

Abstract
Ameloblastin null mice fail to make an enamel layer, but the defects could be due to an absence of functional ameloblastin or to the secretion of a potentially toxic mutant ameloblastin. We hypothesized that the enamel phenotype could be rescued by the transgenic expression of normal ameloblastin in Ambn mutant mice. We established and analyzed 5 transgenic lines that expressed ameloblastin from the amelogenin (AmelX) promoter and identified transgenic lines that express virtually no transgene, slightly less than normal (Tg+), somewhat higher than normal (Tg++), and much higher than normal (Tg+++) levels of ameloblastin. All lines expressing detectable levels of ameloblastin at least partially recovered the enamel phenotype. When ameloblastin expression was only somewhat higher than normal, the enamel covering the molars and incisors was of normal thickness, had clearly defined rod and interrod enamel, and held up well in function. We conclude that ameloblastin is essential for dental enamel formation.
Keywords: amelogenin, ameloblastin, enamel, tooth
Introduction
Amelogenin is a secreted enamel matrix protein that is expressed by ameloblasts starting just before the initial mineralization of dentin and terminating early in the maturation stage (Inai et al., 1991; Bronckers et al., 1993; Wurtz et al., 1996; Wakida et al., 1999; Hu et al., 2001). Amelogenin is the most abundant protein in developing enamel (Fincham et al., 1999), indicating that it is transcribed from a strong promoter. Amelogenin is expressed predominantly by ameloblasts, although trace expression has been reported in other tissues (Gruenbaum-Cohen et al., 2009). Mutations in AMELX cause X-linked amelogenesis imperfecta (AI), an inherited condition featuring “isolated” or “non-syndromic” enamel malformations, that is, having no defects outside of the dentition (Hu et al., 2007; Bailleul-Forestier et al., 2008). Vertebrates, such as birds, that have lost the ability to develop teeth during evolution no longer encode a viable amelogenin gene (Sire et al., 2008). The strength and tissue-specificity of the amelogenin promoter make it an ideal choice to direct expression of transgenes in ameloblasts.
The mouse amelogenin (AmelX) 5′ transcriptional regulatory region has been successfully used to drive transgenic expression specifically in ameloblasts (Snead et al., 1996, 1998; Paine and Snead, 2005; Wen et al., 2008). A 3.5-kb fragment of bovine AmelX 5′ regulatory region up to and including part of exon 1 directed the expression of ß-galactosidase in ameloblasts, while a 5.5-kb segment of bovine AmelX expanded to include intron 1 and part of exon 2 directed expression of an amelogenin transgene in mouse ameloblasts (Gibson et al., 2007), which partially rescued the enamel phenotype in AmelX null mice (Li et al., 2008). Transgenic expression of rat ameloblastin by ameloblasts was successful with 2.3 kb of mouse AmelX promoter sequence (Paine et al., 2003), and human biglycan was expressed in ameloblasts after the mouse AmelX promoter was expanded to include intron 1 (Wen et al., 2008). These studies indicate that the AmelX promoter can direct the expression of transgenes by ameloblasts in a pattern that mimics the normal expression of enamel proteins during tooth development.
Ameloblastin (Ambn) is an enamel matrix protein that is co-secreted with amelogenin by ameloblasts during enamel formation (Zalzal et al., 2008). AmelX and Ambn are members of the secretory calcium-binding phosphoprotein (SCPP) family and evolved from a common ancestral gene (Delgado et al., 2001; Kawasaki and Weiss, 2003). Ameloblastin expression is enamel specific (Krebsbach et al., 1996; Lee et al., 1996). Ambn is reduced to a pseudogene in whales that evolved alternatives to teeth (Demere et al., 2008). Ambn-/- mice exhibit a non-syndromic AI phenotype (Fukumoto et al., 2004). Currently, no AMBN mutations have been identified in persons with amelogenesis imperfecta, although AMBN mutational analyses have been performed on dozens of probands from AI kindreds.
