

Abstract
Von Hippel–Lindau (VHL) disease type 2A is an inherited tumor syndrome characterized by predisposition to pheochromocytoma (pheo), retinal hemangioma (RA), and central nervous system hemangioblastoma (HB). Specific VHL subtypes display genotype–phenotype correlations but, unlike other familial syndromes such as MEN-2, the phenotype in VHL has not yet been stratified at the codon level. Over decades, we have managed two very large VHL type 2A regional kindreds with nearly adjacent but distinct VHL missense mutations. We determined the phenotype of Family 2 and compared the clinical and pathologic parameters of pheo between 30 members of Family 1 (Y112H mutation) and 33 members of Family 2 (Y98H mutation) with mean follow-up of 15.5 and 12.1 years, respectively (P=0.24). In Family 2, pheo was the most frequent VHL manifestation (79%) and all pheo diagnoses occurred by age 50. Age at first diagnosis was younger in Family 2 than in Family 1 (mean 19.7 vs. 28.8 years; P=0.02). Pheo expressivity differed by genotype: Family 1 pheo was more likely to be multifocal (P=0.04), as well as malignant (P<0.01) and lethal (P=0.02). Family 1 pheo was also more likely to secrete vanillylmandelic acid (VMA) alone (P=0.05). This analysis of 130 pheochromocytomas in 63 VHL type 2A patients demonstrates that mutation-specific malignancy and expression patterns exist within the VHL type 2A subtype, and provides information that may help tailor the screening and management algorithms of affected members and those at risk.
Keywords: von Hippel–Lindau syndrome, pheochromocytoma, missense mutation, genotype–phenotype correlation
INTRODUCTION
Von Hippel–Lindau (VHL) disease is an autosomal dominant tumor syndrome characterized by predisposition to the development of pheochromocytoma (pheo), retinal hemangioma (RA), renal cell carcinoma (RCC), and central nervous system hemangioblastoma (HB) [Latif et al., 1993]. The US incidence of VHL is approximately 1 in 32,000 [VHL Family Alliance, 2010] and overall penetrance is >90% by 65 years [Maher et al., 1991]. Molecular diagnostic techniques facilitate a mutation detection rate approaching 100% [Stolle et al., 1998]. Clinical heterogeneity is a hallmark of VHL, with four phenotypes that exhibit widely differing frequencies of pheo and RCC [Neumann and Wiestler, 1991; Chen et al., 1995; Linehan et al., 1995; Zbar et al., 1996]. VHL type 1 and type 2, respectively, show a low (<5%) versus high (>60%) risk of pheo [Chen et al., 1995; Zbar et al., 1996]. Types 2A and 2B are distinguished by rare versus high rates (70%) of RCC, and type 2C carries the risk of pheo alone [Zbar et al., 1996; Woodward et al., 1997]. In VHL type 2 patients with pheo, 96% of germline mutations are of the missense type [Chen et al., 1995]. Pheo is a neoplasm of neural crest origin that secretes one or several catecholamines and it is the most life-threatening and penetrant manifestation of VHL type 2. Up to one-third of pheo patients are both asymptomatic and normotensive at diagnosis [Walther et al., 1999]. If untreated, pheo is fatal primarily due to the complications of severe hypertension [Sutton et al., 1981; Goldfien, 1991]. About 20% of pheo are malignant, and because tumor histology is not predictive of malignancy, long-term postoperative surveillance is required to determine cure.
