

Abstract
Acute mountain sickness (AMS) is a neurological disorder that typically affects mountaineers who ascend to high altitude. The symptoms have traditionally been ascribed to intracranial hypertension caused by extracellular vasogenic edematous brain swelling subsequent to mechanical disruption of the blood–brain barrier in hypoxia. However, recent diffusion-weighted magnetic resonance imaging studies have identified mild astrocytic swelling caused by a net redistribution of fluid from the “hypoxia-primed” extracellular space to the intracellular space without any evidence for further barrier disruption or additional increment in brain edema, swelling or pressure. These findings and the observation of minor vasogenic edema present in individuals with and without AMS suggest that the symptoms are not explained by cerebral edema. This has led to a re-evaluation of the relevant pathogenic events with a specific focus on free radicals and their interaction with the trigeminovascular system. (Part of a multi-author review.)
Keywords: Acute mountain sickness, Blood–brain barrier, Cerebral edema, Brain swelling, Intracranial hypertension, Free radicals, Inflammation, Trigeminovascular system
Perspective
Acute mountain sickness (AMS) and high altitude cerebral edema (HACE) are caused by hypoxia that typically affect mountaineers who ascend rapidly to high altitude [1]. While it is well established that patients with HACE exhibit extracellular (vasogenic) edema subsequent to disruption of the blood–brain barrier (BBB) [2], the situation with AMS is more complex, due in part to difficulties associated with clinical diagnosis. While controversial [3], current opinion suggests that AMS is a mild form of HACE and that both syndromes share a common pathophysiology linked by vasogenic edematous brain swelling and intracranial hypertension [1, 4].
However, the advent of sophisticated neuro-imaging technologies has provided unique insight into the molecular and vascular mechanisms associated with these high altitude illnesses. Since the literature has dealt so comprehensively with the clinical, epidemiological and therapeutic bases of AMS and HACE [1, 3, 5, 6], the purpose of the current paper is to critically review the emerging and often contentious issues surrounding pathophysiology with a focus on the molecular bases of disease.
First, we consider the atomic structure of the diatomic O2 molecule to reveal a more nebulous side to a “free radical” gas traditionally considered the elixir of life, on which the brain has evolved to be so heavily dependent yet can prove paradoxically fatal when in excess. We discuss how redox-sensitive mechanisms are activated by hypoxia, with free radicals assuming a protective rather than a purely destructive role that serves to defend cerebral O2 homeostasis and the neurovascular complications that arise when the brain’s “O2-sensing” apparatus fails to respond adequately. Finally, in the light of recent neuro-imaging findings, we critically examine whether AMS is indeed a mild form of HACE and whether symptoms can be ascribed to intracranial hypertension subsequent to vasogenic edematous brain swelling.
Oxygen and the brain
Photosynthesizing cyanobacteria breathed life into what was, until a billion years ago, considered a reductive atmosphere, thus providing a selective pressure for the evolution of O2-dependent micro-organisms that began with the autotrophic eukaryotes [7]. Since these primordial times, the respiring mammalian cell has become entirely dependent on molecular O2 since it serves as the terminal electron acceptor in mitochondrial oxidative phosphorylation, and multiple enzymes require O2 as a substrate [8].
The human brain exemplifies this reliance on O2 since, unlike most other tissues, it is committed to a continually active state. Weighing less than 2% of body mass, it accounts for an impressive, albeit disproportionate, 25% of the basal O2 budget. This ability to process large amounts of O2 over a relatively small tissue mass is required to support the high rate of adenosine tri-phosphate (ATP) production to maintain an electrically active state for the continual transmission of neuronal signals [9].
However, this obligatory high rate of cerebral O2 consumption is associated with a correspondingly high “vulnerability for failure”. In the human brain, ATP concentrations fall drastically within minutes of anoxia which can result in devastating consequences, as the complications associated with stroke and head trauma clearly demonstrate. However, evolution solved this problem millions of years ago with the emergence of anoxia-tolerant vertebrates such as the freshwater turtle and the carp fish which display extraordinary abilities to cope with functional disturbances in cerebral O2 delivery [10]. Though the human brain’s defense mechanisms against hypoxia are impressive, they pale into insignificance when compared with those exhibited by the ectothermic vertebrates [9].
Bi-radical O2: a thermodynamic quirk of evolution
Before we consider the more subtle aspects of hypoxia and its impact on central nervous system (CNS) function at high altitude, it is worth emphasizing that the brain relies on what is essentially a mutagenic gas. Our pre-occupation with improving O2 delivery to respiring tissue, especially in the sports performance context, often begets the vascular complications caused by “O2 excess” which is toxic to the CNS and can often prove fatal.
This is because the triplet ground state diatomic O2 molecule exists as a free radical with two unpaired electrons each located in a different π* molecular orbital but with the same spin quantum number (Fig. 1). Fortunately, unlike most other oxidizing free radical species, this parallel spin renders O2 less reactive at “normal” concentrations despite its powerful oxidizing nature. This “spin-restriction” is a thermodynamic quirk of evolution, and the fact that O2 prefers to accept one electron at a time protects the organic bio-molecules of the human brain from spontaneous combustion!
Fig. 1.

Molecular orbital diagram of the electronic ground state (most stable form) diatomic oxygen molecule (3∑g−O2). Each line represents a molecular orbital and the arrows represent electrons, the direction of which indicates their spin quantum number. Note that O2 qualifies as a di-radical since it contains two unpaired electrons each occupying different π*2p anti-bonding orbitals (each missing electron is marked X). These unpaired electrons have the same spin quantum number (parallel spin). During the process of oxidation, O2 thermodynamically prefers to accept only one electron at a time into each of the vacant π*2p anti-bonding orbitals to conform with the Pauli Exclusion Principle. Fortuitously, this “spin restriction” means that O2 does not react vigorously with the brain’s organic compounds and instead prefers to react with other radicals
However, at elevated partial pressures of O2 (PO2), the mitochondrial formation of the univalent reductant, the superoxide anion (O−•2), increases and is considered the major source of the biological damage associated with O2 toxicity [11]. Though not especially reactive (E 0 = +940 mV), O−•2 can be converted to hydrogen peroxide (H2O2) through reduction or dismutation and, upon reaction with transition metal ions, can form the hydroxyl species (OH•). This species is at the top of the free radical “pecking order” or “league of reactivity” (E 0 = +2,310 mV), thermodynamically capable of oxidizing any bio-molecule that it collides with at a rate constant very near the diffusion limit [12].
