhibition and 24 h average CETP inhibition were also not consistently predictive of HDL-C levels. Additionally, the largest increases in HDL-C occur over a relatively narrow range of CETP inhibition. Because of the inconsistent exposure–response relationship between CETP activity and relative efficacy of different doses, a mechanistic model relating drug to target engagement biomarker (i.e., CETP activity and concentration), and the pharmacodynamic effect (changes in lipid parameters) was not pursued further.
However, HDL-C levels did display consistent relationships with predicted anacetrapib trough concentrations and 24 h average concentrations. Of these, the predicted anacetrapib trough concentration was most predictive of changes in subject lipid levels.
Final PK/HDL-C and PK/LDL-C Model Development
The final models for HDL-C and LDL-C were developed using a combined dataset that included 72 NHVs and 474 patients. Several structural models were attempted including linear, additive Emax, and proportional Emax. Effects of study population (NHVs vs. patients) and of atorvastatin were also investigated.
The proportional Emax model provided the best fit to the observed HDL-C data. Parameter estimates for the final model fit to the phase I and phase IIb data alone and to the integrated dataset (phase I and phase IIb) are provided in Table II. Relative to the earlier phase I analysis, the integrated dataset includes approximately five times as many observations in almost seven times as many patients. Not surprisingly, Emax and EC50 were more precisely estimated with the larger dataset (i.e., with phase I and phase II data), but the parameter values were similar among the analyses.
Table II.
Parameter Estimates for HDL-C Exposure–Response Model
| Parameter | Estimate (SE) | ||
|---|---|---|---|
| Phase 1 data alone | Phase IIb data alone | Integrated dataset | |
| Baseline HDL-C (mg/dL) | 53.8 (1.6) | 50.6 (0.6) | 50.8 (0.5) |
| E max (dimensionless) | 1.6 (0.10) | 1.6 (0.07) | 1.76 (0.07) |
| EC50 (ng/mL) | 140 (13%a) | 108 (10%) | 135 (8%) |
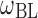
|
13.2 (1) | 11.4 (0.4) | 11.7 (0.4) |
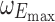
|
— | 0.38 (0.03) | 0.40 (0.04) |
| σ | 6.5 (0.2) | 7.3 (0.1) | 7.6 (0.1) |
| N observations | 617 | 2,515 | 3,089 |
| N subjects | 79 | 473 | 545 |
aThe error for EC50 is an approximate coefficient of variation
The final proportional Emax model for HDL-C (Eq. 8) includes intersubject random effects on baseline HDL-C and on the maximal drug effect. No significant distinctions between patient populations or with respect to statin administration on the baseline HDL-C or the maximum effect of anacetrapib were revealed.
Equation 8 Final HDL-C exposure–response model
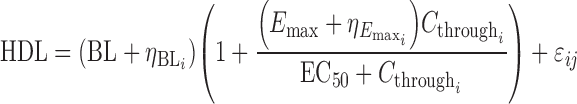 |
8 |
The final parameter estimate for the maximum drug effect, Emax, is 1.76 suggesting that at maximum effect, anacetrapib provides an increase of 176% (95% CI, 162% to 190%) over baseline HDL-C levels. As shown by the visual-predictive checks in Fig. 2, the observed data are mostly very well predicted by the final model (additional diagnostic plots are found in Figs. 1 and 3 in the Electronic Supplementary Material). There is some lack of fit among normal volunteers at the highest exposure levels (C24h > 400 ng/mL; 400 mg dose, six subjects receiving high-fat meal). However, the overall model fit is dominated by the large amount of data at lower doses in both NHV and dyslipidemic patients. As shown in Table II, the parameter estimates changed only slightly when the phase IIb data were added to the initial phase I data.
Fig. 2.