Members of two different VHL type 2A families, each of German descent, have been followed for decades at our institution. Family 1, first described in 1962 [Tisherman et al., 1962], was found to have a T334C germline mutation (T547C in old nomenclature, codon Y112H [Mulvihill et al., 1997]) and is the largest known VHL kindred with pheo (>1,000 members when last described [Tisherman et al., 1993]). We recently performed a 50-year update of Family 1 phenotype (manuscript in preparation) which originates from east central Germany. The Family 2 phenotype has not previously been characterized, although branches have been briefly assessed as part of VHL mapping and study of founder effects and genotype–phenotype correlations [Hosoe et al., 1990; Glenn et al., 1991; Brauch et al., 1995; Chen et al., 1995; Crossey et al., 1995]. Members of Family 2 have a T292C germline VHL mutation (T505C in old nomenclature; codon Y98H) [Brauch et al., 1995; Chen et al., 1995] which originated from the Black Forest region of Germany [Brauch et al., 1995; Chen et al., 1995]. The VHL mutations in these two families occur in the same narrow region of exon 1, but are distinct. Based on initial clinical observations, we hypothesized that these differing genetic mutations correlate with differences in phenotypic expression of pheo.
MATERIALS AND METHODS
Members of Family 1 were initially studied under a prior IRB research protocol (# 951057) which was established by Dr. Tisherman and lapsed in 2002 with his death. Following identification of the VHL gene, genetic testing was available through this protocol free of charge. As Family 2 did not come to investigation until later, its members did not have a similar opportunity for research testing. Prospective screening included yearly or biyearly 24-hr urine catecholamines (epinephrine, norepinephrine, dopamine, and/or vanillylmandelic acid (VMA)) or metabolites (metanephrine and normetanephrine), ophthalmologic examination, brain MRI, abdominal CT, and spine MRI, and was recommended under this protocol as well as in routine clinical care but compliance was inconsistent in both families.
In 2009 under a new protocol (IRB# 09010261), we identified all Family 1 and Family 2 VHL patients treated at our institution and invited their participation in the present study. We retrospectively retrieved VHL-specific medical and genetic counseling records, conducted extensive interviews with family historians, and reviewed other data provided by kindred members to construct a de novo pedigree for Family 2 and an updated and expanded pedigree for Family 1. VHL diagnosis was determined by the presence of ≥1 major manifestation (pheo, RA, HB, and RCC), and was assigned using two categories of veracity: (A) robust or confirmed genetic, medical or pathologic documentation; or (B) status strongly suspected for example by confluent oral history and/or prior pedigree. Obligate carriers were identified by pedigree analysis. Only patients with category A data in support of VHL type 2A were considered informative for analysis of differences in pheo expression. Malignancy was defined as documented regional or metastatic spread or death from disease. Mean follow-up was similar between the two families: 15.5 years for Family 1 (range 1.8–41) and 12.1 years for Family 2 (range 0.1–34; P=0.24). Mean age at last follow-up did not differ (44.3 years Family 1, 41.6 years Family 2; P=0.57). Fisher's exact test was used to compare categorical data and Student's t-test was used to compare continuous data. Significance was set at 0.05 and P values were two tailed.
RESULTS
Family 1 Phenotype
Family 1 presently consists of 108 at-risk members over seven generations. Phenotype was previously described for 19 affected members [Tisherman et al., 1993]. Although an updated description of the current 49 gene carriers is detailed separately (manuscript in preparation), results pertinent to the present family comparison of pheo expression are highlighted below.
Family 2 Phenotype
Family 2 is comprised of 131 individuals over six generations (Fig. 1) including 65 members (50% of those at risk) with VHL type 2A (72% clinically affected, 12% obligate carriers, and 15% positive genetic analysis alone) and nearly equal gender distribution for affected status (54% male). An additional six members (II-3, II-4, II -5, III-1, III-2, and III-3) are suspected to have VHL without confirmation. Of note, the nuclear family (branch IV-13) contains 13 siblings, of whom 12 have been diagnosed with pheo. Family 2 clinical data were assessed as category A in 24 patients, of whom seven had confirmed genetic testing, and as category B in 23 patients of whom four reportedly had genetic testing. VHL has been asymptomatic to date in 18/65 (28%) Family 2 members.
FIG. 1.

Family 2 pedigree, showing members with confirmed and suspected VHL and pheochromocytoma; members diagnosed with RCC and those positive by DNA analysis alone are also noted.
