

Abstract
Hippocampal spine density varies with the estrus cycle. The cyclic change in estradiol levels in serum was hypothesized to underlie this phenomenon, since treatment of ovariectomized animals with estradiol induced an increase in spine density in hippocampal dendrites of rats, as compared to ovariectomized controls. In contrast, application of estradiol to hippocampal slice cultures did not promote spinogenesis. In addressing this discrepancy, we found that hippocampal neurons themselves are capable of synthesizing estradiol de novo. Estradiol synthesis can be suppressed by aromatase inhibitors and by knock-down of Steroid Acute Regulatory Protein (StAR) and enhanced by substrates of steroidogenesis. Expression of estrogen receptors (ERs) and synaptic proteins, synaptogenesis, and long-term potentiation (LTP) correlated positively with aromatase activity in hippocampal cultures without any difference between genders. All effects due to inhibition of aromatase activity were rescued by application of estradiol to the cultures. Most importantly, gonadotropin-releasing hormone (GnRH) increased estradiol synthesis dose-dependently via an aromatase-mediated mechanism and consistently increased spine synapse density and spinophilin expression. As a consequence, our data suggest that cyclic fluctuations in spine synapse density result from pulsative release of GnRH from the hypothalamus and its effect on hippocampal estradiol synthesis, rather than from varying levels of serum estradiol. This hypothesis is further supported by higher GnRH receptor (GnRH-R) density in the hippocampus than in the cortex and hypothalamus and the specificity of estrus cyclicity of spinogenesis in the hippocampus, as compared to the cortex.
Keywords: Estrus cycle, Spinogenesis, GnRH, Estradiol synthesis
Introduction
For more than a decade, it has been known that estrogens influence synaptic plasticity in the hippocampus. Gould et al. (1990) were the first to show that ovariectomy of female rats resulted in a decrease of dendritic spine density in CA1 pyramidal neurons in the hippocampus. Accordingly, systemic estradiol treatment of these ovariectomized animals caused a rescue of impaired spinogenesis in this particular hippocampal region. Consistently, spine synapse density varied in response to fluctuating estrogen levels during the estrus cycle in female rats (Woolley et al. 1990). Along these lines, a decrease in spine density was observed over the 24-h period between the late proestrus and the late estrus phases of the cycle, which is characterized by a distinct decline in plasma estrogen level. Subsequently, during diestrus dendritic spine synapse density cycles back from low values to high values. Further studies have demonstrated that a variety of synaptic markers are also upregulated after systemic estrogen application (for review see: McEwen 2002), confirming the supportive role of estrogens on synapse formation. Along these lines, estradiol increases the magnitude of LTP at CA3-CA1 synapses in the hippocampus (Cordoba Montoya and Carrer 1997; Yun et al. 2007). This has been related to the memory-enhancing effects of this hormone (Walf and Frye 2006). Warren et al. (1995) were the first to demonstrate enhanced synaptic activity in proestrus rats and Cordoba Montoya and Carrer (1997) found that estrogen facilitates the induction of LTP in the hippocampus of awake rats. Acute application of estradiol to native hippocampal slices in vitro increases NMDA and AMPA receptor transmission and LTP (Foy et al. 1999; Good et al. 1999). In more recent studies, Smith and McMahon (2005, 2006) demonstrated that the estrogen-induced increase in the magnitude of LTP occurs only when the ratio of NMDA transmission to AMPA transmission is increased.
The results of these initial studies on estrogen-induced synaptic plasticity and the fact that gonads are the main source of estrogen, strongly suggest an endocrine regulation of spinogenesis in the hippocampus by gonadal estradiol. The latter reaches its target, the hippocampus, via the plasma. We used hippocampal dispersed cultures and hippocampal slice cultures in an attempt to adopt this paradigm to hippocampal culture models for further experimental approaches in vitro. Slice cultures allow preservation of neuronal connectivity and can be maintained for 4 weeks (Frotscher et al. 1995). In these cultures neither high pharmacological nor physiological serum concentrations of estradiol increased the number of spine synapses and spines. These results questioned the endocrine effect of gonadal estrogens on synapse formation (Kretz et al. 2004). Here, we review our previous findings and add novel results in addressing the discrepancy between our in vitro findings and estrus cyclicity of synaptogenesis in animals (Wehrenberg et al. 2001; Rune et al. 2002; Prange-Kiel et al. 2003; Kretz et al. 2004; Rune et al. 2006; Prange-Kiel et al. 2008).
Steroidogenic enzymes in the hippocampus
Neurosteroids are defined as steroids that accumulate in the brain even in the absence of steroidogenic glands and are synthesized in the brain from endogenous precursors by enzymes that are present in situ (Baulieu and Robel 1990). The precursor of all steroid hormones is cholesterol, which is transported into the mitochondria, where steroidogenesis starts by conversion of cholesterol to pregnenolone. This cholesterol transfer across the mitochondrial membranes is considered to be the rate-limiting step of steroid biosynthesis and is mediated by the Steroid Acute Regulatory Protein (StAR). Using in situ hybridization and immunohistochemistry, we showed the expression of StAR mRNA as well as StAR protein in the hippocampus (Wehrenberg et al. 2001). Pyramidal neurons, granule cells of the dentate gyrus, and interneurons showed specific staining with both methods. A comparable expression pattern was demonstrated for StAR in the hippocampus by Furukawa et al. (1998). It is taken for granted that StAR-positive cells always contain side-chain cleavage enzyme and 3β-hydroxysteroid dehydrogenase (Furukawa et al. 1998). These enzymes synthesize the substrates for aromatization of androgen precursors, whose aromatization eventually leads to the synthesis of estrogens. Furthermore, aromatase, the enzyme that converts testosterone to estradiol, and StAR show an overlapping hippocampal expression on mRNA and protein level (Wehrenberg et al. 2001). This coexpression strongly suggests that hippocampal neurons are able to synthesize estrogens de novo.