Visual-predictive check of HDL-C model versus trough anacetrapib concentration a, all data; b, by category. (Abbreviations: NHV normal healthy volunteers, Pt patients)
Exploratory data analyses of the LDL-C response revealed differences between dyslipidemic patients and healthy subjects at baseline, as expected, as well as a significant additional effect of atorvastatin treatment. The relationship between anacetrapib trough concentration and LDL-C in patients remains evident with or without atorvastatin, though patients treated with a combination of atorvastatin and anacetrapib exhibit a larger response than those treated with either drug alone. There was a clear dose–response with respect to anacetrapib in patients treated with atorvastatin. Several structural models were attempted including linear, additive Emax, and proportional Emax. Population-specific parameters (NHVs vs. patients) and the effect of atorvastatin were also investigated. Both the additive and the proportional Emax models produce an acceptable fit to the observed LDL-C data. Though the additive model provided a somewhat better fit on the basis of −2log-Likelihood values, the proportional Emax model is biologically more plausible. The proportional model fit the observed data adequately with a single estimate for Emax across populations (healthy subjects and patients) while the additive model (data not shown) required separate population-specific Emax estimates, and the fit of the proportional model was very similar to that of the additive model.
Table III displays the final parameter estimates for the proportional Emax LDL-C model fit to both population- and individual-predicted anacetrapib exposures; these parameter estimates do not differ meaningfully. As shown in the visual-predictive check Fig. 3, the observed data are reasonably well described by the proportional Emax model (additional diagnostic plots are shown in Figs. 2, 4, and 5 in the Electronic Supplementary Material). The proportional model includes differences in baseline LDL-C between NHV and dyslipidemic patients, an atorvastatin effect and an interaction between the effects of anacetrapib exposure and atorvastatin. The baseline estimates were almost identical to the estimates from the additive model, with a baseline LDL-C about 33 mg/dL (27 to 40 mg/dL) higher in patients than healthy subjects (Fig. 4 in the Electronic Supplementary Material). In the proportional model, no difference in drug treatment effect was found between patients and NHVs. Daily treatment with atorvastatin, 20 mg acts to reduce LDL-C 44.2% (95% CI, 42.5% to 46%) from baseline, which is comparable to a published value of 42.7% reduction for the effect of a 20 mg atorvastatin dose (6). The estimate of the interaction parameter in the proportional model is 0.99 (0.88 to 1.1), which suggests pharmacologic independence between the effects of atorvastatin and anacetrapib. When given in combination with atorvastatin, the anacetrapib reduces LDL-C a further 43.8% (38% to 50%) (Fig. 5 Electronic Supplementary Material).
Table III.
Parameter Estimates for the Proportional E max LDL-C Exposure–Response Model
| Parameter | Symbol | Estimate (SE) based on | |
|---|---|---|---|
| Population-predicted PK | Individual-predicted PK | ||
| Baseline-volunteers (mg/dL) | BLNHV | 107 (3) | 103 (3) |
| Baseline LDL-C-patients (mg/dL) | BLPts | 140 (1) | 141 (1) |
| Effect of atorvastatin, 20 mg/d |
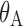
|
−0.442 (0.009) | −0.445 (0.008) |
| E max for anacetrapib in monotherapy | E max | −0.80 (0.04) | −0.78 (0.04) |
| EC50 (ng/mL) | EC50 | 237 (25) | 240 (24) |
| Anacetrapib–atorvastatin interaction effect | γ | 0.99 (0.06) | 0.95 (0.05) |
| Between-subject variability in baseline LDL-C |
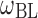
|
25 (0.9) | 24 (0.8) |
| Residual variability | σ | 16 (0.2) | 16 (0.2) |
| N obervations | 3,078 | 3,078 | |
| N subjects | 544 | 544 | |
Fig. 3.