Contemporary physiology emphasizes the conceptual significance of the “O2-cascade”, which describes an ever-decreasing PO2 gradient to provide a “pressure-head” to maintain diffusive O2 flux from the ambient to the (cerebral) mitochondrion [13]. What is often overlooked, however, is the concept that the endogenous resistances offered to O2 transport may have evolved, by chance or by fate, to protect the cell from the full force of O2, an in-built antioxidant defence system. The fact that the Michaelis constant (Km) of the terminal reductant, cytochrome c oxidase, for O2 is so extraordinarily low (0.03–0.3 mmHg) [14] shows how important it is to harness this molecule and maintain cellular PO2 within “safe” manageable biological limits.
Molecular targets of oxidative stress
Our reliance on this toxic gas is matched by an equally paradoxical finding; despite its limited regenerative capacity, the brain is poorly equipped to cope with these potentially damaging free radical reactions [15]. Nervous tissue seems to out-perform other tissues in that it is more prone to O−•2 formation with advancing age [16].
Several features related to the molecular composition of the brain and biochemical milieu it supports render it particularly vulnerable to free radical attack. For example, antioxidant defenses are modest at best, and neuronal membrane lipids rich in eicosapentaenoic (C20:5) and docosahexaenoic (C22:6) polyunsaturated fatty-acid side chains are especially susceptible to lipid peroxidation. Second to adipose tissue, nerve tissue contains the highest concentration of these highly peroxidizable lipids. A dense network of mitochondria exposed to a high mass-specific O2 flux, autoxidizable neurotransmitters, cytochrome P450 and reactive microglia also serve to compound O−•2 formation. Excito-toxic amino acids and highly active neuronal Ca2+ trafficking further contribute to the brain’s “oxidant burden” [17].
However, what makes the brain especially vulnerable to oxidation is the fact that it is rich in iron, particularly the globus pallidus (up to 370 μg/g tissue wet weight), basal ganglia, substantia nigra, red nucleus, caudate nucleus, and putamen [18] that, if not adequately regulated, can promote damage [15]. Unlike human plasma, cerebrospinal fluid (CSF) has no significant iron-binding capacity since transferrin content is remarkably low and thus considered at or close to saturation [19]. Thus, the high rate of O−•2 formation coupled with mounting potential for cerebrovascular tissue damage could potentially unlock Fe2+ to catalyze Fenton/Haber–Weiss-driven generation of OH• to initiate neuronal apoptosis and further compound membrane destabilization and vascular damage. Ironically, ascorbate, the most effective aqueous-phase chain-breaking antioxidant, is present in high concentrations in the white and the grey matter and has the capacity to further aggravate oxidative catalysis [15].
The concept that the human brain is “oxidatively-primed” with a poor ability to contain these potentially damaging free radical chain reactions [17] has important implications for the morphological changes that occur in response to hypoxia which is the focus of subsequent discussion. The reader is encouraged to consult several excellent reference texts that deal comprehensively with the source, mechanisms, and functional consequences of biologically-relevant free radical species [12, 20, 21].
The adaptive response to hypoxia: O2 “sensing”
This reliance on O2 renders the human brain exquisitely sensitive to hypoxia and, as a consequence, elaborate cellular oxygen-sensing mechanisms have evolved to defend cerebral O2 homeostasis to ultimately prevent brain damage in the face of a blunted PO2. The search for a “unifying” O2 sensor has stimulated enormous research interest with numerous candidate proteins proposed which have been discussed in detail by Drs Pugh and Simone in this issue.
One elegant theory that is gaining prominence is the notion that the mitochondria function as the primary “O2 sensors” [22]. This makes intuitive sense since they bind O2 at the cytochrome oxidase and represent the primary site of cellular O2 consumption. However, the “cornerstone” of the mitochondrial model, the concept that hypoxia accelerates free radical formation represents “yet another O2 paradox” [23] and remains the subject of on-going controversy. Though elegant in design, this model has been met with some resistance mostly because cytochrome oxidase activity does not become O2 limited until concentrations fall to pathophysiologically low levels in the order of ≈1 mmHg or <1% [24].
The mitochondrial model contends that hypoxia triggers free radical release mostly from Complex III and that these act as signaling molecules that ultimately activate O2 “salvage” genes through stabilization of the transcription factor hypoxia-inducible factor (HIF)-1α [24]. Hypoxia is known to increase the expression of both astrocytic and neuronal erythropoietin (EPO) [25] and vascular endothelial growth factor (VEGF) mRNA [26], which serve to not only increase O2 transport but also reduce neuronal apoptosis and counteract inflammation. Thus, the brain walks a biochemical tight-rope; it needs to carefully titrate free radical formation to optimize O2 delivery while limiting the potential for vascular damage. It would appear that the brain pivots delicately between a bimodal distribution of free radicals that can cause damage when in excess under both hypoxic and hyperoxic PO2’s (Fig. 2). Understanding what constitutes a physiologically “appropriate” concentration of free radicals in the hypoxic human brain and how this is achieved remains the Holy Grail for molecular biologists and physiologists alike.
Fig. 2.

The healthy human brain pivots delicately between a bi-modal distribution of free radicals poised to increase in both hypoxia (that serve to defend cerebral oxygenation) and hyperoxia (capable of causing structural damage) subsequent to mitochondrial superoxide (O−•2) formation. The rate of increase in O−•2 increases in hypoxia (primarily the Q cycle) and hyperoxia due to an increase in the concentration of electron donors (R•) and O2 concentration, respectively. The resting intracellular partial pressure of oxygen is based on measurements recently documented in resting normoxic human skeletal muscle [70]
When the molecular and physiological adaptations unite, global cerebral energetics remain remarkably well preserved at least during moderate hypoxia when arterial hemoglobin O2 saturations (SaO2) fall to 80% even in the face of hypocapnia (PaCO2 ≈ 25 mmHg) and attendant cerebral vasoconstriction [27]. The cerebral metabolic rate for oxygen and glucose are also maintained at normal levels for up to 21 days of hypoxia [27]. However, while existing measurement techniques fail to identify any evidence for a “global” O2 deficit in the human brain, positron-emission tomography (PET) measurements of [18F]2-deoxy-2-fluoro-d-glucose uptake have revealed striking spatio-temporal differences in the brain’s “regional” energy consumption [28]. The hippocampus, and specifically the CA1 neurons, the corpus striatum, and Purkinje cells of the cerebellum, are especially sensitive to the severe hypoxia associated with ischemia [29]. Furthermore, neurotransmitters, the ubiquitous regulators of neuronal activity, are particularly vulnerable in light of their correspondingly high Km for O2 [30].