The capacity of hippocampal neurons to synthesize estrogens de novo
In order to prove the hypothesis of a de novo estradiol synthesis in hippocampal neurons, we established a pure neuronal hippocampal dispersion culture, obtained from hippocampi of adult (10-week-old) rats (Prange-Kiel et al. 2003). These neurons were cultivated under serum- and steroid-free conditions. An RIA revealed the presence of estradiol in the medium, indicating the ability of hippocampal cell cultures to synthesize and release estradiol. Subsequent experiments with hippocampal slice cultures and hippocampal dispersion cultures obtained from postnatal rats confirmed these findings (Kretz et al. 2004). 17β-Estradiol must be synthesized de novo from cholesterol, the precursor of all steroid hormones, since the medium of the dispersion cultures contained neither serum nor steroids.
The use of an aromatase inhibitor, letrozole, confirmed the aromatase dependency of estrogen synthesis. Supplementation of the medium with letrozole resulted in a significant decrease in the amount of synthesized estrogen. The effect was most prominent in dispersion cultures obtained from adult hippocampi, where the estradiol synthesis was almost completely downregulated (Prange-Kiel et al. 2003). In cultures originating from postnatal (p5) animals, the inhibiting effect of letrozole on estrogen synthesis was demonstrated to be dose-dependent (Kretz et al. 2004).
Transfection of the cells with siRNA against StAR was performed to confirm the results after letrozole treatment. We chose siRNA against StAR, instead of siRNA against aromatase, to ensure that the effects seen after aromatase inhibition are due to the lack of estrogen rather than to elevated levels of precursors in estrogen synthesis (see below). The transfection did not influence StAR expression, as immunostaining of StAR in siRNA-StAR control cells did not differ from non-transfected cells. Measurement of estradiol in media collected after 7 days revealed a downregulation of estradiol synthesis by approximately 20% in siRNA against StAR transfected cells compared to siRNA controls (Fester et al. 2006; Fester et al. 2009). These experiments prove that hippocampal neurons synthesize estradiol de novo from cholesterol.
In order to stimulate aromatase activity by offering elevated levels of precursors of estrogen synthesis, which serve as substrates of aromatase, we treated dispersed cultures with cholesterol and testosterone. To demonstrate the effects of cholesterol, we used water-soluble, methyl-β-cyclodextrin-encapsuled cholesterol, which is able to pass through the plasma membrane (Daniel et al. 2004; Daniel et al. 2007). This is a common approach to load cells with cholesterol without using lipoproteins. Testosterone was used as a control as it is aromatized to estradiol by aromatase and, as a consequence, increases spine density in the rat hippocampus (Leranth et al. 2004). Treatment of our cultures with testosterone or cholesterol resulted in a highly significant increase in estradiol release in the supernatant, showing that elevated substrate levels do indeed increase estradiol synthesis (Fester et al. 2009).
We used the same dose of cholesterol and testosterone in order to prove the hypothesis that the dose of cholesterol required for affecting synaptic plasticity has to be far higher than the doses of steroid metabolites (Pfrieger 2003). At first glance, this hypothesis seems to be plausible, considering the twofold function of cholesterol: it serves as building material for different synaptic components and alternatively, as a precursor for steroid biosynthesis. The degree of upregulation was notably higher in response to cholesterol than after testosterone treatment, which very likely results from cholesterol depletion under serum-free conditions, such as our dispersed cultures. Cholesterol depletion has severe consequences in neuronal culture preparations (de Chaves et al. 1997; Michikawa and Yanagisawa 1999; Thiele et al. 2000; Chamberlain et al. 2001; Lang et al. 2001; Fan et al. 2002). We did not observe any effect on cell viability, namely because neurons cultivated under serum- and glia-free conditions produce enough cholesterol to survive and to differentiate axons and dendrites (Mauch et al. 2001). Supplementation of the cultures with cholesterol, however, compensates for cholesterol depletion under serum-free conditions. Thus, it is not unexpected that the increase in estradiol synthesis in dispersed cultures is very dramatic in response to cholesterol and less strong in response to testosterone (Fester et al. 2009).
The effect of hippocampus-derived estradiol on synaptic plasticity
The fact that hippocampal neurons are capable of producing estrogens raises the question of the functional significance of this endogenous steroid. As an internal control we studied the expression of ERα and β. In dispersion cultures, ERα was upregulated, whereas ERβ expression was decreased in response to estradiol. Vice versa, ERα was downregulated and ERβ upregulated in response to letrozole. Since ERs are ligand-inducible transcription factors, these data show that the cultures are responsive to estrogen-treatment (Prange-Kiel et al. 2003).