The presenting tumor types in Family 2 were pheo (70%), HB (17%), and RA (13%). No Family 2 member had RCC as the initial manifestation. The cumulative tumor manifestations showed similar patterns; pheo was the most penetrant, occurring in 79% of patients. Table I provides the age 50 years and cumulative penetrance among informative Family 2 carriers with either category A or category B data. Because this was not a prospective study, the number of informative carriers differed by type of manifestation. Pheo was the most common manifestation in Family 2 with 66% penetrance by age 50. HB and RA penetrance were 21% and 13% by age 50, respectively. RCC was the least penetrant manifestation in Family 2 (2%), with only one case observed in a woman (IV-8) who developed RCC at age 44 following RA at age 33.
TABLE I.
Penetrance of VHL Type 2A Manifestations Among Family 2 Informative Carriers
| Penetrance by age 50 | Cumulative penetrance | |
|---|---|---|
| Pheo (n=56) | 37 (66%) | 37 (66%) |
| HB (n=53) | 11 (21%) | 12 (23%) |
| RA (n=54) | 7 (13%) | 9 (17%) |
| RCC (n=53) | 1 (2%) | 1 (2%) |
Pheo, pheochromocytoma; HB, hemangioblastoma; RA, retinal angioma; RCC, renal cell carcinoma.
Expression Patterns of Pheochromocytoma in Family 1 Compared to Family 2
Thirty Family 1 individuals and 33 from Family 2 met criteria for analysis of pheo expression, representing a total of 130 pheos (65 from each family). Pheo was diagnosed at a lower mean age in Family 2 (mean 19.7 years, range 8–47) than in Family 1 (mean 28.8 years, range 6–72; P=0.02; Table II). Kaplan-Meier analysis of age at first pheo diagnosis suggested a higher probability of pheo diagnosis at almost every age range up to 50 years in Family 2 versus Family 1 (P=0.55; Fig. 2). All Family 2 members with pheo were diagnosed by age 50, whereas five Family 1 members (17%) were diagnosed after age 50 (P=0.02). The mean age at last follow-up did not differ significantly, being 45.4 years for Family 1 members with pheo (range 14–77 years) compared to 38.0 years (range 10–69 years) in Family 2 (P=0.09).
TABLE II.
Pheo Parameters Compared Between VHL Type 2A Families
| Family 1 | Family 2 | P value | |
|---|---|---|---|
| Clinical parameters | |||
| Age first pheo diagnosis (y) (n=30; 33) | 28.8 | 19.7 | 0.02a |
| Diagnosis by symptoms (n=52; 49) | 42 (81%) | 33 (67%) | 0.17 |
| Secretion of Norepi only (n=30; 33) | 19 (63%) | 16 (49%) | 0.31 |
| Secretion of VMA only (n=30; 33) | 4 (13%) | 0 | 0.05a |
| Metachronous pheo (n=65; 65) | 21 (32%) | 13 (20%) | 0.16 |
| Time to new occurrence (y) (n=21; 12) | 7.8 | 12.8 | 0.08 |
| Pathologic parameters | |||
| Size (cm) (n=41; 33) | 3.60 | 3.20 | 0.33 |
| Weight (g) (n=29; 18) | 33.3 | 30.1 | 0.81 |
| R Laterality (n=63; 54) | 20 (32%) | 19 (35%) | 0.84 |
| L Laterality (n=63; 54) | 22 (35%) | 17 (31%) | 1.00 |
| Extra-adrenal (n=63; 54) | 9 (14%) | 15 (28%) | 0.11 |
| Abdominal (n=9; 15) | 7 (78%) | 11 (73%) | 1.00 |
| Organ of zuckerkandl (n=7; 11) | 2 (29%) | 7 (64%) | 0.33 |
| Chest (n=9; 15) | 0 | 3 (20%) | 0.27 |
| Neck (n=9; 15) | 1 (11%) | 0 | 0.37 |
| Synchronous (multifocal at presentation) (n=65; 55) | 26 (40%) | 33 (60%) | 0.04a |
Pheo, pheochromocytoma; Norepi, norepinephrine; VMA, vanillylmandelic acid; y, years.