Visual-predictive check of LDL-C model in normal healthy volunteers (NHV) and dyslipidemic patients treated with and without atorvastatin (Abbreviations: NHV normal healthy volunteers, Pt patients)
Despite the statistically better fit of the additive Emax model to the data (not shown), the proportional model was used for subsequent simulations. The proportional model is biologically plausible and was able to fit the observed data with a single Emax estimate rather than the population-specific estimates required for the additive model. This model is given by Eq. (9):
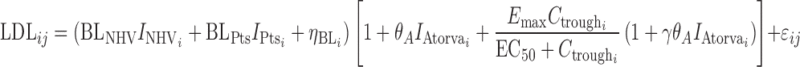 |
9 |
where 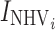 and
and 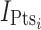 are indicator variables for healthy subjects (NHV) or patient with dyslipdemia, respectively.
are indicator variables for healthy subjects (NHV) or patient with dyslipdemia, respectively. 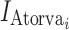 is an indicator for atorvastatin treatment. The parameters Emax and EC50 have their usual definitions for the effect of anacetrapib trough concentration. The parameter, γ, is the value for the interaction effect between anacetrapib and atorvastatin. A value of γ = 1 represents pharmacologic independence, i.e., that each drug has its full effect, irrespective of the effect of the other drug. A value γ > 1 represents a synergistic effect, where the effects of the drugs together are amplified, while γ < 1 suggests that the drugs, while possibly having effect greater than each alone, may have a smaller effect than would be suggested by examining monotherapy alone. For anacetrapib and atorvastatin, the estimated value of γ = 0.99 suggests that each agent acts essentially independently.
is an indicator for atorvastatin treatment. The parameters Emax and EC50 have their usual definitions for the effect of anacetrapib trough concentration. The parameter, γ, is the value for the interaction effect between anacetrapib and atorvastatin. A value of γ = 1 represents pharmacologic independence, i.e., that each drug has its full effect, irrespective of the effect of the other drug. A value γ > 1 represents a synergistic effect, where the effects of the drugs together are amplified, while γ < 1 suggests that the drugs, while possibly having effect greater than each alone, may have a smaller effect than would be suggested by examining monotherapy alone. For anacetrapib and atorvastatin, the estimated value of γ = 0.99 suggests that each agent acts essentially independently.
Simulations of Efficacy Endpoints
The final HDL-C model and the final proportional Emax LDL-C model were used in population simulations to understand the predicted effects of covariates and model uncertainty on the expected (mean) exposure–response. The simulations estimated the size of the drug effect alone or in combination with atorvastatin on HDL-C and LDL-C. Population simulations were conducted using SPLUS version 8.0 (Insightful, Seattle, USA).
The only covariates impacting HDL-C response are diet and anacetrapib formulation. The largest HDL-C response is seen in high-fat regimens which results in significantly increased exposure to anacetrapib, while fasted patients are predicted to experience the smallest response in HDL-C (Fig. 4). Low-fat and patient-selected regimens reflect similar responses (Fig. 4). Because neither atorvastatin nor the patient population impact the HDL-C response, these simulation results hold true for a range of atorvastatin dosing regimens.
Fig. 4.

Population mean predicted HDL-C and LDL-C effects. The population mean predicted effect of fed state and meal type on HDL-C in patients treated with anacetrapib monotherapy (a, top left). The population mean predicted effect of fed state and meal type on LDL-C in patients treated with anacetrapib monotherapy (b, top right) or in combination with 20 mg atorvastatin (c, bottom left). The population mean predicted effect of atorvastatin dose on LDL-C in patients treated with anacetrapib in combination with atorvastatin (d, bottom right)
The LDL-C response is influenced not only by diet and anacetrapib formulation, but is also strongly influenced by patient population and co-administration with atorvastatin. Figure 5 in the Electronic Supplementary Material shows the mean predicted % decrease in LDL-C versus dose for anacetrapib monotherapy and in combination with 20 mg atorvastatin. The impact of fed state and fat content of meals is similar to that for HDL-C, with fasted patients experiencing the smallest LDL-C decrease and patients on high-fat regimens experiencing the largest decrease in LDL-C (Fig. 4). The expected response in combination with atorvastatin is substantially greater than for anacetrapib alone (Fig. 4). In a fasted patient, the expected response of 50 mg anacetrapib, for example, in combination with 20 mg atorvastatin is a 52.4% (46.3% to 58.8% and 90% uncertainty interval) decrease in LDL-C, compared with 16.3% (13.7% to 19.8%) without atorvastatin. In a patient on a high-fat regimen, the expected response with atorvastatin is a 67.3% (62.5% to 72.4%) decrease in LDL-C, compared with 42.2% (39.3% to 45.3%) without atorvastatin. The expected response for atorvastatin, 20 mg, alone is a 43.1% (36.1% to 50.4%) decrease in LDL-C without regard to diet.