Synaptic proteins
To further substantiate the role of endogenous estrogens on synaptic plasticity, we studied the effect of endogenous estrogen on synaptic proteins (Kretz et al. 2004) such as synaptophysin. This protein is a component of the transmitter vesicle membrane, and is commonly used as a presynaptic marker (for review see: Sudhof and Jahn 1991). As an example of a postsynaptic marker we studied spinophilin, a cytoskeleton associated protein, which has been shown to be enriched in spines (Allen et al. 1997; Feng et al. 2000). In hippocampal slice cultures and dispersion cultures, immunoreactivity of both proteins was found in somata and dendrites, but was also present as punctate staining, indicating the labeling of spines (spinophilin) or presynaptic boutons (synaptophysin). When we treated the cultures with letrozole, we found a clear-cut downregulation of both proteins; this could be verified by subsequent image analysis of the immunolabeling. The letrozole-induced downregulation was rescued by simultaneous application of estradiol to the cultures (Rune et al. 2002; Rune et al. 2006). The findings on synaptophysin confirm observations by other groups under in vivo (Yankova et al. 2001; Choi et al. 2003) and in vitro (Yokomaku et al. 2003) conditions.
In siRNA against StAR transfected cells, thus in cells in which estrogen synthesis was blocked from its beginning, the expression of spinophilin as well as of synaptophysin was significantly downregulated. A rescue of the knock-down in these transfected cultures was achieved by administration of estradiol to the cultures, but not by application of cholesterol. This finding strongly indicates that cholesterol is ineffective regarding synapse formation when the access of cholesterol to the first steroidogenic enzyme in the inner mitochondrial membrane is blocked. A slight increase in synaptic protein expression was found after cholesterol treatment. This slight increase was expected, since non-transfected cells in the culture dish, which continue to synthesize estradiol, are stimulated by cholesterol.
Supplementation of the medium with cholesterol, testosterone, or estradiol resulted in clear-cut upregulation of synaptic proteins. Notably, in dispersed cultures, where cholesterol synthesis, testosterone synthesis, and estradiol synthesis were simultaneously inhibited, a rescue of synaptic protein expression was achieved solely by estradiol, but neither by cholesterol nor by testosterone, pointing to the high specificity of aromatase activity in steroid-regulated synaptic plasticity in the hippocampus. The simultaneous inhibition of cholesterol synthesis, testosterone synthesis, and estradiol synthesis resulted in a decrease in the expression of synaptic proteins. Remarkably, neither treatment with cholesterol nor with testosterone abolished the negative effect of this combined inhibition. The rescue of synaptic protein expression was solely achieved by treatment with estradiol. This finding points to the high specificity of aromatase activity in steroid-regulated synaptic plasticity in the hippocampus.
Hippocampal spine synapses
As a next step, we investigated the effect of endogenous estradiol on spine and spine synapse formation (Kretz et al. 2004). By inhibiting aromatase activity with letrozole, we lowered the endogenous estradiol levels in hippocampal slice cultures. In electron micrographs we analyzed the number of spine synapses, shaft synapses, and the number of boutons by using unbiased stereological methods. Inhibition of estrogen synthesis resulted in a significant downregulation of spine synapses and of presynaptic boutons. The results of this analysis were confirmed by spine counts in Lucifer yellow-filled neurons in dispersion cultures (Kretz et al. 2004). The effects of letrozole on synaptogenesis were rescued by simultaneous application of 17β-estradiol to the cultures (Zhou et al. 2007).
In agreement with previous findings, stereological determination of spine synapse number in organotypic slice cultures revealed that the number of spine synapses increased significantly in response to cholesterol (Mauch et al. 2001). Testosterone treatment, performed for control purposes, also resulted in an increase in spine synapse number. Most strikingly, when we supplemented the medium with cholesterol together with letrozole, cholesterol-induced synapse formation was abolished, strongly suggesting that cholesterol does not induce synapse formation directly but serves as a substrate for estrogen synthesis. Similarly, after blockade of ERs using ICI 182 780, no increase in spine synapse number was found in response to cholesterol in hippocampal slice cultures. A rescue of spine synapse number after letrozole treatment was not achieved with testosterone either.
In order to test the effect of exogenous estradiol we also treated slice cultures and dispersion cultures with estradiol at a dose of 10−7 M (Kretz et al. 2004), which induced a significant increase in ERα immunostaining in slice cultures (Rune et al. 2002).
A change in spine synapse number was not found, which raises the question as to why cholesterol increases the number of spine synapses while estradiol does not?
Although application of estradiol to the cultures did not induce spine synapse formation, the pre and postsynaptic markers synaptophysin and spinophilin were upregulated in response to estradiol (Fester et al. 2009). In view of these data the reliability of synaptophysin immunoreactivity and spinophilin immunoreactivity as indicative of the presence of spine synapses, which is taken for granted in many studies (Spencer et al. 2008), appears questionable. An increase in transmitter vesicles in preexisting boutons could provide an alternative explanation for synaptophysin upregulation. Similarly, spinophilin, a protein phosphatase-1 and actin-binding protein localized adjacent to the postsynaptic membrane, plays a role in glutamatergic neurotransmission and dendritic spine morphology. With both proteins the upregulation may serve to increase individual synaptic strength.
To find out why estradiol does not induce spine formation, we further compared the regulation of ERα in response to 17β-estradiol and cholesterol. It has been shown that this ER subtype mediates estrogen-induced spine formation in the CA1 region of the hippocampus (Mukai et al. 2007), and we have shown that ERα is upregulated by 17β-estradiol (Prange-Kiel et al. 2003). In our study, the upregulation of ERα was stronger after treatment of the cultures with cholesterol than after estradiol treatment. This finding suggests that cholesterol-enhanced estrogen synthesis results in higher spine synapse density via upregulation of ERα. Uptake of exogenously applied estradiol from the extracellular space, however, appears to be limited. The degree of ERα upregulation in response to estradiol appears to be insufficient to induce spine synapse formation.