Not all pheos were informative for each parameter, thus the specific n (Family 1; Family 2) is denoted under each category.
Statistical significance.
FIG. 2.
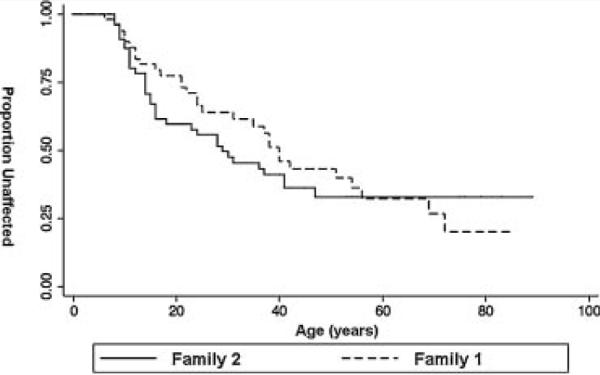
Kaplan–Meier survival curve comparing age at diagnosis of first pheochromocytoma in Family 1 and Family 2. Although Family 2 has a significantly and consistently lower probability of being unaffected at each major age range, the entire curve did not reach statistical significance (P=0.55).
Comparison of the clinical and pathologic features of pheo showed that Family 1 had a higher likelihood of synchronous multifocality (60% vs. 40%; P=0.04, Table II). In Family 1, pheo also had a lower likelihood of secreting only VMA (0 vs. 4; P=0.05). Pheo in Family 1 was also significantly more morbid across most measures including biochemical cure with initial resection (76% vs. 100% in Family 2; P=0.0002), metastasis (20% vs. 5%; P<0.01), and death from disease (5 vs. 0; P=0.02; Table III). In Family 2, extra-adrenal pheo was present somewhat more often (14% in Family 1 vs. 32% in Family 2; P=0.11). Time to metachronous pheo occurrence did not differ significantly (7.8 vs. 12.8 years; P=0.08) and there were no observed differences by genotype in pheo symptomatology (unexplained headaches, “spells”, and/or constipation), laterality, size, weight, extra-adrenal location, or norepinephrine secretion.
TABLE III.
Measures of Morbidity and Mortality of Pheo Compared Between VHL Type 2A Families
| Family 1 | Family 2 | P value | |
|---|---|---|---|
| Biochemical cure (n=50; 49) | 38 (76%) | 49 (100%) | 0.0002a |
| Recurrence (n=63; 62) | 1 (2%) | 3 (5%) | 0.36 |
| Metastasis (n=64; 62) | 13 (20%) | 3 (5%) | 0.01a |
| DOD (n=30;33) | 5 (17%) | 0 | 0.02a |
DOD, dead of disease.
Not all pheo were informative for each parameter, thus the specific n (Family 1; Family 2) is denoted under each category.
Statistical significance.
DISCUSSION
This analysis of a two large Western Pennsylvania VHL type 2A kindreds with nearly adjacent but distinct VHL exon 1 mutations identifies specific phenotypic differences that impact morbidity and mortality of pheochromocytoma, the most serious manifestation of VHL type 2A. Our findings support the idea that low rates of RA and HB distinguish VHL subtype 2A from other subtypes, while confirming that pheo is its heralding feature. Our data must be viewed against the limitations that some kindred members have not undergone prospective evaluation and many members have not complied with consistent follow-up (this issue is of particular concern for Family 2, for whom we have anecdotally observed greater barriers to care). In this regard, the RA prevalence found through routine ophthalmologic evaluation was recently reported to be 75% among 16 individuals carrying the same mutation as Family 2, suggesting that incomplete evaluation may underlie the lower rate observed herein [Allen et al., 2001]. Family 2 did not have access to research genetic testing under the prior protocol and has undergone a lower rate of confirmatory genetic testing (31% vs. 88% Family 1; P<0.0001); conceivably this may be associated with lower rates of ophthalmologic evaluation.