Higher doses of atorvastatin in combination with anacetrapib were also evaluated, with the atorvastatin response based on the model developed by Mandema et al. (6). Increasing doses of atorvastatin is predicted to result in an increased LDL-C response, as shown in Fig. 4; however, the increase in predicted effect is small. The 20 mg atorvastatin dose is very close to the maximum in the dose–response curve. The predicted LDL-C response increases from 37% (30.3% to 43.7%) at 10 mg to 54.5% (46.6% to 62.2%) at 80 mg atorvastatin. With increasing doses of anacetrapib, this increase is somewhat mitigated and increasing atorvastatin produces only a modest effect on LDL-C decrease.
DISCUSSION
A model-based strategy was employed in the development of anacetrapib, and this strategy contributed significantly to understanding the effects of meals and formulation on drug exposure, to describing dose–response of lipids to anacetrapib treatment and the combined effects of anacetrapib and atorvastatin, and to the selection of the phase III anacetrapib dose. The objective of the initial population PK and PK/PD modeling was primarily descriptive, with the goal of integrating a diverse set of studies and data in a unified, quantitative framework. Population and clinical trial simulations used these models predictively to inform key development decisions. Both the models and the simulations evolved with emerging data as exemplified by the phase IIb study results. The new information served an informal check on the overall structure of the models, and was ultimately incorporated in the modeling framework to further refine the predictions.
Although most of the PK and PD data were generated using an Imwitor/Tween LFC formulation (six studies and 474 subjects for LFC), the intended late phase development and commercial formulation was a HME formulation (two trials, a single 78 subject bridging study and 24 subject food effect study for HME; Table I in the Electronic Supplementary Material). In addition, food has a large effect on anacetrapib exposure for both formulations. The models developed here included all of these data in order to determine the impact of formulation and meals on drug exposure and lipid-altering effects. Biopharmaceutics studies performed in support of formulation development activities revealed that the PK of the two formulations were generally similar (unpublished data). Early results with LFC indicated that a standard low-fat meal increased the exposure of anacetrapib by approximately 3.6-fold and increased the Cmax by approximately 6-fold relative to the fasted state. A high-fat meal increased the AUC of anacetrapib by approximately 9-fold and increased Cmax by approximately 18-fold. The effect of food on AUC was generally similar for both formulations; the fed/fasted AUC geometric mean ratios at the HME, 150 mg dose was approximately the same as that observed at the LFC, 125 mg dose. In contrast, the fed/fasted Cmax geometric mean ratio for the HME, 150 mg formulation were somewhat higher than that for the LFC, 125 mg dose due to a lower Cmax in the fasted state with the HME formulation. For the single doses of the HME formulation, the 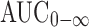 , Cmax, and C24h increased in a less than dose proportional manner in the 50 to 300 mg dose range. Multiple dose accumulation in the 50 to 300 mg dose range when administered with a low-fat meal as an HME tablet was approximately 1.2–1.9-fold, roughly as expected from single dose data. A subsequent biopharmaceutics study revealed that the HME formulation had a slightly (∼25%) greater exposure than the LFC formulation when 200 mg anacetrapib were administered with a high-fat meal (unpublished data). The results also indicated that in the fasted state, the
, Cmax, and C24h increased in a less than dose proportional manner in the 50 to 300 mg dose range. Multiple dose accumulation in the 50 to 300 mg dose range when administered with a low-fat meal as an HME tablet was approximately 1.2–1.9-fold, roughly as expected from single dose data. A subsequent biopharmaceutics study revealed that the HME formulation had a slightly (∼25%) greater exposure than the LFC formulation when 200 mg anacetrapib were administered with a high-fat meal (unpublished data). The results also indicated that in the fasted state, the 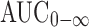 and C24h following administration of 200 mg LFC or HME formulations, HME formulation showed a smaller Cmax than the LFC formulation (∼40% decrease). Although these modest differences in overall drug exposure and food effects were apparent between the two formulations, the population PK model was able to describe these differences, and was subsequently used in the PK/PD modeling to provide a common framework to examine the lipid-altering effects of the two formulations, and to integrate this information in support of later development decisions.