This hypothesis is supported by recent studies, which show that estradiol is actively transported through the plasma membrane (Hammes et al. 2005; Lin and Scanlan 2005). The regulation of this transport is unknown so far. The rescue of the effects after inhibition of estrogen synthesis by estradiol that we found in our study, suggests that this transport may be regulated by intracellular estradiol concentrations. Alternatively, at the plasma membrane high doses of estradiol, such as 10−7 M estradiol, induce a Ca2+ influx via membrane-bound non-genomic signaling, which results in a release of Ca2+ from intracellular stores (Zhao et al. 2005 and our own unpublished observations). Ca2+ release from intracellular stores, in turn, downregulates aromatase activity via Ca2+-dependent kinases (Balthazart et al. 2005, 2006) Thus, high doses of estradiol would finally inhibit estradiol synthesis in the neurons. This mechanism of estradiol synthesis inhibition by its product was consistently shown in permanent cell lines (Shimizu et al. 1993).
Long-term potentiation
Very recently, Zhou et al. (unpublished observations) demonstrated that LTP is no longer inducible in hippocampal slice cultures after letrozole treatment. In letrozole-treated cultures, TBS of CA3-CA1 Schaffer collaterals failed to induce LTP. Consistently, immunoreactivity of NR2B NMDA receptors, which mediate estradiol-induced LTP, was downregulated in the stratum radiatum of the CA1 region in the letrozole-treated slice cultures. All effects in response to letrozole were rescued by estradiol. Estradiol increases the magnitude of LTP at CA3-CA1 synapses in the hippocampus. This has been related to the memory-enhancing effects of this hormone (Walf and Frye 2006). Warren et al. (1995) were the first to demonstrate enhanced synaptic activity in proestrus rats and Cordoba Montoya and Carrer (1997) found that estrogen facilitates the induction of LTP in the hippocampus of awake rats. Acute application of estradiol to native hippocampal slices in vitro increases NMDA and AMPA receptor (R) transmission and LTP (Foy et al. 1999; Good et al. 1999). In more recent studies, Smith and McMahon (2005, 2006) demonstrated that the estrogen-induced increase in the magnitude of LTP occurs only when the ratio of NMDA transmission to AMPA transmission is increased (Smith and McMahon 2005). They further showed that blocking NR2B NMDARs prevented the increase in LTP magnitude in response to estradiol (Smith and McMahon 2006). In hippocampal slice cultures, application of 17β-estradiol increased the magnitude of LTP. This effect was, however, not significant.
Gender differences
In view of the estrus cycle dependency of spinogenesis (Woolley et al. 1990), gender differences in hippocampal synaptogenesis should be a logical consequence. In vitro we did not find differences between hippocampal cultures that originated from males and females (Fig. 1). The expression pattern of aromatase mRNA is similar in the hippocampi of males and females, as shown by in situ hybridization (Wehrenberg et al. 2001). ERs density differed neither in vivo nor in vitro (Rune et al. 2002) and in hippocampal cultures obtained from males and females, the estradiol levels in the medium were in the same order of magnitude. Furthermore, spine synapse density did not differ in hippocampal slice cultures originating from male and female animals. Finally, estradiol treatment of hippocampal slices does not result in an increase in spine synapse density, irrespective of the gender of the animals providing the tissue (Kretz et al. 2004).
Fig. 1.
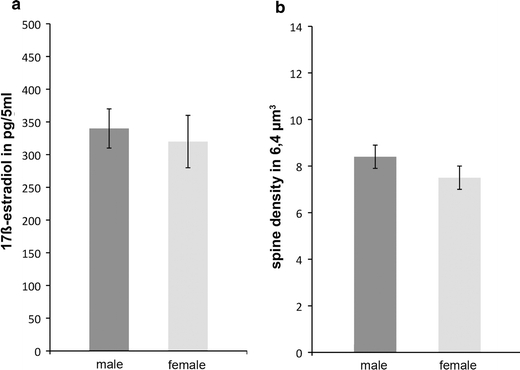
In organotpyic slice cultures the release of estradiol into the medium and spine synapse density did not differ in slices that originated from male and female animals (SEM, n = 5 cultures)
Why is synaptogenesis estrus cycle-dependent?
Obviously, there is a discrepancy between our findings on the unique role of endogenous estradiol in the regulation of spinogenesis and the fact that spine synapse density in females varies with the estrus cycle. We therefore hypothesized that hippocampal and ovarian estradiol synthesis are synchronized by a common factor. In view of abundant GnRH-R mRNA expression in the hippocampus (Bard and Pelletier 1987; Reubi et al. 1987; Jennes et al. 1988) and the direct effect of GnRH on estradiol synthesis in gonadal cells (for review: Janssens et al. 2000), we tested GnRH as a potential regulator of hippocampal estradiol synthesis. GnRH is the key regulator of reproduction, as its pulsative release from the hypothalamus controls the secretion of follicle stimulating hormone and luteinizing hormone in the pituitary, which, in turn, regulate steroid hormone synthesis in the gonads. In recent years, however, numerous in vitro and in vivo studies in mammals have revealed that GnRH also influences estradiol synthesis directly in the ovary (for review: Janssens et al. 2000). Treatment of dispersed hippocampal neurones with GnRH affected the release of estradiol into the cell culture medium in a specific dose-dependent manner (Fig. 2a). Intermediate doses of 10−8 and 10−7 M GnRH resulted in a significant increase in estradiol synthesis as compared to controls. At the highest dose of GnRH (5 × 10−7 M), estradiol synthesis was inhibited as compared to the intermediate doses and did not differ from the untreated control. Notably, an inverted U-shaped dose–response curve of estradiol synthesis has also been described for cultured granulosa cells that were treated with the GnRH agonist buserelin (Parinaud et al. 1988). This type of dose–response curve, caused by receptor desensitization, is typical of G-protein-coupled receptors such as GnRH-R. Receptor desensitization, mainly due to receptor internalization, accounts for this phenomenon (for review: McArdle et al. 2002).