Mice normally secrete 2 ameloblastin isoforms, having 381 or 396 amino acids. The 2 isoforms differ by the alternative mRNA splicing of 15 codons at the beginning of exon 6 (Hu et al., 1997). The best evidence that ameloblastin is essential for dental enamel formation comes from the absence of normal enamel in Ambn-/- mice (Fukumoto et al., 2004). However, the Ambn knockout construct deleted exons 5 and 6 and resulted in the expression of an ameloblastin protein having 279 amino acids (missing 102 or 117 amino acids, depending upon the isoform) (Wazen et al., 2009). The expression of truncated amelogenins by ameloblasts, even in the presence of normal amelogenin expression, induced enamel malformations in transgenic mice (Paine et al., 2000). Therefore, the enamel phenotype in Ambn mutant mice might be due to the absence of functional ameloblastin and/or to a negative effect caused by secretion of the mutant ameloblastin protein (Smith et al., 2009; Wazen et al., 2009). If the phenotype is caused predominantly by the lack of functional ameloblastin, we hypothesize that the enamel phenotype in Ambn mutant mice should be rescued by transgenic expression of normal ameloblastin by ameloblasts. We tested this hypothesis with a transgene that drives expression of the mouse ameloblastin 396-amino-acid isoform from the mouse amelogenin promoter in Ambn mutant mice.
Materials & Methods
Protocol Approval
Animal protocols were reviewed and approved by the University of Michigan Institutional Animal Care and Use Committee.
Fabricating the Ambn Transgene
C57BL/6 mice were sacrificed on post-natal day 5 (PN5), and the developing first molars were removed by dissection under a stereoscopic microscope. Total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA) and chloroform, converted into cDNA with SuperScript II reverse transcriptase (Invitrogen), and used as a template to amplify Ambn transcripts by polymerase chain-reaction (PCR). We isolated genomic DNA from the tail tissues of C57BL/6 mice with the DNeasy Tissue kit (Qiagen, Valencia, CA, USA) and used it as a template to amplify 5′ and 3′ AmelX sequences. The amplification products were ligated into the pCR2.1-TOPO cloning vector (Invitrogen), transfected into bacteria, and selected for by ampicillin resistance. The orientations of the inserts were determined by restriction analyses of DNA mini-preps. Clones with their 5′ ends on the NotI side of the multiple cloning site were characterized by DNA sequencing and used to fabricate the Ambn transgene (see online Appendix Figs. 1-4 for details).
Generation of Ambn Transgenic Mice and Breeding with Ambn-/- Mice
The 7.5-kb Ambn transgene was excised from the vector by restriction digestion with NotI-SrfI purified with a Qiaquick gel extraction kit (Qiagen, Germantown, MD, USA) and micro-injected into fertilized C57BL/6 X SJL F2 oocytes and surgically transferred to recipients at the Transgenic Animal Model Core at the University of Michigan. In total, 17 independent lines were generated and mated with C57BL/6 mice. Germline transmission was determined by PCR analyses of genomic DNA obtained from tail biopsies. Offspring carrying the Ambn transgene (Tg) were mated with Ambn mutant mice (Fukumoto et al., 2004). F1 offspring positive for the transgene and heterozygous for ameloblastin (Ambn+/-) were mated, yielding 4 genotypes in the F2 generation (Appendix Fig. 1A). Tail biopsies were analyzed by PCR with primers specific for the Ambn Tg, wild-type Ambn, or the Ambn knockout gene (Appendix Figs. 1B, 1C).
Assessment of Ambn Tg Expression Levels by Western Blotting
To obtain an antibody that would specifically recognize the ameloblastin protein expressed from the wild-type gene and the ameloblastin transgene, but not the smaller mutant ameloblastin expressed from the Ambn knockout gene, we raised rabbit antibodies against the KLH-conjugated peptide MRPREHETQQYEYS, which is encoded by the exon 5–6 region deleted in the ameloblastin knockout. Specific anti-peptide antibodies were purified from the final bleed with an affinity column containing the immobilized, unconjugated ameloblastin peptide and designated Ambn-89 (YenZym, Burlingame, CA, USA).