This high rate of metabolic activity in the human brain contrasts with that observed in hypoxic-tolerant ectothermic species and diving mammals, particularly the aquatic turtle, which exhibit a remarkable ability to suppress cerebral metabolism and thus optimize energy demand–supply coupling as part of a co-ordinated defense strategy [31]. Preliminary indications suggest that a similar strategy exists in humans born and bred at high altitude (native highlanders) particularly in regions classically associated with higher cortical functions (frontal cortex and angular gyri) intimating that cerebral hypometabolism likely represents a universal adaptation of the CNS in hypoxia-tolerant species [32].
Thus, at least in the native lowlander, it is conceivable that focal metabolism and neuronal communication may become impaired even with subtle reductions in the inspired PO2. This certainly appears to be the case in those individuals who, for reasons that remain unclear, appear more “hypoxia-sensitive” and, as a consequence, less able to defend systemic and cerebral oxygenation. This phenotype is the focus of subsequent discussion since these individuals appear to be more susceptible to the high altitude syndromes that affect the brain, specifically AMS and HACE.
AMS and HACE
AMS is a syndrome of non-specific symptoms typically experienced by non-acclimatized mountaineers within 6–12 h of arrival to altitudes above 2,500 m [1, 3]. It is defined by the presence of headache plus at least one of the following symptoms: anorexia, nausea, vomiting, fatigue, dizziness, and insomnia. AMS is generally benign though may progress to HACE in more severe cases or during continued ascent above 4,000 m, when symptoms of AMS are present. HACE is characterized by altered consciousness, ataxia, or both in a subject with AMS or high altitude pulmonary edema (HAPE) and, if left untreated, can result in death subsequent to brain herniation [1].
Pathophysiology: the traditional paradigm
Current opinion suggests that AMS is a self-limiting mild form of HACE and that both syndromes share a common pathophysiology linked by vasogenic edematous brain swelling that ultimately leads to intracranial hypertension [1]. Intracranial hypertension has been believed to be the primary cause of AMS since the displacement and stretching of pain-sensitive unmyelinated fibres that reside within the trigeminovascular system (TVS) provides an elegant mechanism for the headache [33]. The events that conspire to increase intracranial volume (ICV) have since become the focus of detailed investigation since this is considered the key to understanding the underlying pathophysiology [4].
However, recently published diffusion-weighted magnetic resonance imaging (MRI) studies [34–37] have challenged the traditional paradigm which has led to a revised hypothesis and subsequent modification of the underlying molecular, metabolic, hemodynamic, and morphological events traditionally considered important in AMS and HACE.
Morphological response to hypoxia: the normal (adaptive) phenotype
Figure 3 summarizes the “typical” sequence of events that occur in the human brain in response to hypoxia, which is considered the “upstream” stimulus responsible for activation of the adaptive molecular apparatus that serves to maintain cerebral O2 homeostasis. However, there are rebound consequences “downstream” that increase the potential for cerebral hyper-perfusion (mechanical forces) and capillary leak (chemical forces), which can encourage the formation of extracellular cerebral edema. Cerebral blood flow (CBF) measured using the Kety-Schmidt [38], Xenon-133 [39], or Doppler sonography of the middle cerebral artery [40] techniques has been shown to increase, and disordered cerebral autoregulation has also been documented, in response to high altitude hypoxia [41, 42]. Furthermore, the circulating concentration of free radicals [34, 42–44] and free VEGF also increase in hypoxia [45] thus providing a chemical stress with the potential to increase BBB permeability. These responses eventually conspire to increase cerebral capillary hydrostatic pressure and promote, albeit mild, extracellular (vasogenic) edema and brain swelling subsequent to hydrostatic disruption of the BBB independent of AMS.
Fig. 3.

Normal response of the cerebral circulation to hypoxia. T 2 rt T2 relaxation time, ADC apparent diffusion co-efficient
Two recently published MRI studies (1.5–3T) provided convincing evidence for mild vasogenic edematous brain swelling following 6–18 h passive exposure to normobaric hypoxia in a simulated high altitude chamber (FIO2 ~ 12%) [35, 36]. Vasogenic edema was detected both in subjects diagnozed with clinical AMS (AMS+) and in healthy subjects who were free of symptoms (AMS−) [35, 36]. In contrast, intracellular edema was confined solely to AMS+ subjects, in particular those with more severe symptoms [35, 36].
One of these two studies [36] identified extracellular edema defined by a significant increase of both apparent diffusion coefficients (ADC derived from diffusion-weighted images) and B0 values [T2 relaxation times (T2rt) derived from T2-weighted images] in the white matter. In the other study [35], extracellular edema was confined to the splenium of the corpus callosum, the same predilection site in patients with HACE [2]. The predilection for the corpus callosum may be a consequence of its rather unique vascular anatomy; densely packed horizontal fibers characterized by short arterioles that lack adrenergic tone may render it more susceptible to hyper-perfusion edema in the setting of hypoxic cerebral vasodilatation [2]. However, it is important to note that the MRI examinations of AMS+ and AMS− subjects was performed during exposure to hypoxia, whereas MRI of patients with HACE was performed up to 6 days following evacuation to lower altitudes (i.e., in normoxia). Thus, the possibility that both vasogenic and cytotoxic edema “co-exist” in HACE remains to be examined [2, 46].
In one study which focused on the molecular determinants of altered morphology, there was no evidence of “gross” barrier dysfunction or neuroglial damage, as indicated by the lack of change in the serum and CSF concentration of S100β, neuron-specific enolase (NSE) and CSF-blood protein concentration quotients [34]. Furthermore, there has been a general failure to detect any visual signs consistent with cerebral edema such as midline shift, mass effect, effacement of sulci, ventricle collapse, or compression of cisterns (Fig. 4), and lumbar pressure also appears to be normal [34]. The astrocytic protein, S100β, was incorporated as a specific marker of BBB integrity since recent studies incorporating osmotic opening of the BBB have identified that extravasation of this astrocytic protein in blood can occur in the absence of or preceding neuroglial damage [47–49]. Thus, the MR evidence to date indicates a minor as opposed to a major breach of barrier function which is so subtle that it is indeed beyond standard molecular detection limits.
Fig. 4.