Fig. 2.
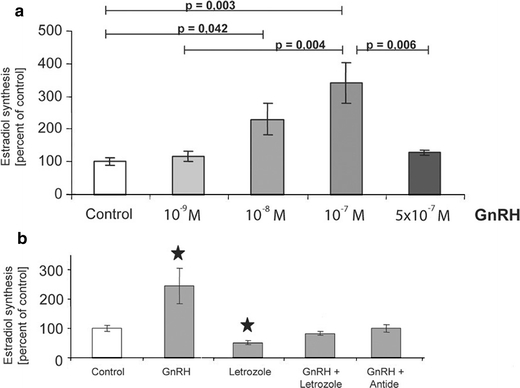
Hippocampal estradiol synthesis is regulated by GnRH. a Estradiol synthesis of dispersion cultures of hippocampal neurons was measured by RIA after treatment with GnRH for 8 days. Estradiol synthesis increased significantly after treatment with intermediate doses of 10−8 and 10−7 M GnRH, as compared to the control. No such increase was seen with the highest dose of 5 × 10−7 M (mean ± SEM; n = 5). b Treatment of hippocampal neurons with the aromatase inhibitor letrozole (10−7 M) inhibited GnRH-induced estradiol synthesis. Treatment of the neurons with 10−7 M letrozole alone resulted in a significant downregulation of estradiol synthesis, as demonstrated earlier. The increase in estradiol synthesis induces by 10−8 M GnRH was blocked by simultaneous treatment of the dispersion cultures with the GnRH antagonist antide (10−7 M) (mean ± SEM; n = 5, *P ≤ 0.05 compared with control)
Our next step was to test whether the effect of GnRH on estradiol synthesis is aromatase-dependent. We applied therefore, GnRH at a dose that would increase estradiol synthesis in combination with the aromatase inhibitor letrozole. The co-treatment with letrozole blocked the positive effect of GnRH on estradiol synthesis, demonstrating that the effect of GnRH is mediated by aromatase (Fig. 2b. The effect of GnRH on estradiol synthesis was abolished by co-treatment of cell cultures with the GnRH antagonist antide (Fig. 2b). This further validated the specificity of the GnRH effect.
In subsequent experiments we demonstrated that GnRH affects the expression of the postsynaptic protein spinophilin and the formation of spine synapses by regulating estrogen synthesis. The application of GnRH to hippocampal cultures increased the expression of spinophilin (Fig. 3) and the formation of spine synapses in a dose-dependency that mirrors that of estradiol synthesis. Moreover, when GnRH-induced hippocampal estradiol synthesis was blocked by the aromatase inhibitor letrozole, the spinophilin-stimulating effect of GnRH was abolished (Fig. 4). This finding shows that the GnRH effect on spine formation is mediated by its influence on estradiol synthesis.
Fig. 3.
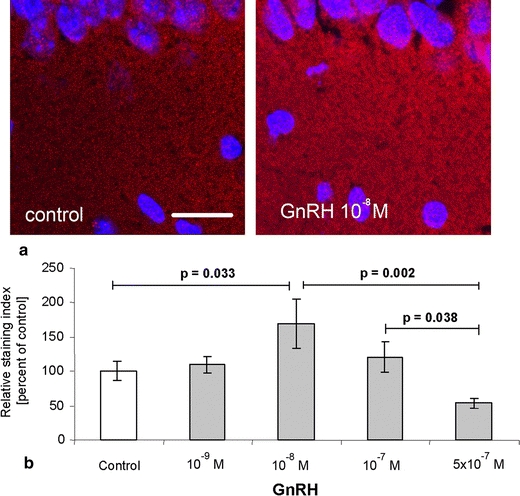
GnRH regulates spinophilin expression in hippocampal slice cultures. a Cy3-couples anti-spinophilin was used to detect spinophilin in the CA1 region of hippocampal slice cultures after treatment with GnRH. The staining intensity varied depending on the treatment. The nuclei were counterstained with DAPI (blue; scale bar 20 μm). b Image analysis of IHC for the postsynaptic protein spinophilin in hippocampal slice cultures after 8 days of GnRH treatment demonstrated that an intermediate dose of GnRH (10 nM) resulted in a significant increase in the staining index compared to the control. The staining index was significantly lower after treatment with 500 nM GnRH and was not significantly altered compared to the control (mean ± SEM, n = 10)
Fig. 4.