The results described here further refine genotype-phenotype correlation of pheochromocytoma within the subset of VHL type 2A to the level of the codon. Pheo in association with Y112H (Family 1) is significantly more morbid, lethal, synchronous/multifocal, and also presents later in life than pheo in association with Y98H (Family 2). We hypothesize that the Y98H Family 1 mutation may be less able to promote apoptosis of adrenal medulla precursor cells than the Y112H or other type 2A mutations. If genetic modifiers and environmental factors are playing a role, then the young age of pheo diagnosis may not be unique to Family 2; confirming studies in other kindreds with these mutations would be ideal. Various rates of multifocality in VHL pheo expression have been reported, from 29% [Baghai et al., 2002] to 36% [Erlic et al., 2009] to 58% [Walther et al., 1999]. Our rates of synchronous disease were high in both families (40% in Family 2 and 60% in Family 1) and we further suspect that in Family 1 the subclinical synchronous rate may be even higher since 24% continued to secrete detectable catecholamines after resection. The 20% rate of metastasis observed in Family 1 (vs. 5% in Family 2) is another important finding and is consistent with the prior 1993 report wherein 3/16 (19%) of pheos were malignant.
Because it is well established that VHL pheos tend to secrete norepinephrine [Walther et al., 1999; Eisenhofer et al., 2001], it is thought that its less potent vascular and metabolic effects (compared to epinephrine) contribute to a lower rate of hypertension and symptoms than in sporadic pheo [Hjemdahl, Belfrage, Daleskog, 1979; Walther et al., 1999; Eisenhofer et al., 2001]. We confirmed the prevalent biochemical profile and also confirmed the important fact that, although pheo in Family 1 was more likely to secrete VMA only, pheo biochemical profile does not otherwise assist with determination of malignant potential preoperatively.
As the molecular mechanisms that cause pheo have yet to be elucidated, protein modeling or gene expression profiling studies may help discern clues that underlie codon-specific phenotypes [Bryant et al., 2003]. Amino acids Y98 and Y112 are found in the S3 and S4 β sheets, respectively, in the VHL protein β domain [Stebbins et al., 1999]. The VHL protein three-dimensional structure indicates that Y98 and Y112 are separated in space by roughly 0.8 nm and are part of a cluster of surface mutations likely to be involved in substrate recognition and binding [Stebbins et al., 1999]. VHL protein mutations at Y98 or Y112 result in the loss of regulation of hypoxia-inducible factor. Interestingly, while tyrosine to histidine VHL mutations are associated with the type 2A phenotype, tyrosine to asparagine mutations are associated with VHL type 1 disease as well as sporadic RCC [Bradley et al., 1999]. It is not currently understood how mutation of the aromatic tyrosine side group to the positively charged histidine or the polar, uncharged asparagine may affect VHL protein structure or substrate recognition.
We offer several clinical recommendations based on these findings. First, the high rates of metastatic, synchronous, and extra-adrenal pheo observed here in both families have important implications for genetic screening. Current testing algorithms for a patient with apparent sporadic pheo that is synchronous, meta-static, or extra-adrenal, lead to SDHB/SDHD mutation analysis rather than VHL testing. Half of patients in both our families presented with extra-adrenal pheo, and in Family 2, 73% of extra-adrenal pheos were multifocal. Genetic screening algorithms in such patients should incorporate this important presentation of VHL. Second, it is critical that biochemical screening for VHL type 2A pheo begin in early childhood as the earliest age of pheo diagnosis was age 6 years in Family 1. Third, we suggest that patients with the Y98H mutation undergoing evaluation after a biochemical diagnosis of pheo receive Metaiodobenzylguanidine scanning since they are likely to have both extra-adrenal and synchronous disease. Fourth, our findings confirm that all VHL type 2A patients with pheo require long-term postoperative surveillance. For individuals with the Y112H mutation, it may also be prudent to perform initial open resection of what is more likely to be malignant tumor. Finally, the rarity of RCC in VHL type 2A, plus the lack of reports indicating a reduction of morbidity or mortality through surveillance, suggests that such screening may be unnecessary in VHL type 2A. Additional studies are needed to confirm and extend these findings.