Tg Expression Assessment by Western Blotting
Mice of four genotypes from the F2 generation (Appendix Fig. 1A) and wild-type were terminated on PN5. The first molars were surgically extracted under a dissecting microscope and the soft-tissue removed. Tails were collected for PCR genotyping. The hard tissues from a maxillary and a mandibular first molar were incubated in 1 mL of 0.5% formic acid at 4°C overnight, centrifuged briefly to remove residual insoluble material, transferred to an Ultracel YM-3 filter (3 kDa cut-off, Millipore, Billerica, MA, USA), and centrifuged, and the enamel protein on the filter was raised in sample buffer, run on SDS-PAGE, and transferred to a membrane, and ameloblastin bands were visualized by Western blotting.
Scanning Electron Microscopy (SEM)of Mandibular Incisors
The soft tissue was removed from left and right half-mandibles at 7 wks. We measured the distances from the incisor tip to the labial alveolar crest and then extracted the incisors. We used a rotating diamond disc to cut through just the dentin (at the measured distance from the incisor tip) and fractured the incisor at the notch. SEM imaging of the broken surface highlighted the decussating rods. The broken edge of the incisor was polished with a diamond disc and lightly etched with 0.1% nitric acid. The surface was sputter-coated with gold and analyzed by SEM for determination of enamel thickness.
Results
We introduced rare (8 bp) restriction sites at the edges of mouse AmelX 5′ and 3′ genomic sequences and the Ambn cDNA by PCR amplification, cloned the products, and fabricated an ameloblastin transgene construct (Appendix Figs. 2, 3). Multiple independent transgenic lines were evaluated for their levels of ameloblastin transgene expression (Fig. 1A). We identified transgenic lines that expressed virtually no transgene, slightly less than normal (Tg+), somewhat higher than normal (Tg++), and much higher than normal (Tg+++) levels of ameloblastin, and then compared their gross enamel phenotypes (Fig. 1B). Whenever detectable levels of ameloblastin protein were expressed in the Ambn mutant background, the enamel phenotype was at least partially recovered. In the case where the ameloblastin transgene expression was only somewhat higher than normal (Tg++), the enamel covering the molars and incisors appeared identical to that of the wild-type mouse. The mandibular incisors were extracted and examined under a dissecting microscope. The enamel layer of the extracted mandibular incisor from the Tg++ mouse was analogous to that of the wild-type mouse along its entire length (Fig. 2).
Figure 1.

Ameloblastin expression levels and associated enamel phenotypes. (A) SDS-PAGE stained with Coomassie brilliant blue (CBB), and Western blots immunostained with an antibody raised against recombinant mouse amelogenin (Amel-179), or against an ameloblastin peptide absent from the smaller ameloblastin expressed by the Ambn knockout (Ambn-89). The apparent molecular weights (in kDa) of protein bands in the samples are indicated on the left. Equal fractions of enamel proteins extracted from day 5 molars from Ambn wild-type (lanes ++), Ambn heterozygotes (lanes +-), Ambn heterozygotes expressing an ameloblastin transgene (lanes +-T), Ambn homozygous knockout mice (lanes—), and Ambn knockout mice expressing an ameloblastin transgene (lanes—T) were applied to each lane. The yield of enamel proteins from the Ambn-/- mice was always much lower than that from other samples because of the virtual absence of an enamel layer. Except for the wild-type (++) lanes, all samples on a single gel or blot are from littermates. The only ameloblastin band reacting against the Ambn-89 antibody migrated at 17 kDa. The ladder of immunopositive bands in Western for Line 14 is due to aggregation of the 17-kDa ameloblastin cleavage product when loaded at the high concentrations expressed. (B) Oral photographs at 7 wks of the incisors in situ and a lingual view of molars following removal of soft tissue. The molars and incisors from the Ambn-/-Tg++ mice held up to occlusal forces, did not chip or abrade, and could not be distinguished from teeth from wild-type mice.
Figure 2.