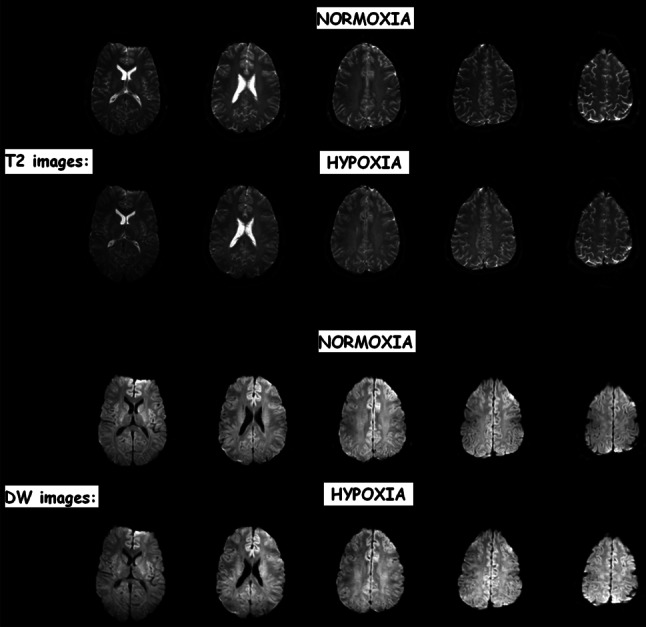
3T-T2 and diffusion-weighted (DW) images of a subject with clinical AMS. Note the lack of any visual difference between normoxia and following 6 h passive exposure to hypoxia (12%O2) indicating that the edema detected by MRI is very subtle indeed. Baumgartner et al. (unpublished findings) based on [36]
Priming of the extracellular space (ECS) may actually prove of functional significance and represent an adaptive response to hypoxia. An increase in the ECS would serve to buffer any rise in the interstitial concentration of neurotransmitters, such as glutamate and glycine, thus reducing the risk of a tonic activation of NMDA receptors [50]. Furthermore, it may also attenuate extracellular K+ that could threaten to depolarize the brain [51]. Interestingly, neonates, who are far more hypoxia-resistant than adults, exhibit an enlarged ECS (15–25% of the whole brain compared to 10–20% in the adult) [52]. Though speculative, vasogenic edematous brain swelling may prove the adaptive phenotypical response in the hypoxia-tolerant human.
AMS: the maladaptive phenotype?
One would anticipate that these changes would be more pronounced in AMS+ subjects consistent with the vasogenic hypothesis. However, no relationship was identified between the hypoxia-induced increase in brain volume, T2rt, or ADC and cerebral AMS scores [35, 36]. Furthermore, post hoc analysis indicated that there were no differences in volumetrics or relaxometry when AMS+ were compared against AMS− subjects. The morphological findings were further complemented by the lack of any molecular-biochemical evidence for altered BBB function or neuronal-parenchymal damage. These important studies emphasize three conceptually important findings: (1) mild vasogenic edematous brain swelling represents the typical, arguably adaptive phenotypical response to hypoxia that has little bearing on the symptoms of AMS, (2) edema occurs in the absence of a “major” breach of the BBB, and (3) the “AMS brain” is not characterized by any additional volume overload thus excluding intracranial hypertension as a significant pathophysiological event (Fig 5).
Fig. 5.

Recent evidence suggesting that subjects diagnozed with clinical acute mountain sickness (AMS+) do not exhibit any additional increment in brain swelling (a), (vasogenic) edema (b), alteration in blood–brain barrier (BBB) permeability (c), or lumbar pressure (d). Data modified from [34]; values represent mean ± SD calculated as the difference between hypoxia relative to the normoxic control
Anatomy: intracranial-intraspinal buffering capacity
Borne out of an original hypothesis developed over 20 years ago [53], the lack of correlation between brain swelling and AMS symptoms has since been ascribed to anatomical differences that determine how effectively the human skull can accommodate swelling through displacement of cranial CSF to extra-cranial compartments [1]. Thus, individuals characterized by a smaller ratio of cranial CSF to brain volume (popularized as the “tight-fit” brain) would be expected to be more prone to intracranial hypertension and thus, by consequence, AMS, since they are less capable of “buffering” the volume increase through changes in CSF dynamics due to an inadequate cranio-spinal axis CSF reserve.
To date, only one human study has provided indirect evidence to support the “tight-fit” hypothesis with AMS+ subjects retrospectively identified as having comparatively larger brain volume to ICV ratios even at sea-level [35]. Though elegant by design, we are not entirely convinced that this has any bearing on the pathophysiology of AMS and likely represents an incidental finding. It would seem highly unlikely that the volumetric changes imposed would translate into any physiologically meaningful changes in the mechanical displacement of pain-sensitive structures that could activate the TVS [3]. The small increases in ICV documented in the literature likely occupy the flat part of the ICP-volume curve, eminently well-buffered by semi-elastic membranes and changes in CSF dynamics [30].
Reduced ADC score associated with increased T2rt in AMS: interpretation and significance
Arguably one of the most important findings in recent years relates to the combined observation that AMS+ subjects exhibited a selective decrease in ADC scores in hypoxia and that this was not associated with any additional evidence of vasogenic edematous brain swelling beyond the mild changes incurred by hypoxia per se [35, 36]. This finding was unexpected in light of the prevailing vasogenic hypothesis. Both groups interpreted this as evidence for an additional, albeit mild, intracellular (cytotoxic) edema superimposed on a pre-existing extracellular vasogenic edema provoked by hypoxia.
The lack of any difference in the 3T-MRS signal of lactate in AMS+ compared to AMS− (unpublished findings) confirms that the reduction in ADC was indeed a genuine finding that was not confounded by intra-cerebral lactacidosis which has been raised as a potential concern since the lactate anion can interfere with the ADC measurements [35]. Since both studies failed to identify any evidence for additional edema or brain swelling, the decrease in ADC likely reflects “fluid re-distribution” from within the ECS as intracellular (astrocytic) swelling proceeds without any additional increment in brain volume, edema, or ICP [54].
However, the critical question that remains unanswered is whether AMS is caused by such a “minor” shift of water into the cells of the corpus callosum that entered as a result of mild vasogenic edema in hypoxia. Edema of the corpus callosum would be expected to cause a disconnection syndrome [55] but no symptom of AMS.
The observation that intracellular swelling occurs in hypoxia is not new with early studies documenting up to a 0.25 L (+12%) increase in the intracellular fluid compartment, albeit in skeletal as opposed to cerebral tissue [56]. This has been attributed to a down-regulation of (muscle) cell membrane ionic pumping subsequent to inactivation of the Na+/K+-ATPase [57]. The hypoxic suppression of BBB ion-pumping activity may therefore provide an alternative explanation for the accumulation of extracellular fluid recently observed in the brain independent of barrier disruption [34]. Other mechanisms such as increased Na+ and K+ permeability of the cell membrane and a sustained uptake of osmotically-active solutes may have also contributed to what has been coined “pump-leak-equilibrium disturbance” [58].
Intracranial pressure
While the technical challenges posed by the accurate, non-invasive assessment of ICP are acknowledged, the human evidence in support of elevated ICP in AMS remains limited despite preliminary indications of decreased compliance (for a review, see [30]). This contrasts with the findings reported by Krasney et al. [59] in the hypoxic sheep model which lends itself to a more invasive approach. However, it is important to recognize the limitations associated with this model, most notably the unavoidable difficulties associated with the clinical definition of AMS.