ACKNOWLEDGMENTS
The Masters thesis work of Ms. Nielsen was supported by the Carrie L. Hughes Endocrine Genetics Research Fund of the University of Pittsburgh. The authors gratefully recognize the therapeutic and academic relationships of Drs. Samuel E. Tisherman, Samuel A. Tisherman, and John J. Mulvihill in the evaluation of both VHL type 2A families.
Grant sponsor: Carrie L. Hughes Endocrine Genetics Research Fund of the University of Pittsburgh.
REFERENCES
- Allen RC, Webster AR, Sui R, Brown J, Taylor CM, Stone EM. Molecular characterization and ophthalmic investigation of a large family with type 2A von Hippel–Lindau disease. Arch Ophthalmol. 2001;119:1659–1665. doi: 10.1001/archopht.119.11.1659. [DOI] [PubMed] [Google Scholar]
- Baghai M, Thompson GB, Young WF, Jr, Grant CS, Michels VV, van Heerden JA. Pheochromocytomas and paragangliomas in von Hipple-Lindau disease: a role for laparoscopic and cortical-sparing surgery. Arch Surg. 2002;137:682–688. doi: 10.1001/archsurg.137.6.682. [DOI] [PubMed] [Google Scholar]
- Bradley JF, Collins DL, Schimke RN, Parrott HN, Rothberg PG. Two distinct phenotypes caused by two different missense mutations in the same codon of the VHL gene. Am J Med Genet. 1999;87:163–167. doi: 10.1002/(sici)1096-8628(19991119)87:2<163::aid-ajmg7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Brauch H, Kishida T, Glavac D, Chen F, Pausch F, Hoöfler H, Latif F, Lerman MI, Zbar B, Neumann HP. Von Hippel–Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: Evidence of a founder effect. Hum Genet. 1995;95:551–556. doi: 10.1007/BF00223868. [DOI] [PubMed] [Google Scholar]
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: The expanding genetic differential diagnosis. J Natl Cancer Inst. 2003;95:1196–1204. doi: 10.1093/jnci/djg024. [DOI] [PubMed] [Google Scholar]
- Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, Green J, Hsia Y, Lamiell J, Li H, Wei M, Schmidt L, Tory K, Kuzmin I, Stackhouse T, Latif F, Linehan W, Lerman M, Zbar B. Germline mutations in the von Hippel–Lindau disease tumor suppressor gene: Correlations with phenotype. Hum Mutat. 1995;5:66–75. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- Crossey PA, Eng C, Ginalska-Malinowska M, Lennard TW, Wheeler DC, Ponder BA, Maher ER. Molecular genetic diagnosis of von Hippel–Lindau disease in familial pheochromocytoma. J Med Genet. 1995;32:885–886. doi: 10.1136/jmg.32.11.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlic Z, Rybicki L, Peçzkowska M, Golcher H, Kann PH, Brauckhoff M, Müssig K, Muresan M, Schäaffler A, Reisch N, Schott M, Fassnacht M, Opocher G, Klose S, Fottner C, Forrer F, Plöckinger U, Petersenn S, Zabolotny D, Kollukeh O, Yaremchuk S, Januszewicz A, Walz MK, Eng C, Neumann HP. European-American Pheochromocytoma Study Group. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15:6378–6385. doi: 10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Walther MM, Huynh T, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in von Hippel–Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86:1999–2008. doi: 10.1210/jcem.86.5.7496. [DOI] [PubMed] [Google Scholar]
- Glenn GM, Daniel LN, Choyke P, Linehan WM, Oldfield E, Gorin MB, Hosoe S, Latif F, Weiss G, Walther MM, Lerman MI, Zbar B. Von Hippel–Lindau (VHL) disease: Distinct phenotypes suggest more than one mutant allele at the VHL locus. Hum Genet. 1991;87:207–210. doi: 10.1007/BF00204184. [DOI] [PubMed] [Google Scholar]