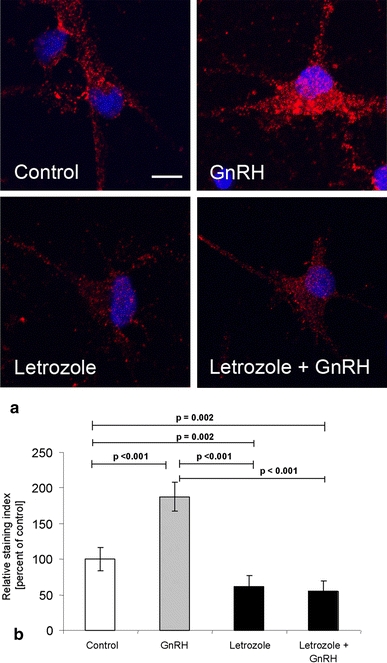
Effects of GnRH on spinophilin expression is mediated by estrogen synthesis in hippocampal dispersion cultures. a Punctate spinophilin immunoreactivity was detected on single pyramidal neurons (red, Cy3). The stained area and its staining intensity varied with the different treatments. The nuclei were counterstained with DAPI (blue), scale bar 10 μm. b The GnRH-induced increase of spinophilin expression was blocked when E2 synthesis was inhibited by letrozole (10−7 M). In contrast to the treatment with GnRH alone, the combination of GnRH (10−8 M) and letrozole (10−7 M) did not result in an increase in the spinophilin staining (mean ± SEM, n = 5)
Previous studies have shown that estradiol decreases GnRH-R expression in ovarian cells and in a gonadotrope-derived cell line (Nathwani et al. 2000; McArdle and Poch 1992). This raises the question of whether the expression of GnRH-R in the hippocampus is influenced by endogenous estradiol. The treatment of hippocampal neurons with letrozole resulted in an increase in GnRH-R immunoreactivty in the cells, compared to untreated cells. These findings argue for an inhibition of hippocampal GnRH-R expression by endogenously produced estradiol and may indicate a negative-feedback mechanism that prevents excessive estradiol production, thus, balancing the system.
Spine synapse counting in the brains of females at estrus and proestrus revealed that estrus cyclicity of spine density occurs specifically in the hippocampus, but not in the frontal cortex. Quantitative RT-PCR proved that GnRH-R expression is fivefold higher in the hippocampus than in the cortex (Fig. 5), which points to the specific responsiveness of the hippocampus to GnRH. A recent study by Quintanar et al. (2007) further supports this idea, as it shows that only 7% of the cortical neurons are immunoreactive for GnRH-R, whereas the GnRH binding capacity in the hippocampus is considerably high (Bard and Pelletier 1987; Reubi et al. 1987; Jennes et al. 1988).
Fig. 5.
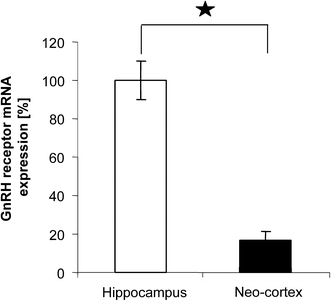
GnRH-R mRNA expression is strongest in the hippocampus. Quantitative RT-PCR demonstrated that the amount of GnRH-R mRNA in the hippocampus was significantly higher than in the neocortex of adult female rats (mean ± SEM, *P < 0.0001, n = 10)
Our data strongly suggest that estrus cyclic synaptogenesis in the female hippocampus results from the cyclic regulation of hippocampal estradiol synthesis in response to the pulsative release of GnRH. Thus, GnRH may synchronize both gonadal and hippocampal estradiol synthesis, and as a consequence, estradiol serum levels and hippocampal spine density change in parallel.
Acknowledgment
This work was supported by the Deutsche Forschungsgemeinschaft.
Footnotes
J. Prange-Kiel and L. Fester contributed equally to this work.
References
- Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthazart J, Baillien M, Ball GF. Interactions between kinases and phosphatases in the rapid control of brain aromatase. J Neuroendocrinol. 2005;17:553–559. doi: 10.1111/j.1365-2826.2005.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthazart J, Baillien M, Ball GF. Rapid control of brain aromatase activity by glutamatergic inputs. Endocrinology. 2006;147:359–366. doi: 10.1210/en.2005-0845. [DOI] [PubMed] [Google Scholar]
- Bard M, Pelletier G. Characterization and autoradiographic localization of Lhrh receptors in the rat-brain. Synapse. 1987;1(6):567–571. doi: 10.1002/syn.890010609. [DOI] [PubMed] [Google Scholar]
- Baulieu EE, Robel P. Neurosteroids: a new brain function? J Steroid Biochem Mol Biol. 1990;37:395–403. doi: 10.1016/0960-0760(90)90490-C. [DOI] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD, Gould GW. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc Natl Acad Sci USA. 2001;98:5619–5624. doi: 10.1073/pnas.091502398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JM, Romeo RD, Brake WG, Bethea CL, Rosenwaks Z, McEwen BS. Estradiol increases pre- and post-synaptic proteins in the CA1 region of the hippocampus in female rhesus macaques (Macaca mulatta) Endocrinology. 2003;144:4734–4738. doi: 10.1210/en.2003-0216. [DOI] [PubMed] [Google Scholar]
- Cordoba Montoya DA, Carrer HF. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats. Brain Res. 1997;778:430–438. doi: 10.1016/S0006-8993(97)01206-7. [DOI] [PubMed] [Google Scholar]
- Daniel EE, Bodie G, Mannarino M, Boddy G, Cho WJ. Changes in membrane cholesterol affect caveolin-1 localization and ICC-pacing in mouse jejunum. Am J Physiol Gastrointest Liver Physiol. 2004;287:G202–G210. doi: 10.1152/ajpgi.00356.2003. [DOI] [PubMed] [Google Scholar]