and C24h following administration of 200 mg LFC or HME formulations, HME formulation showed a smaller Cmax than the LFC formulation (∼40% decrease). Although these modest differences in overall drug exposure and food effects were apparent between the two formulations, the population PK model was able to describe these differences, and was subsequently used in the PK/PD modeling to provide a common framework to examine the lipid-altering effects of the two formulations, and to integrate this information in support of later development decisions.
Because of the variation in PK with food and formulation, and in order to identify an appropriate dose for further development, it was important to understand the relationship between anacetrapib exposure and lipid response. Initial efforts in this regard focused on the use of the proximal target engagement biomarker for anacetrapib, namely CETP activity. The goal was first to describe the relationship between drug exposure and CETP activity, and then to relate CETP inhibition to lipid effects. Accordingly, PK/PD models were constructed between anacetrapib exposure and CETP activity. However, changes in anacetrapib’s effect on CETP activity between days 1 and 14 (steady state) were readily apparent in multiple dose studies, with drug effect on the biomarker apparently diminishing with repeated dosing, possibly due to an increase in CETP concentration due to treatment with anacetrapib. In the same studies, the lipid effects on both HDL-C and LDL-C remained approximately constant at steady state, suggesting an inconsistent relationship between the biomarker and lipid levels. These effects were also noted in reports of torcetrapib’s effects on CETP activity, suggesting that the increases in CETP concentrations was mechanism related (7).
For these reasons, the focus of the analysis was on examining the direct relationship between drug exposure and lipid effects. Of the various metrics of drug exposure, trough anacetrapib concentration (C24h) proved to be the most predictive of lipid effects. Models with trough concentrations based on individual-predicted PK parameter values were consistent with results obtained using population-predicted PK parameter estimates. Use of the population-predicted values was selected for use in the final models since this allows for prediction of future lipid effects, given the treatment population and fed state, in subjects who may or may not have PK data available.
The pharmacokinetic data generated in the fasting state and with meals, low-fat and high-fat meals, allowed the simulations to span a wide range of exposures with the high-fat meal representing a highest exposure scenario. Based on developed PK/LDL-C or PK/HDL-C models, it was determined that trough plasma concentrations were a predictor of efficacy Of the three key pharmacokinetic parameters, the C24h parameter was relatively less influenced both in magnitude and variability, in the presence of food. The findings from the phase IIb study, wherein patients were instructed to take anacetrapib with a meal, lend credibility to the hypothesis, in that the effects on HDL-C and LDL-C were as predicted. In the modeling strategy, “patient-selected” meal was used to reflect conditions wherein patients were instructed to take anacetrapib with a prescribed AHA TLC diet with an assumption that patients stayed compliant and not deviating significantly from the prescribed diet (approximating to fat intake slightly higher than a low-fat meal). Simulations based on the PK/PD model, inclusive of the phase IIb data, suggests that the predicted response following a patient-selected meal is similar to the predicted response following a low-fat or a high-fat meal.
The pharmacodynamic models showed consistent increases in HDL-C following treatment with anacetrapib, and there were no apparent differences in either baseline values or drug effects between healthy subjects and dyslipidemic patients. The maximal increase in HDL-C was estimated to be 176%, and plasma level for half-maximal effect (EC50) was estimated at 140 ng/mL. HDL-C is predicted to increase approximately 120% at a dose of 100 mg in subjects consuming a patient-selected, low-fat meal.