- Goldfien A. Phaechromocytoma. Clin Endocrinol Metab. 1991;10:606. doi: 10.1016/s0300-595x(81)80014-x. [DOI] [PubMed] [Google Scholar]
- Hjemdahl P, Belfrage E, Daleskog M. Vascular and metabolic effects of circulating epinephrine and norepinephrine. Concentration-effect study in dogs. J Clin Invest. 1979;64:1221–1228. doi: 10.1172/JCI109576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoe S, Brauch H, Latif F, Glenn G, Daniel L, Bale S, Choyke P, Gorin M, Oldfield E, Berman A, Goodman J, Orcutt M, Hampsch K, Delisio J, Modi W, McBride W, Anglard P, Weiss G, Walther MM, Linehan WM, Lerman MI, Zbar B. Localization of the von Hippel–Lindau disease gene to a small region of chromosome 3. Genomics. 1990;8:634–640. doi: 10.1016/0888-7543(90)90249-t. [DOI] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, Schmidt L, Zhou F, Li H, Wei MH, Chen F, Glenn G, Choyke P, Walther MM, Weng Y, Duan DR, Dean M, Glavac D, Richards FM, Crossey PA, Ferguson-Smith MA, Palier DL, Chumakov I, Cohen D, Chinault AC, Maher ER, Linehan WM, Zbar B, Lerman MI. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- Linehan WM, Lerman MI, Zbar B. Identification of the von Hippel–Lindau (VHL) gene: Its role in renal cancer. J Am Med Assoc. 1995;273:564–570. [PubMed] [Google Scholar]
- Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, Sampson J, Williams A, Ferguson-Smith MA, Morton N. Von Hippel–Lindau disease: A genetic study. J Med Genet. 1991;28:443–447. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill JJ, Ferrell RE, Carty SE, Tisherman SE, Zbar B. Familial pheochromocytoma due to mutant von Hippel–Lindau disease gene. Arch Intern Med. 1997;157:1390–1391. [PubMed] [Google Scholar]
- Neumann HP, Wiestler OD. Clustering of features of von Hippel–Lindau disease: Evidence of a complex genetic locus. Lancet. 1991;337:1052–1054. doi: 10.1016/0140-6736(91)91705-y. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Pavletich NP. Structure of the VHL–ElonginC–ElonginB complex: Implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R, Linehan WM. Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–423. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected phaeochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56:354. [PubMed] [Google Scholar]
- Tisherman SE, Gregg FJ, Danowski TS. Familial pheochromocytoma. J Am Med Assoc. 1962;182:152–156. doi: 10.1001/jama.1962.03050410048010. [DOI] [PubMed] [Google Scholar]
- Tisherman SE, Tisherman BG, Tisherman SA, Dunmire S, Levey GS, Mulvihill JJ. Three-decade investigation of familial pheochromocytoma. Arch Intern Med. 1993;153:2550–2556. [PubMed] [Google Scholar]
- VHL Family Alliance [Accessed June 16, 2010];Basic facts about VHL. 2010 Available at: http://www.vhl.org.
- Walther MM, Reiter R, Keiser HR, Choyke PL, Venzon D, Hurley K, Gnarra JR, Reynolds JC, Glenn GM, Zbar B, Linehan WM. Clinical and genetic characterization of pheochromocytoma in von Hippel–Lindau families: Comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162:659–664. doi: 10.1097/00005392-199909010-00004. [DOI] [PubMed] [Google Scholar]
- Woodward ER, Eng C, McMahon R, Voutilainen R, Affara NA, Ponder BA, Maher ER. Genetic predisposition to phaeochromocytoma: analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet. 1997;6:1051–1056. doi: 10.1093/hmg/6.7.1051. [DOI] [PubMed] [Google Scholar]
- Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Lerman M. Germline mutations in the von Hippel–Lindau (VHL) disease gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]