- Daniel S, Diaz AJ, Martinez KM, Bench BJ, Albertorio F, Cremer PS. Separation of membrane-bound compounds by solid-supported bilayer electrophoresis. J Am Chem Soc. 2007;129:8072–8073. doi: 10.1021/ja0720816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves EI, Rusinol AE, Vance DE, Campenot RB, Vance JE. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem. 1997;272:30766–30773. doi: 10.1074/jbc.272.49.30766. [DOI] [PubMed] [Google Scholar]
- Fan QW, Yu W, Gong JS, Zou K, Sawamura N, Senda T, Yanagisawa K, Michikawa M. Cholesterol-dependent modulation of dendrite outgrowth and microtubule stability in cultured neurons. J Neurochem. 2002;80:178–190. doi: 10.1046/j.0022-3042.2001.00686.x. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fester L, Ribeiro-Gouveia V, Prange-Kiel J, Schassen C, Bottner M, Jarry H, Rune GM. Proliferation and apoptosis of hippocampal granule cells require local oestrogen synthesis. J Neurochem. 2006;97:1136–1144. doi: 10.1111/j.1471-4159.2006.03809.x. [DOI] [PubMed] [Google Scholar]
- Fester L, Zhou L, Butow A, Huber C, Lossow R, Prange-Kiel J, Jarry H, Rune GM. Cholesterol-promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus. 2009;19:692–705. doi: 10.1002/hipo.20548. [DOI] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Zafirov S, Heimrich B. Development of identified neuronal types and of specific synaptic connections in slice cultures of rat hippocampus. Prog Neurobiol. 1995;45:143–164. doi: 10.1016/0301-0082(94)00040-O. [DOI] [PubMed] [Google Scholar]
- Furukawa A, Miyatake A, Ohnishi T, Ichikawa Y. Steroidogenic acute regulatory protein (StAR) transcripts constitutively expressed in the adult rat central nervous system: colocalization of StAR, cytochrome P-450SCC (CYP XIA1), and 3beta-hydroxysteroid dehydrogenase in the rat brain. J Neurochem. 1998;71:2231–2238. doi: 10.1046/j.1471-4159.1998.71062231.x. [DOI] [PubMed] [Google Scholar]
- Good M, Day M, Muir JL. Cyclical changes in endogenous levels of oestrogen modulate the induction of LTD and LTP in the hippocampal CA1 region. Eur J Neurosci. 1999;11:4476–4480. doi: 10.1046/j.1460-9568.1999.00920.x. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, Metzger J, Schweigert FJ, Luppa PB, Nykjaer A, Willnow TE. Role of endocytosis in cellular uptake of sex steroids. Cell. 2005;122:751–762. doi: 10.1016/j.cell.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Janssens RM, Brus L, Cahill DJ, Huirne JA, Schoemaker J, Lambalk CB. Direct ovarian effects and safety aspects of GnRH agonists and antagonists. Hum Reprod Update. 2000;6:505–518. doi: 10.1093/humupd/6.5.505. [DOI] [PubMed] [Google Scholar]
- Jennes L, Dalati B, Conn PM. Distribution of gonadotropin releasing hormone agonist binding-sites in the rat central nervous-system. Brain Res. 1988;452(1–2):156–164. doi: 10.1016/0006-8993(88)90020-0. [DOI] [PubMed] [Google Scholar]
- Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci. 2004;24:5913–5921. doi: 10.1523/JNEUROSCI.5186-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. Embo J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Hajszan T, MacLusky NJ. Androgens increase spine synapse density in the CA1 hippocampal subfield of ovariectomized female rats. J Neurosci. 2004;24:495–499. doi: 10.1523/JNEUROSCI.4516-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin BC, Scanlan TS. Few things in life are “free”: cellular uptake of steroid hormones by an active transport mechanism. Mol Interv. 2005;5:338–340. doi: 10.1124/mi.5.6.5. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Poch A. Dependence of gonadotropin-releasing hormone-stimulated luteinizing hormone release upon intracellular Ca2+ pools is revealed by desensitization and thapsigargin blockade. Endocrinology. 1992;130:3567–3574. doi: 10.1210/en.130.6.3567. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Franklin J, Green L, Hislop JN. Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J Endocrinol. 2002;173(1):1–11. doi: 10.1677/joe.0.1730001. [DOI] [PubMed] [Google Scholar]
- McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res. 2002;57:357–384. doi: 10.1210/rp.57.1.357. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Yanagisawa K. Inhibition of cholesterol production but not of nonsterol isoprenoid products induces neuronal cell death. J Neurochem. 1999;72:2278–2285. doi: 10.1046/j.1471-4159.1999.0722278.x. [DOI] [PubMed] [Google Scholar]
- Mukai H, Tsurugizawa T, Murakami G, Kominami S, Ishii H, Ogiue-Ikeda M, Takata N, Tanabe N, Furukawa A, Hojo Y, Ooishi Y, Morrison JH, Janssen WG, Rose JA, Chambon P, Kato S, Izumi S, Yamazaki T, Kimoto T, Kawato S. Rapid modulation of long-term depression and spinogenesis via synaptic estrogen receptors in hippocampal principal neurons. J Neurochem. 2007;100:950–967. doi: 10.1111/j.1471-4159.2006.04264.x. [DOI] [PubMed] [Google Scholar]