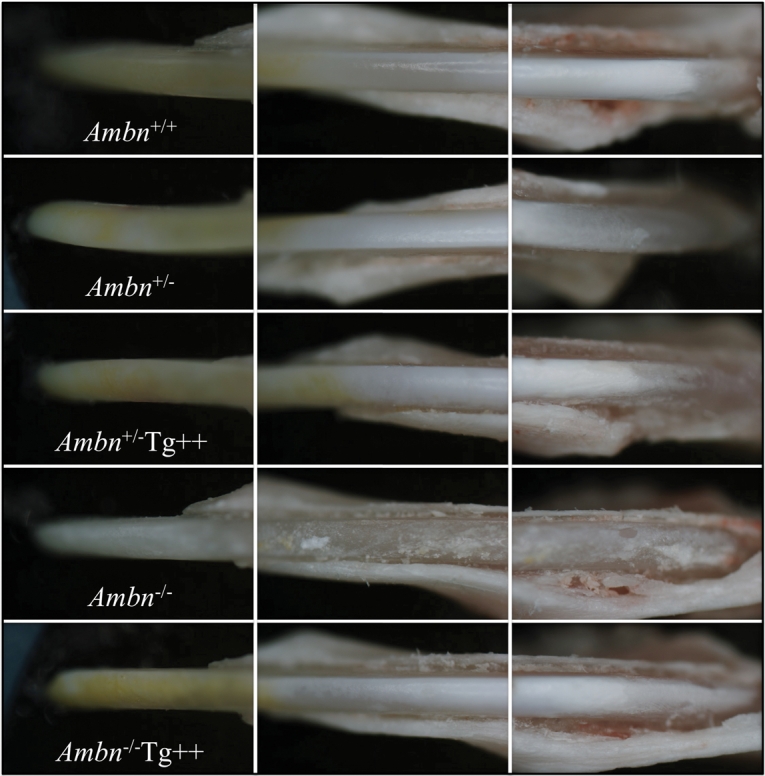
Exposed mandibular incisors. These photos were taken through a dissecting microscope and show the labial surfaces of mandibular incisors at 7 wks. The incisors from the transgenic line expressing somewhat higher than normal amounts of the ameloblastin transgene (Tg++) appeared similar to those of the wild-type mouse when expressed in either the Ambn heterozygous or Ambn null mice.
Mandibular incisors at 7 wks were cut or fractured at the level of the labial alveolar bone crest and analyzed by scanning electron microscopy. The enamel thickness and decussating pattern of enamel rods were significantly recovered, even with less than normal or much greater than normal expression of ameloblastin (Fig. 3). Recovery was most complete in the central region, where the enamel forms earliest and reaches the greatest thickness. The area most sensitive to ameloblastin levels was the distal enamel surface, where low or high levels of ameloblastin expression resulted in reduced enamel thickness and a rough surface. The enamel thickness and rod patterns looked normal in the mouse expressing somewhat higher than normal (Tg++) levels of ameloblastin, although the SEM images seemed to lack definition, which might be attributable to increased residual enamel protein.
Figure 3.

Scanning electron micrographs of mandibular incisors at 9 wks. All images are of cross-sections obtained at the level of the buccal alveolar crest. Row 1: Low-magnification view of polished cross-section showing the entire enamel layer. The Ambn+/+ and Ambn+/- mice show no enamel defects. The Ambn-/- mice have no true enamel, but a thin, rough crust of mineral covers the surface of dentin. The enamel phenotype is recovered in the central region when ameloblastin transgenes provide less than (Tg+) and more than (Tg+++) optimal amounts of ameloblastin, but the distal enamel is thinner than normal and has a rough surface. Row 2: Higher-magnification view of polished enamel showing the enamel thickness in the central region. The line spanning the enamel layer is where measurements of enamel thickness were made. The enamel layer in the Tg++ mice was the same thickness as in the wild-type. Row 3: High magnification of polished, cut, lightly etched enamel showing the underlying rod architecture. Only the Ambn-/- mice show a complete absence of enamel rods. Rod and interrod enamel is clearly distinguished in the Tg++ mice. Row 4: The enamel layer after being fractured reveals the decussating patterns of enamel rods. Although a normal pattern of decussating enamel rods is evident in the mice expressing different levels of the ameloblastin transgene, the fractured surface of the mice expressing the smaller Ambn from the knockout gene was not as well-defined as the wild-type, possibly due to an increased level of, or residual, enamel protein.