The models of LDL-C showed an expected difference between healthy subjects and dyslipidemic patients at baseline, with patients having approximately 30% higher LDL-C levels than healthy subjects. The final model for LDL-C described the decrease in LDL-C as a proportional change relative to the baseline level, and showed no distinction in effect size between healthy subjects and patients. Interestingly, the EC50 for changes in LDL-C was 70% higher than that estimated for an increase in HDL-C, suggesting that LDL-C is somewhat less sensitive to changes with anacetrapib than is HDL-C. The model also described the effects of combination treatment with atorvastatin. The interaction effect, with estimated value γ = 0.99, was suggestive of pharmacologic independence of the two agents. Treatment with 20 mg atorvastatin is predicted to reduce LDL-C approximately 45%, while treatment with 100 mg anacetrapib under a patient-selected meal is expected to yield approximately 42% lowering in LDL-C. Pharmacologic independence suggests that additional treatment with anacetrapib will further reduce LDL-C from 55% of baseline (following statin treatment) to 32% of the original baseline LDL-C value, for a net reduction of 68% in LDL-C. The model, and approximate pharmacologic independence, also suggests a 42% (90% prediction intervals, 40.9–43.8%) reduction due to anacetrapib in patients already under treatment with atorvastatin at the start of observation. Clinical trial simulations (Fig. 5) with these models showed that, although both LDL-C lowering and HDL-C raising effects are expected to be sensitive to diet, neither of these endpoints is expected to be overly sensitive to random, dietary indiscretions as long as patients generally adhere to a low-fat diet, and take anacetrapib with meals.
Fig. 5.

Simulation based assessment of the exposure/response relationship under compliant and noncompliant conditions
One of the key decisions in any drug development program is the selection of a dose or doses for progression to phase III study and, ultimately, for registration. The models developed here were used for this purpose. To enable dose selection for phase III, assumptions were made of how patients will be taking their dose of anacetrapib with respect to food. This assumption was categorized into a meal type (fasting, low fat, patient-selected, or high fat) and entered into the updated PK/PD model (based on the integration of the phase Ib and IIb data) to generate the 100 mg dose that was selected for phase III. The population simulations rationalized that a 100 mg dose will result in lipid-altering effects that are at or near the pharmacodynamic plateau and a dose level or two lower than 100 mg at the midpoint of the pharmacodynamic range. A threshold for a simulated observed LDL-C lowering effect of 40% was selected as the minimum target lipid effect. Based on population simulations at the 100 mg dose there was a mean predicted ∼42% decrease in LDL-C and a ∼116% mean predictive increase in HDL-C. The dose of 100 mg is also supported for anacetrapib when coadministered with atorvastatin. A dose of 100 mg as a HME formulation appears to produce LDL-C lowering effects that are at or near the pharmacodynamic plateau. Specifically, 100 mg anacetrapib/20 mg atorvastatin results in ∼67% mean predictive decrease (95% CI, 62–72). From Fig. 4, it was noted that the 100 mg dose was predicted to meet this threshold, and that further increases in dose were not expected to lead to substantial additional efficacy. Because anacetrapib is somewhat more potent with respect to increases in HDL-C than it is for its LDL-C lowering effect, it is not surprising that the 100 mg dose was also predicted to result in HDL-C increases that were very close to the maximum expected for a given meal condition.