- Nathwani PS, Kang SK, Cheng KW, Choi KC, Leung PCK. Regulation of gonadotropin-releasing hormone and its receptor gene expression by 17beta-estradiol in cultured human granulosa-luteal cells. Endocrinology. 2000;141(5):1754–1763. doi: 10.1210/en.141.5.1754. [DOI] [PubMed] [Google Scholar]
- Parinaud J, Beaur A, Bourreau E, Vieitez G, Pontonnier G. Effect of a luteinizing hormone-releasing hormone agonist (Buserelin) on steroidogenesis of cultured human preovulatory granulosa cells. Fertil Steril. 1988;50:597–602. doi: 10.1016/s0015-0282(16)60190-5. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60:1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange-Kiel J, Wehrenberg U, Jarry H, Rune GM. Para/autocrine regulation of estrogen receptors in hippocampal neurons. Hippocampus. 2003;13:226–234. doi: 10.1002/hipo.10075. [DOI] [PubMed] [Google Scholar]
- Prange-Kiel J, Jarry H, Schoen M, Kohlmann P, Lohse C, Zhou L, Rune GM. Gonadotropin-releasing hormone regulates spine density via its regulatory role in hippocampal estrogen synthesis. J Cell Biol. 2008;180:417–426. doi: 10.1083/jcb.200707043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanar JL, Salinas E, Gonzalez R. Expression of gonadotropin-releasing hormone receptor in cerebral cortical neurons of embryos and adult rats. Neurosci Lett. 2007;411:22–25. doi: 10.1016/j.neulet.2006.06.077. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Palacios JM, Maurer R. Specific luteinizing-hormone-releasing hormone receptor-binding sites in hippocampus and pituitary—an autoradiographical study. Neuroscience. 1987;21(3):847–856. doi: 10.1016/0306-4522(87)90041-8. [DOI] [PubMed] [Google Scholar]
- Rune GM, Wehrenberg U, Prange-Kiel J, Zhou L, Adelmann G, Frotscher M. Estrogen up-regulates estrogen receptor alpha and synaptophysin in slice cultures of rat hippocampus. Neuroscience. 2002;113:167–175. doi: 10.1016/S0306-4522(02)00152-5. [DOI] [PubMed] [Google Scholar]
- Rune GM, Lohse C, Prange-Kiel J, Fester L, Frotscher M. Synaptic plasticity in the hippocampus: effects of estrogen from the gonads or hippocampus? Neurochem Res. 2006;31:145–155. doi: 10.1007/s11064-005-9004-8. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Yarborough C, Osawa Y. Competitive product inhibition of aromatase by natural estrogens. J Steroid Biochem Mol Biol. 1993;44:651–656. doi: 10.1016/0960-0760(93)90274-Z. [DOI] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci. 2006;26:8517–8522. doi: 10.1523/JNEUROSCI.5279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008;29:219–237. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Jahn R. Proteins of synaptic vesicles involved in exocytosis and membrane recycling. Neuron. 1991;6:665–677. doi: 10.1016/0896-6273(91)90165-V. [DOI] [PubMed] [Google Scholar]
- Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol. 2000;2:42–49. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA. A review and update of mechanisms of estrogen in the hippocampus and amygdala for anxiety and depression behavior. Neuropsychopharmacology. 2006;31:1097–1111. doi: 10.1038/sj.npp.1301067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–30. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- Wehrenberg U, Prange-Kiel J, Rune GM. Steroidogenic factor-1 expression in marmoset and rat hippocampus: co-localization with StAR and aromatase. J Neurochem. 2001;76:1879–1886. doi: 10.1046/j.1471-4159.2001.00207.x. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankova M, Hart SA, Woolley CS. Estrogen increases synaptic connectivity between single presynaptic inputs and multiple postsynaptic CA1 pyramidal cells: a serial electron-microscopic study. Proc Natl Acad Sci USA. 2001;98:3525–3530. doi: 10.1073/pnas.051624598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomaku D, Numakawa T, Numakawa Y, Suzuki S, Matsumoto T, Adachi N, Nishio C, Taguchi T, Hatanaka H. Estrogen enhances depolarization-induced glutamate release through activation of phosphatidylinositol 3-kinase and mitogen-activated protein kinase in cultured hippocampal neurons. Mol Endocrinol. 2003;17:831–844. doi: 10.1210/me.2002-0314. [DOI] [PubMed] [Google Scholar]
- Yun SH, Lee DS, Lee H, Baeg EH, Kim YB, Jung MW. LTP induction modifies functional relationship among hippocampal neurons. Learn Mem. 2007;14:190–194. doi: 10.1101/lm.466307. [DOI] [PubMed] [Google Scholar]
- Zhao L, Chen S, Ming Wang J, Brinton RD. 17beta-estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism. Neuroscience. 2005;132:299–311. doi: 10.1016/j.neuroscience.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lehan N, Wehrenberg U, Disteldorf E, Lossow R, Mares U, Jarry H, Rune GM. Neuroprotection by estradiol: a role of aromatase against spine synapse loss after blockade of GABA(A) receptors. Exp Neurol. 2007;203:72–81. doi: 10.1016/j.expneurol.2006.07.020. [DOI] [PubMed] [Google Scholar]