Discussion
We designed and fabricated a plasmid construct with rare restriction enzyme sites positioned at key places to direct transgenic expression of target proteins specifically in ameloblasts. This construct allows for one-step directional cloning of a desired coding sequence between the 5′ and 3′ sequences from AmelX and is a useful vector for specifically expressing transgenic proteins in ameloblasts. Transgenic mice were generated so that ameloblasts secreted the 396-amino-acid isoform of mouse ameloblastin. We characterized 5 transgenic lines and assessed ameloblastin protein levels in developing first molars at PN5. Two lines did not secrete detectable levels of ameloblastin. Three lines expressed progressively increasing amounts of ameloblastin, ranging from less than normal amounts to levels well above normal. The thickness of the enamel layer and the decussating patterns of enamel rods were assessed by SEM. The enamel phenotype varied in each of the transgenic lines. When no ameloblastin was expressed, there was no recovery of the enamel phenotype in Ambn mutant mice. There was partial recovery when ameloblastin expression was below normal (Tg+) or much greater than normal (Tg+++), with the best recovery being in the central portion of the incisor enamel. Ameloblastin transgene expression at somewhat higher than normal levels (Tg++) showed full recovery of enamel thickness and rod architecture.
These results prove that the severe enamel phenotype in Ambn mutant mice is predominantly or entirely due to the absence of functional ameloblastin and not due to toxic effects caused by secretion of the shortened 279-amino-acid ameloblastin protein, which appears to be relatively inert, but might contribute to higher levels of residual protein in the rescued enamel. The recovery of the enamel phenotype in Ambn mutant mice by transgenic expression of functional ameloblastin confirms the previous conclusion that ameloblastin is essential for proper dental enamel formation, and that Ambn mutant mice are a valid animal model for investigation of the absence of functional ameloblastin on dental enamel formation.
We were surprised to observe so much variation in ameloblastin expression in the various transgenic lines. Clearly, integration site and copy number have a profound effect on transgene expression levels in ameloblasts, and expression levels are a major determinant of how transgenic expression affects enamel formation. Interpretations of the effects on enamel formation of transgenic protein expression by ameloblasts are meaningful only in the context of transgenic protein expression levels in the matrix.
For this study, we used an anti-peptide antibody that recognizes an epitope from the exon 5-exon 6 encoded portion of ameloblastin. This antibody specifically detected a 17-kDa ameloblastin cleavage product in mouse tooth extracts. A homologous 17-kDa ameloblastin cleavage product having this epitope was previously isolated from porcine enamel and characterized (Fukae et al., 2006). It consists of the ameloblastin N-terminal region (Val1 to Arg170) and is generated by Mmp-20 cleavage (Chun et al., 2010) of ameloblastin following its secretion. Despite its relatively small size, the 17-kDa ameloblastin cleavage product elutes in the first chromatographic peak on size exclusion columns, suggesting that the protein tends to aggregate (Fukae and Tanabe, 1987). A ladder of immunopositive bands was observed on our Western blot of tooth proteins extracted from the transgenic line that overexpresses ameloblastin. These bands extend well above the apparent molecular weight of uncleaved ameloblastin (~ 65 kDa) and are likely to be aggregates of the 17-kDa ameloblastin cleavage product. Apparently, Mmp-20 is able to process ameloblastin completely, even though significantly more ameloblastin than normal is being secreted by the ameloblastin transgene. When ameloblastin expression levels are highest, a smaller immunoreactive band appears below the 17-kDa band on the Western blot. The porcine 17-kDa ameloblastin is O-glycosylated (Kobayashi et al., 2007). We suspect that this smaller band is the unglycosylated form of the 17-kDa protein and that excessive ameloblastin expression might allow some ameloblastin molecules to escape glycosylation.