It should be noted that only a 20 mg dose of atorvastatin was used in the phase IIb study. Thus, it is unclear whether dose/response observed between anacetrapib and 20 mg atorvastatin will differ meaningfully relative to different statins and different doses of statins. Notably, Mandema et al. have modeled the exposure/LDL-C lowering relationships of statins (atorvastatin, lovastatin, pravastatin, simvastatin, rosuvastatin) and various doses of statins (5 to 80 mg) (6). In their work, the reduction in LDL-C with statins was best described by a dose–response model with a common Emax (maximal effect) and a different ED50 (potency) for each of the statins. There were no statistically significant difference in Emax or Hill coefficient (n) found between the statins. This indicates that all of the statins, sharing common mechanism of action, are expected to share a similar shape of the dose–response relationship with a similar maximal effect of about ∼79% LDL-C decrease over placebo (estimated maximum effect at infinite dose). If atorvastatin is considered as a reference, then the relative potencies of rosuvastatin, simvastatin, lovastatin, and pravastatin were found to be 0.33, 2.3, 6.3, and 7.4, respectively (6). This model was leveraged in the model-based strategy for anacetrapib where the response to atorvastatin was found to be close to maximum at 20 mg and any additional increases in atorvastatin doses may result in modest incremental benefit. Thus, selection of anacetrapib dose on top of atorvastatin as a representative statin is considered reasonable based on these simulations. For these reasons, a dose of 100 mg was selected for further study following the characterization of different sources of variation due to the formulation, diet, and study population. This selection was made prospectively, and based on the recently completed add-on phase III study, DEFINE (published during the review of this manuscript), the lipid-altering effects at the 100 mg anacetrapib dose are remarkably similar to those predicted in this model based approach (8,9). Specifically, LDL-C was reduced by 39.8% and HDL-C was increased by 138.1% at an anacetrapib dose of 100 mg once daily in patients with high risk coronary heart disease for 18 months (9). The modeling and simulation strategy supported that anacetrapib be dosed with a meal to increase compliance and preserve the efficacy of anacetrapib. Clinical trial simulations have showed that the variability was not overt when individual patients undertake dietary indiscretion consistent with the hypothesis that intrasubject day-to-day variability in meal content is unlikely to have an impact on efficacy given the pharmacokinetic and pharmacodynamic half life of anacetrapib. This is supported by data from the phase IIb study (3) as well as the recently reported phase III study (9), which showed that the variability in pharmacodynamic responses were not as large as those associated typically with pharmacokinetics. Whereas the phase I studies were controlled clinical studies where the subjects consumed prespecified and prescribed meals, the phase IIb and III studies was more realistic of the clinical and real world condition, where patients eat differently from each other and from day to day.
In conclusion, a 100 mg dose and a formulation (HME) have been selected for continued development of anacetrapib. The model-based strategy utilized here enabled the development team to successfully pursue a dose and formulation in phase III that were not specifically studied in a phase IIb study.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
Additional model diagnostic plots for HDL-C exposure/response model. The dashed line is a smooth through the data. The solid line is the line of unity in the upper left hand panel, and a horizontal line with intercept zero in the remaining panels. (TIFF 1,647 kb) (GIF 130 kb)
{kind=link}
Additional model diagnostic plots for LDL-C exposure/response model (TIFF 1,685 kb) (GIF 131 kb)
{kind=link}
Observed and predicted HDL-C versus predicted trough concentration. The patient population includes patients in phase Ib and phase IIb studies treated with and without atorvastatin. The solid line represents the model best fit for the whole population. (TIFF 705 kb) (GIF 42 kb)
{kind=link}
Observed and predicted LDL-C versus trough concentration for the proportional E max model. (TIFF 689 kb) (GIF 38 kb)
{kind=link}
Observed and predicted decrease in LDL-C versus predicted trough concentration for patients treated with anacetrapib or anacetrapib + atorvastatin (proportional E max model). The solid and dashed lines represent the model best fit for the population treated with anacetrapib + atorvastatin and anacetrapib alone, respectively. (TIFF 702 kb) (GIF 41 kb)
Summary of final dataset for population PK model development (DOC 42 kb) (DOC 42 kb)
Summary of final dataset for PD model development (DOC 41 kb) (DOC 41 kb)
Acknowledgments
The authors like to acknowledge the input of several team members including Alan Hartford and Bo Jin from biostatistics, Daniel Bloomfield from cardiovascular clinical research, Julie Stone and Amit Garg from clinical PK/PD in the model development process and/or interpretation of the results.
Current affiliations: Arthur J. Bergman is presently at Pfizer, Inc., Groton, Connecticut, USA; Marissa F. Dockendorf is at Johnson & Johnson, Inc., Jacksonville, Florida, USA; and Kevin Dykstra is at qPharmetra LLC, Andover, Massachusetts, USA.