More recently, B-scan ultrasound of the eye was employed to assess the optic sheath nerve diameter (OSND) to provide non-invasive insight into ICP changes during an active ascent of Mt. Everest [60]. The authors demonstrated an association between OSND and AMS symptom scores, thus arguing a pathogenic role for intracranial hypertension. However, closer inspection of their data reveals a physiologically insignificant increase in OSND (+0.02 mm) at the intermediate altitudes of 2,000–3,700 m where symptoms of AMS typically present. Furthermore, and by the authors’ own admission, at the higher altitude of 6,400 m, OSND actually increased as symptoms tended to resolve. In summary, there is no clear human evidence to suggest that ICP is raised in AMS [3].
This makes intuitive sense when one considers that intracranial hypertension would only be expected to occur when the ECS has been compressed to its maximum degree with the volume of the swollen astrocytes exceeding the volume translocated from the ECS [54]. In the two most recent neuro-imaging studies, the water entering the astrocytes likely originated from the plasma in the ECS which had already expanded during the phase of mild vasogenic edema observed in hypoxia (illustrated in Fig. 3). However, the volume available for “re-distribution” was very small indeed in light of the modest changes observed in brain volume (+0.6 ± 0.4%) [35] and lack of BBB disruption [34]. Recent estimates suggest that astrocytes need to swell by up to as much as 60% before cytotoxic edema and thus brain swelling can occur [54].
An emerging hypothesis: astrocytic swelling and redox-activation of the TVS
These findings clearly fail to support the traditional theory that AMS is caused by intracranial hypertension as a result of vasogenic edematous brain swelling [1], and have led to the formulation of an alternative hypothesis with a specific focus on redox-activation of the TVS. The reader is referred to an excellent review for a more thorough discussion of the anatomy and subsequent modulation of trigeminovascular nociceptive input from the meningeal vessels albeit during the pathophysiology of migraine pain [61].
Figure 6 presents the revised sequence of events that warrant future investigation, and compares and contrasts these with the mechanisms currently supported by the literature. The schema describes how inspiratory hypoxia catalyzes mitochondrial O−•2 and subsequent OH• formation through Fenton and Haber–Weiss chemistry. These “upstream” radicals are eminently capable of causing neurovascular endothelial dysfunction and depressing hypoxic ventilatory control and pulmonary gas exchange [62], and thus provide a molecular basis to explain why AMS+ subjects are typically more hypoxemic for any given FIO2 [17].
Fig. 6.

Revised schema of events implicated in the pathophysiology of acute mountain sickness (AMS) and high-altitude cerebral edema (HACE). The traditional model (A) (stippled lines) contends that vasogenic edematous brain swelling combined with a reduced CSF buffering capacity may predispose to intracranial hypertension and thus by consequence, AMS. The revised model (B) suggests that AMS is not associated with any “additional” volume overload thus arguing against a role for intracranial hypertension. It describes how free radicals can directly activate the trigeminovascular system to trigger neurovascular headache and AMS. HACE may reflect the more extreme spectrum of “osmotic-oxidative stress” resulting in gross barrier dysfunction and cerebral capillary “stress failure”. ICV Intracranial volume, ECS/ICS extra/intra-cellular space
Free radicals may be responsible for “pump failure” since they can reduce Na+/K+-ATPase activity which may prove the mechanism responsible for ECF re-distribution and subsequent expansion of the astrocytic space independent of any global volumetric changes to the brain [35, 36]. It is tempting to speculate that AMS may represent an “ionopathy” characterized by free radical-mediated ion channel dysfunction whose activity and distribution in the hypoxic brain determine the clinical phenotype.
Astrocyte swelling can directly activate p47phox-dependent NADPH oxidase [63] and further compound “osmotic-oxidative stress”. It can also increase cerebral nitric oxide formation [64] that, in combination with the increase in free radical-mediated lipid peroxidation and subsequent membrane destabilization further compounded by local HIF-1α and VEGF activation and inflammation, can directly activate the TVS, the final common pathway. The recent observation that AMS did not influence the (steady-state) cerebral metabolism of the “migraine-molecule”, calcitonin gene-related peptide (CGRP) [65], tends to complicate this hypothesis and argues against “sustained activation” of the TVS as an important event. However, this finding does not exclude acute release from trigeminal perivascular nerve fibers, the site of nociception during the early phase of hypoxia, and the lack of major breach in the BBB may have also prevented intrathecally-formed CGRP from entering the extracranial circulation in sufficient amounts to permit molecular detection. This important finding also argues a need to focus on regional changes in alternative “non-classical” biomarkers that provide insight into cerebral redox-homeostasis.
For example, direct inhibition of TVS activity with sumatriptan, a selective 5-hydroxytryptamine1B/1D agonist and cerebral vasoconstrictor, has recently been shown to provide effective prophylaxis and to reduce the relative risk of AMS by over 50% [66]. In addition to its ability to attenuate cell excitability in trigeminal nuclei via stimulation of 5-HT1B/1D receptors within the brainstem and vasoconstriction of meningeal, dural, cerebral, or pial vessels via stimulation of vascular 5-HT1B/1D receptors, sumatriptan can directly scavenge O−•2, NO, and OH• [66]. We have recently provided direct electron paramagnetic resonance (EPR) spectroscopic evidence for increased OH• and downstream formation of lipid-derived free radicals in the CSF of a subject diagnozed with severe AMS (Fig. 7). Furthermore, the neuroprotective benefits conferred by dietary antioxidant vitamin prophylaxis [67] and relationship between the increased net cerebral output of free radical-mediated lipid peroxidants in hypoxia and symptoms of AMS add further support to what is rapidly evolving as the redox-hypothesis [17, 68].
Fig. 7.