The ameloblastin transgene was able to recover the thickness of the enamel layer as a whole, the decussating pattern enamel rods, and the overall appearance of normal enamel. SEMs of the fractured incisor enamel, however, seemed to be less well-defined than those of the wild-type. This suggests that the enamel in the transgenic rescue might contain more residual protein than normal. Increased protein retention may be due to the expression of the 279-amino-acid non-functional form of ameloblastin, or to overexpression of the ameloblastin transgene. Expression of the ameloblastin transgene in a total knockout might help distinguish between these explanations.
Supplementary Material
Acknowledgments
We thank Thom Saunders, PhD, and the University of Michigan Transgenic Animal Model Core for generation of the transgenic mice.
Footnotes
This research was supported by USPHS Research Grant DE015846 (NIDCR/NIH). All authors declare that there are no conflicting interests.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Bailleul-Forestier I, Molla M, Verloes A, Berdal A. (2008). The genetic basis of inherited anomalies of the teeth. Part 1: Clinical and molecular aspects of non-syndromic dental disorders. Eur J Med Genet 51:273-291 [DOI] [PubMed] [Google Scholar]
- Bronckers AL, D’Souza RN, Butler WT, Lyaruu DM, van Dijk S, Gay S, et al. (1993). Dentin sialoprotein: biosynthesis and developmental appearance in rat tooth germs in comparison with amelogenins, osteocalcin and collagen type-I. Cell Tissue Res 272:237-247 [DOI] [PubMed] [Google Scholar]
- Chun YP, Yamakoshi Y, Yamakoshi F, Fukae M, Hu JC, Bartlett JD, et al. (2010). Cleavage site specificity of Mmp-20 for secretory stage ameloblastin. J Dent Res 89:785-790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado S, Casane D, Bonnaud L, Laurin M, Sire JY, Girondot M. (2001). Molecular evidence for precambrian origin of amelogenin, the major protein of vertebrate enamel. Mol Biol Evol 18:2146-2153 [DOI] [PubMed] [Google Scholar]
- Demere TA, McGowen MR, Berta A, Gatesy J. (2008). Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Syst Biol 57:15-37 [DOI] [PubMed] [Google Scholar]
- Fincham AG, Moradian-Oldak J, Simmer JP. (1999). The structural biology of the developing dental enamel matrix. J Struct Biol 126:270-299 [DOI] [PubMed] [Google Scholar]
- Fukae M, Tanabe T. (1987). Nonamelogenin components of porcine enamel in the protein fraction free from the enamel crystals. Calcif Tissue Int 40:286-293 [DOI] [PubMed] [Google Scholar]
- Fukae M, Kanazashi M, Nagano T, Tanabe T, Oida S, Gomi K. (2006). Porcine sheath proteins show periodontal ligament regeneration activity. Eur J Oral Sci 114(Suppl 1):212-218 [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G, et al. (2004). Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol 167:973-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CW, Yuan ZA, Li Y, Daly B, Suggs C, Aragon MA, et al. (2007). Transgenic mice that express normal and mutated amelogenins. J Dent Res 86:331-335 [DOI] [PubMed] [Google Scholar]
- Gruenbaum-Cohen Y, Tucker AS, Haze A, Shilo D, Taylor AL, Shay B, et al. (2009). Amelogenin in cranio-facial development: the tooth as a model to study the role of amelogenin during embryogenesis. J Exp Zool B Mol Dev Evol 312(B):445-457 [DOI] [PubMed] [Google Scholar]
- Hu CC, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. (1997). Sheathlin: cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel sheath proteins. J Dent Res 76:648-657 [DOI] [PubMed] [Google Scholar]
- Hu JC, Sun X, Zhang C, Simmer JP. (2001). A comparison of enamelin and amelogenin expression in developing mouse molars. Eur J Oral Sci 109:125-132 [DOI] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, Simmer JP. (2007). Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 186:78-85 [DOI] [PubMed] [Google Scholar]