Financial Disclosures
This work is sponsored/funded by Merck & Co., Inc. Authors who are employees of Merck & Co., may hold stock or stock options in the company.
References
- 1.Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, Rosko K, Chavez-Eng C, Lutz R, Bloomfield D, Gutierrez M, Doherty J, Bieberdorf F, Chodakewitz J, Gottesdiener KM, Wagner JA. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomized placebo-controlled phase I studies. Lancet. 2007;370:1907–14. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 2.Krishna R, Bergman AJ, Jin B, Fallon M, Cote J, Van Hoydonck P, Laethem T, Gendrano IN, Van Dyck K, Hilliard DA, Laterza OF, Snyder KM, Chavez-Eng CM, Lutz R, Chen J, Bloomfield DM, De Smet M, Van Bortel L, Gutierrez M, Al-Huniti N, Dykstra K, Gottesdiener KM, Wagner JA. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008;84(6):679–83. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- 3.Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, 3rd, Sisk CM, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(2):352–60. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Krishna R, Garg A, Jin B, Cote J, Bergman A, Von Hoydonck P, Laethem T, Van Dyck K, Chavez-Eng C, Archer L, Lutz R, Hilliard D, Snyder K, Panebianco D, Bortel L, Lasseter K, Al-Huniti N, Dykstra K, Gottesdiener K, Wagner JA. Single-dose pharmacokinetics and pharmacodynamics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009;68(4):535–45. doi: 10.1111/j.1365-2125.2009.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheiner LB, Beal SL. NONMEM Users Guide. San Francisco: Division of Pharmacology, University of California; 1979.
- 6.Mandema JW, Hermann D, Wang W, Sheiner T, Milad M, Bakker-Arkema R, Hartman D. Model-based development of gemcabene, a novel lipid altering agent. AAPS J. 2005;7(3):E513–22. doi: 10.1208/aapsj070352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark RW, Sutfin TA, Suggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ. Raising high-density lipoprotein in humans through inhibition of cholestryl ester transfer protein: an initial multidose study of torceterapib. Aterioscler Thromb Vasc Biol. 2004;24:490–7. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- 8.Cannon CP, Dansky HM, Davidson M, Gotto AM, Jr, Brinton EA, Gould AL, Stepanavage M, Liu SX, Shah S, Rubino J, Gibbons P, Hermanowski-Vosatka A, Binkowitz B, Mitchel Y, Barter P. DEFINE investigators. Design of the DEFINE trial: determining the EFficacy and tolerability of CETP INhibition with AnacEtrapib. Am Heart J. 2009;158(4):513–519. doi: 10.1016/j.ahj.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 9.Cannon CP, Shah S, Dansky H, Davidson M, Brinton E, Gotto AM, Jr, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional model diagnostic plots for HDL-C exposure/response model. The dashed line is a smooth through the data. The solid line is the line of unity in the upper left hand panel, and a horizontal line with intercept zero in the remaining panels. (TIFF 1,647 kb) (GIF 130 kb)
Additional model diagnostic plots for LDL-C exposure/response model (TIFF 1,685 kb) (GIF 131 kb)
Observed and predicted HDL-C versus predicted trough concentration. The patient population includes patients in phase Ib and phase IIb studies treated with and without atorvastatin. The solid line represents the model best fit for the whole population. (TIFF 705 kb) (GIF 42 kb)
Observed and predicted LDL-C versus trough concentration for the proportional E max model. (TIFF 689 kb) (GIF 38 kb)
Observed and predicted decrease in LDL-C versus predicted trough concentration for patients treated with anacetrapib or anacetrapib + atorvastatin (proportional E max model). The solid and dashed lines represent the model best fit for the population treated with anacetrapib + atorvastatin and anacetrapib alone, respectively. (TIFF 702 kb) (GIF 41 kb)
Summary of final dataset for population PK model development (DOC 42 kb) (DOC 42 kb)
Summary of final dataset for PD model development (DOC 41 kb) (DOC 41 kb)