X-band electron paramagnetic (EPR) spectroscopic detection of a hydroxyl (OH•: a N = a H = 14.9 Gauss) and b mixture of lipid-derived alkyl (LC•: a N1 = 15.8G, a H1 = 2.8G), •OCH3 (a N2 = 14.5G, a H2 = 2.1G) and nitroxide (a N3 = 16.0G) free radicals in the cerebrospinal fluid of one subject in normoxia without acute mountain sickness (AMS−) and following 18 h exposure to hypoxia (12%O2) when severe acute mountain sickness (AMS+) was diagnozed. EPR spectroscopy is considered the most sensitive, specific, and direct molecular technique for the detection and subsequent identification of free radicals sine qua non [17]. The spectra illustrated were obtained using an ex vivo spin-trapping technique [34]. The spin-traps 5,5-dimethylpyrroline-N-oxide (DMPO-1 mM) and α-(4-pyridyl 1-oxide)-N-tert-butylnitrone (POBN-50 mM) were employed to detect proximal •OH and distal lipid-derived species, respectively. Note the marked increase in free radical-mediated lipid peroxidation in AMS+
The link to HACE
While HACE has traditionally been ascribed to vasogenic edema [1], it is reasonable to assume that cytotoxic edema may also be present [2, 46] since hypoxaemia is likely more marked compared to AMS. Therefore, cell swelling should even be even more enhanced in HACE. However, support for this hypothesis by measurements of ADC immediately after evacuation from altitude in patients with HACE is lacking and thus at present remains speculative. Thus, it is conceivable that both AMS and HACE share similar features in that there is an underlying vasogenic component (albeit mild and not responsible for symptoms in AMS) that, subsequent to a more pronounced regional O2 diffusion limitation, may trigger intracellular swelling that is yet to be confirmed in HACE. In this sense, AMS may be considered a mild form of HACE.
In addition, HACE may also reflect the more extreme spectrum of osmotic-oxidative stress resulting in gross BBB dysfunction, cerebral capillary “stress failure”, and erythrocyte extravasation. This was confirmed in a recent study that combined conventional T2* with a novel, highly-sensitive susceptibility-weighted MRI technique to reveal multiple “microbleeds” detected as hemosiderin deposits confined to the genu and splenium of the corpus callosum (Fig. 8) of HACE patients [69]. The failure to detect any hemosiderin deposits in subjects diagnosed with severe AMS supports the concept that vasogenic edema is minor in AMS and does not account for symptoms, and further points to a novel diagnostic feature that may discriminate between AMS and HACE.
Fig. 8.
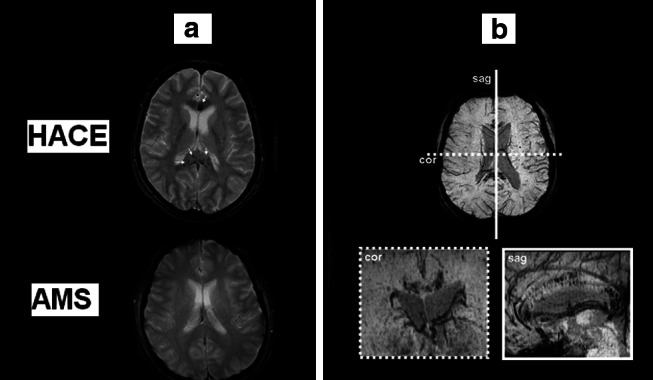
Susceptibility-weighted T2*-images (a) obtained in one subject 7 weeks after CT diagnosis of HACE following ascent to 3,860 m and shortly following clinical diagnosis of severe AMS in a separate subject at 5,895 m. Note the multiple patchy hypo-intensities (arrows) present in the HACE subject only that correspond to micro-hemorrhages that result in hemosiderin [insoluble Fe(III) oxide hydroxide] deposition secondary to blood–brain barrier disruption and cerebral capillary “stress failure”. These “micro-bleeds” were located predominantly in the genu and splenium of the corpus callosum (b) which have previously been identified as predilection sites for vasogenic edema in HACE [2]. Data modified from [69]
References
- 1.Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345:107–114. doi: 10.1056/NEJM200107123450206. [DOI] [PubMed] [Google Scholar]
- 2.Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J. High-altitude cerebral edema evaluated with magnetic resonance imaging: clinical correlation and pathophysiology. J Am Med Assoc. 1998;280:1920–1925. doi: 10.1001/jama.280.22.1920. [DOI] [PubMed] [Google Scholar]
- 3.Bartsch P, Bailey DM, Berger MM, Knauth M, Baumgartner RW. Acute mountain sickness; controversies, advances and future directions. High Alt Med Biol. 2004;5:110–124. doi: 10.1089/1527029041352108. [DOI] [PubMed] [Google Scholar]
- 4.Roach RC, Hackett PH. Frontiers of hypoxia research: acute mountain sickness. J Exp Biol. 2001;204:3161–3170. doi: 10.1242/jeb.204.18.3161. [DOI] [PubMed] [Google Scholar]
- 5.Basnyat B, Murdoch DR. High altitude illness. Lancet. 2003;361:1967–1974. doi: 10.1016/S0140-6736(03)13591-X. [DOI] [PubMed] [Google Scholar]
- 6.Wilson MH, Newman S, Imray CH. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009;8:175–191. doi: 10.1016/S1474-4422(09)70014-6. [DOI] [PubMed] [Google Scholar]
- 7.Massabuau JC. From low arterial- to low tissue-oxygenation strategy. An evolutionary theory. Respir Physiol. 2001;128:249–261. doi: 10.1016/S0034-5687(01)00305-X. [DOI] [PubMed] [Google Scholar]
- 8.Erecinska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–276. doi: 10.1016/S0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- 9.Lutz PL, Nilsson GE, Prentice HM. The brain without oxygen: causes of failure-physiological and molecular mechanisms for survival. Dordrecht: Kluwer; 2003. [Google Scholar]
- 10.Nilsson GE. Surviving anoxia with the brain turned on. News Physiol Sci. 2001;16:217–221. doi: 10.1152/physiologyonline.2001.16.5.217. [DOI] [PubMed] [Google Scholar]
- 11.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 12.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, [alpha]-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 13.Wagner PD. Determinants of maximal oxygen transport and utilization. Annu Rev Physiol. 1996;58:21–50. doi: 10.1146/annurev.ph.58.030196.000321. [DOI] [PubMed] [Google Scholar]
- 14.Vanderkooi JM, Erecinska M, Silver IA. Oxygen in mammalian tissue: methods of measurement and affinities of various reactions. Am J Physiol Cell Physiol. 1991;260:C1131–C1150. doi: 10.1152/ajpcell.1991.260.6.C1131. [DOI] [PubMed] [Google Scholar]