- Inai T, Kukita T, Ohsaki Y, Nagata K, Kukita A, Kurisu K. (1991). Immunohistochemical demonstration of amelogenin penetration toward the dental pulp in the early stages of ameloblast development in rat molar tooth germs. Anat Rec 229:259-270 [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Weiss KM. (2003). Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci USA 100:4060-4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamakoshi Y, Hu JC, Gomi K, Arai T, Fukae M, et al. (2007). Splicing determines the glycosylation state of ameloblastin. J Dent Res 86:962-967 [DOI] [PubMed] [Google Scholar]
- Krebsbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada K, Yamada Y. (1996). Full-length sequence, localization, and chromosomal mapping of ameloblastin: a novel tooth-specific gene. J Biol Chem 271:4431-4435 [DOI] [PubMed] [Google Scholar]
- Lee SK, Krebsbach PH, Matsuki Y, Nanci A, Yamada KM, Yamada Y. (1996). Ameloblastin expression in rat incisors and human tooth germs. Int J Dev Biol 40:1141-1150 [PubMed] [Google Scholar]
- Li Y, Suggs C, Wright JT, Yuan ZA, Aragon M, Fong H, et al. (2008). Partial rescue of the amelogenin null dental enamel phenotype. J Biol Chem 283:15056-15062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine ML, Snead ML. (2005). Tooth developmental biology: disruptions to enamel-matrix assembly and its impact on biomineralization. Orthod Craniofac Res 8:239-251 [DOI] [PubMed] [Google Scholar]
- Paine ML, Zhu DH, Luo W, Bringas P, Jr, Goldberg M, White SN, et al. (2000). Enamel biomineralization defects result from alterations to amelogenin self-assembly. J Struct Biol 132:191-200 [DOI] [PubMed] [Google Scholar]
- Paine ML, Wang HJ, Luo W, Krebsbach PH, Snead ML. (2003). A transgenic animal model resembling amelogenesis imperfecta related to ameloblastin overexpression. J Biol Chem 278:19447-19452 [DOI] [PubMed] [Google Scholar]
- Sire JY, Delgado SC, Girondot M. (2008). Hen’s teeth with enamel cap: from dream to impossibility. BMC Evol Biol 8:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Wazen R, Hu Y, Zalzal SF, Nanci A, Simmer JP, et al. (2009). Consequences for enamel development and mineralization resulting from loss of function of ameloblastin or enamelin. Eur J Oral Sci 117:485-497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead ML, Paine ML, Chen LS, Luo BY, Zhou DH, Lei YP, et al. (1996). The murine amelogenin promoter: developmentally regulated expression in transgenic animals. Connect Tissue Res 35:41-47 [DOI] [PubMed] [Google Scholar]
- Snead ML, Paine ML, Luo W, Zhu DH, Yoshida B, Lei YP, et al. (1998). Transgene animal model for protein expression and accumulation into forming enamel. Connect Tissue Res 38:279-286 [DOI] [PubMed] [Google Scholar]
- Wakida K, Amizuka N, Murakami C, Satoda T, Fukae M, Simmer JP, et al. (1999). Maturation ameloblasts of the porcine tooth germ do not express amelogenin. Histochem Cell Biol 111:297-303 [DOI] [PubMed] [Google Scholar]
- Wazen RM, Moffatt P, Zalzal SF, Yamada Y, Nanci A. (2009). A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol 28:292-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Zou Y, Luo W, Goldberg M, Moats R, Conti PS, et al. (2008). Biglycan overexpression on tooth enamel formation in transgenic mice. Anat Rec (Hoboken) 291:1246-1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtz T, Lundmark C, Christersson C, Bawden JW, Slaby I, Hammarström L. (1996). Expression of amelogenin mRNA sequences during development of rat molars. J Bone Miner Res 11:125-131 [DOI] [PubMed] [Google Scholar]
- Zalzal SF, Smith CE, Nanci A. (2008). Ameloblastin and amelogenin share a common secretory pathway and are co-secreted during enamel formation. Matrix Biol 27:352-359 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.