- 15.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 16.Sawada M, Carlson JC. Changes in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat. Mech Ageing Dev. 1987;41:125–137. doi: 10.1016/0047-6374(87)90057-1. [DOI] [PubMed] [Google Scholar]
- 17.Bailey DM. Radical dioxygen: from gas to (unpaired!) electrons. Adv Exp Med Biol. 2003;543:201–221. [PubMed] [Google Scholar]
- 18.Haacke EM, Cheng NY, House MJ, Liu Q, Neelavalli J, Ogg RJ, Khan A, Ayaz M, Kirsch W, Obenaus A. Imaging iron stores in the brain using magnetic resonance imaging. Magn Reson Imaging. 2005;23:1–25. doi: 10.1016/j.mri.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Gruener N, Gozlan O, Goldstein T, Davis J, Besner I, Iancu TC. Iron, transferrin, and ferritin in cerebrospinal fluid of children. Clin Chem. 1991;37:263–265. [PubMed] [Google Scholar]
- 20.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJA. Free radical biology and medicine: it’s a gas, man! Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–R511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 21.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clanton T. Yet another oxygen paradox. J Appl Physiol. 2005;99:1245–1246. doi: 10.1152/japplphysiol.00609.2005. [DOI] [PubMed] [Google Scholar]
- 24.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 25.Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I, Mackenzie ET, Petit E. Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia. 2000;30:271–278. doi: 10.1002/(SICI)1098-1136(200005)30:3<271::AID-GLIA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moller K, Paulson OB, Hornbein TF, Colier WNJM, Paulson AS, Roach RC, Holm S, Knudsen GM. Unchanged cerebral blood flow and oxidative metabolism after acclimatization to high altitude. J Cereb Blood Flow Metab. 2002;22:118–126. doi: 10.1097/00004647-200201000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy C, Sakurada O, Shinohara M, Jehle J, Sokoloff L. Local cerebral glucose utilization in the normal conscious macaque monkey. Ann Neurol. 1978;4:293–301. doi: 10.1002/ana.410040402. [DOI] [PubMed] [Google Scholar]
- 29.Pulsinelli WA (1985) Selective neuronal vulnerability: morphological and molecular characteristics. In: Kogure K, Hossman KA, Siesjo BK (eds) Molecular mechanisms of ischemic brain damage. Elsevier, Amsterdam, pp 29–37 [DOI] [PubMed]
- 30.Hackett PH. The cerebral etiology of high-altitude cerebral edema and acute mountain sickness. Wilderness Environ Med. 1999;10:97–109. doi: 10.1580/1080-6032(1999)010[0097:tceoha]2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Suarez RK, Doll CJ, Buie AE, West TG, Funk GD, Hochachka PW. Turtles and rats: a biochemical comparison of anoxia-tolerant and anoxia-sensitive brains. Am J Physiol Regul Integr Comp Physiol. 1989;257:R1083–R1088. doi: 10.1152/ajpregu.1989.257.5.R1083. [DOI] [PubMed] [Google Scholar]
- 32.Hochachka PW, Clark CM, Brown WD, Stanley C, Stone CK, Nickles RJ, Zhu GG, Allen PS, Holden JE. The brain at high altitude: hypometabolism as defence against chronic hypoxia? J Cereb Blood Flow Metab. 1994;14:671–679. doi: 10.1038/jcbfm.1994.84. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez del Rio M, Moskowitz MA. High altitude headache. Lessons from headaches at sea level. Adv Exp Med Biol. 1999;474:145–153. [PubMed] [Google Scholar]
- 34.Bailey DM, Roukens R, Knauth M, Kallenberg K, Christ S, Mohr A, Genius J, Storch-Hagenlocher B, Meisel F, McEneny J, Young IS, Steiner T, Hess K, Bartsch P. Free radical-mediated damage to barrier function is not associated with altered brain morphology in high-altitude headache. J Cereb Blood Flow Metab. 2006;26:99–111. doi: 10.1038/sj.jcbfm.9600169. [DOI] [PubMed] [Google Scholar]
- 35.Kallenberg K, Bailey DM, Christ S, Mohr A, Roukens R, Menold E, Steiner T, Bärtsch P, Knauth M. Magnetic resonance imaging evidence of cytotoxic cerebral edema in acute mountain sickness. J Cereb Blood Flow Metab. 2007;27:1064–1071. doi: 10.1038/sj.jcbfm.9600404. [DOI] [PubMed] [Google Scholar]
- 36.Schoonman GG, Sandor PS, Nirkko AC, Lange T, Jaermann T, Dydak U, Kremer C, Ferrari MD, Boesiger P, Baumgartner RW. Hypoxia-induced acute mountain sickness is associated with intracellular cerebral edema: a 3 T magnetic resonance imaging study. J Cereb Blood Flow Metab. 2008;28:198–206. doi: 10.1038/sj.jcbfm.9600513. [DOI] [PubMed] [Google Scholar]
- 37.Fischer R, Vollmar C, Thiere M, Born C, Leitl M, Pfluger T, Huber R. No evidence of cerebral edema in severe acute mountain sickness. Cephalalgia. 2004;24:66–71. doi: 10.1111/j.1468-2982.2004.00619.x. [DOI] [PubMed] [Google Scholar]
- 38.Sorensen SC, Lassen NA, Severinghaus JW, Coudert J, Zamora MP. Cerebral glucose metabolism and cerebral blood flow in high-altitude residents. J Appl Physiol. 1974;37:305–310. doi: 10.1152/jappl.1974.37.3.305. [DOI] [PubMed] [Google Scholar]
- 39.Jensen JB, Wright AD, Lassen NA, Harvey TC, Winterborn MH, Raichle ME, Bradwell AR. Cerebral blood flow in acute mountain sickness. J Appl Physiol. 1990;69:430–433. doi: 10.1152/jappl.1990.69.2.430. [DOI] [PubMed] [Google Scholar]
- 40.Baumgartner RW, Bartsch P, Maggiorini M, Waber U, Oelz O. Enhanced cerebral blood flow in acute mountain sickness. Aviat Space Environ Med. 1994;65:726–729. [PubMed] [Google Scholar]
- 41.Van Osta A, Moraine J-J, Melot C, Mairbaurl H, Maggiorini M, Naeije R. Effects of high altitude exposure on cerebral hemodynamics in normal subjects. Stroke. 2005;36:557–560. doi: 10.1161/01.STR.0000155735.85888.13. [DOI] [PubMed] [Google Scholar]
- 42.Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L, Gutowski M, Kewley E, McCord JM, Moller K, Ainslie PN. Altered free radical metabolism in acute mountain sickness: implications for dynamic cerebral autoregulation and blood–brain barrier function. J Physiol. 2009;587:73–85. doi: 10.1113/jphysiol.2008.159855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey DM, Davies B, Young IS. Intermittent hypoxic training: implications for lipid peroxidation induced by acute normoxic exercise in active men. Clin Sci (Lond) 2001;101:465–475. doi: 10.1042/CS20010065. [DOI] [PubMed] [Google Scholar]
- 44.Bailey DM, Ainslie P, Jackson S, Richardson R, Ghatei M. Evidence against redox regulation of energy homoeostasis in humans at high altitude. Clin Sci (Lond) 2004;107:589–600. doi: 10.1042/CS20040085. [DOI] [PubMed] [Google Scholar]
- 45.Tissot van Patot MC, Leadbetter G, Keyes LE, Bendrick-Peart J, Beckey VE, Christians U, Hackett P. Greater free plasma VEGF and lower soluble VEGF receptor-1 in acute mountain sickness. J Appl Physiol. 2005;98:1626–1629. doi: 10.1152/japplphysiol.00589.2004. [DOI] [PubMed] [Google Scholar]
- 46.Hackett PH, Roach RC. High altitude cerebral edema. High Alt Med Biol. 2004;5:136–146. doi: 10.1089/1527029041352054. [DOI] [PubMed] [Google Scholar]
- 47.Kapural M, Krizanac-Bengez L, Barnett G, Perl J, Masaryk T, Apollo D, Rasmussen P, Mayberg M, Janigro D. Serum S-100B as a possible marker of blood–brain barrier disruption. Brain Res. 2002;940:102–104. doi: 10.1016/S0006-8993(02)02586-6. [DOI] [PubMed] [Google Scholar]
- 48.Marchi N, Rasmussen P, Kapural M, Fazio V, Kight K, Mayberg M, Kanner A, Ayumar B, Albensi B, Cavaglia M, Janigro D. Peripheral markers of brain damage and blood–brain barrier dysfunction. Restor Neurol Neurosci. 2003;21:109–121. [PMC free article] [PubMed] [Google Scholar]
- 49.Kanner A, Marchi N, Fazio V, Mayberg M, Koltz M, Siomin V, Stevens G, Masaryk T, Ayumar B, Vogelbaum M, Barnett G, Janigro DS. Serum S100beta: a noninvasive marker of blood–brain barrier function and brain lesions. Cancer. 2003;97:2806–2813. doi: 10.1002/cncr.11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linden AVd, Verhoye M, Nilsson GE. Does anoxia induce cell swelling in carp brains? In vivo MRI measurements in crucian carp and common carp. J Neurophysiol. 2001;85:125–133. doi: 10.1152/jn.2001.85.1.125. [DOI] [PubMed] [Google Scholar]
- 51.McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- 52.Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- 53.Ross R. The random nature of cerebral mountain sickness. Lancet. 1985;1:990–991. doi: 10.1016/S0140-6736(85)91771-4. [DOI] [PubMed] [Google Scholar]
- 54.Rosenblum W. Cytotoxic edema: monitoring its magnitude and contribution to brain swelling. J Neuropathol Exp Neurol. 2007;66:771–778. doi: 10.1097/nen.0b013e3181461965. [DOI] [PubMed] [Google Scholar]
- 55.Catani M, Ffytche DH. The rises and falls of disconnection syndromes. Brain. 2005;128:2224–2239. doi: 10.1093/brain/awh622. [DOI] [PubMed] [Google Scholar]
- 56.Hannon JP, Chinn KS, Shields JL. Effects of acute high-altitude exposure on body fluids. Fed Proc. 1969;28:1178–1184. [PubMed] [Google Scholar]
- 57.Green H, Roy B, Grant S, Burnett M, Tupling R, Otto C, Pipe A, McKenzie D. Downregulation in muscle Na+-K+-ATPase following a 21-day expedition to 6,194 m. J Appl Physiol. 2000;88:634–640. doi: 10.1152/jappl.2000.88.2.634. [DOI] [PubMed] [Google Scholar]
- 58.Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 59.Krasney J. A neurogenic basis for acute altitude illness. Med Sci Sports Exerc. 1994;26:195–208. doi: 10.1249/00005768-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 60.Sutherland AI, Morris DS, Owen CG, Bron AJ, Roach RC. Optic nerve sheath diameter, intracranial pressure and acute mountain sickness on Mount Everest: a longitudinal cohort study. Br J Sports Med. 2008;42:183–188. doi: 10.1136/bjsm.2007.045286. [DOI] [PubMed] [Google Scholar]
- 61.Goadsby P, Lipton R, Ferrari M. Migraine-current understanding and treatment. N Engl J Med. 2002;346:257–270. doi: 10.1056/NEJMra010917. [DOI] [PubMed] [Google Scholar]
- 62.Hildebrandt W, Alexander S, Bartsch P, Droge W. Effect of N-acetyl-cysteine on the hypoxic ventilatory response and erythropoietin production: linkage between plasma thiol redox state and O2 chemosensitivity. Blood. 2002;99:1552–1555. doi: 10.1182/blood.V99.5.1552. [DOI] [PubMed] [Google Scholar]
- 63.Reinehr R, Gorg B, Becker S, Qvartskhava N, Bidmon H, Selbach O, Haas H, Schliess F, Haussinger D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia. 2007;55:758–771. doi: 10.1002/glia.20504. [DOI] [PubMed] [Google Scholar]
- 64.Master S, Gottstein J, Blei A. Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology. 1999;30:876–880. doi: 10.1002/hep.510300428. [DOI] [PubMed] [Google Scholar]
- 65.Bailey DM, Taudorf S, Berg RMG, Jensen LT, Lundby C, Evans KA, James PE, Pedersen BK, Moller K. Transcerebral exchange kinetics of nitrite and calcitonin gene-related peptide in acute mountain sickness. Evidence against trigeminovascular activation? Stroke. 2009;40:2205–2208. doi: 10.1161/STROKEAHA.108.543959. [DOI] [PubMed] [Google Scholar]
- 66.Ikeda Y, Jimbo H, Shimazu M, Satoh K. Sumatriptan scavenges superoxide, hydroxyl, and nitric oxide radicals: in vitro electron spin resonance study. Headache. 2002;42:888–892. doi: 10.1046/j.1526-4610.2002.02208.x. [DOI] [PubMed] [Google Scholar]
- 67.Bailey DM, Davies B. Acute mountain sickness: prophylactic benefits of antioxidant vitamin supplementation at high altitude. High Alt Med Biol. 2001;2:21–29. doi: 10.1089/152702901750067882. [DOI] [PubMed] [Google Scholar]
- 68.Bailey DM. Ascorbate, blood–brain barrier function and acute mountain sickness: a radical hypothesis. Wilderness Environ Med. 2004;15:231–233. doi: 10.1580/1080-6032(2004)15[234:ltte]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 69.Kallenberg K, Dehnert C, Dorfler A, Schellinger PD, Bailey DM, Knauth M, Bartsch PD. Microhemorrhages in nonfatal high-altitude cerebral edema. J Cereb Blood Flow Metab. 2008;28:1635–1642. doi: 10.1038/jcbfm.2008.55. [DOI] [PubMed] [Google Scholar]
- 70.Richardson RS, Duteil S, Wary C, Wray DW, Hoff J, Carlier PG. Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol. 2006;571:415–424. doi: 10.1113/jphysiol.2005.102327. [DOI] [PMC free article] [PubMed] [Google Scholar]