

Abstract
Objective
A recessively transmitted fatal hypertonic infantile muscular dystrophy has been described in Canadian Aboriginals. The affected infants present with progressive limb and axial muscle stiffness, develop severe respiratory insufficiency, and most die in the first year of life. We sought to determine the genetic basis of this disease.
Methods
We performed histochemical, immunocytochemical, electron microscopy and molecular genetic studies in a cohort of 12 patients affected by this disease.
Results
Conventional histochemical and electron microscopy studies suggested myofibrillar myopathy (MFM). Therefore we searched for ectopic expression of multiple proteins typical of MFM. αB-crystallin (αBC) expression was absent from all fibers using a monoclonal antibody raised against the entire protein. However, a monoclonal antibody directed against the first 10 residues of αBC immunostained portions of abnormal fibers. Pursuing this clue, we searched for mutations in the gene for αBC (CRYAB) in available DNA samples of 8 patients. All harbor a homozygous deletion, c.60C, predicting a Ser to Ala change at codon 21 and a stop codon after 23 missense residues (p.Ser21AlafsX24). Clinically unaffected parents are heterozygous for this mutation.
Interpretation
The homozygous c.60delC in CRYAB pinpoints the genetic basis of the fatal infantile hypertonic muscular dystrophy of Canadian Aboriginals. MFMs are typically transmitted by dominant inheritance but in this disease the parental phenotype is rescued by limited expression of the highly truncated nonfunctional mutant gene product. The severe patient phenotype is due to homozygosity for the markedly hypomorphic allele.
In 1994, Lacson and coworkers described 11 Canadian Aboriginal infants of Cree ancestry who developed rigid muscles and respiratory insufficiency shortly after birth; all died in infancy except one child who survived to 3 years of age. The disease appeared to be transmitted by autosomal recessive inheritance. The pathological features included severe variation in myofiber size, endomysial fibrosis, homogeneous eosinophilic inclusions and, at the ultrastructural level, focal Z-disk abnormalities in continuity with coalescing electron dense granular material. Although presumed to be an abnormality of myofilaments in the vicinity of Z-disks, the mutant protein remained unidentified.1 Examination of recent cases from central Canada revealed that the pathological features were essentially identical with those found in the myofibrillar myopathies (MFMs).2, 3 However, most MFMs present after the first decade or later in life, the weakness frequently involves distal limb muscles, and in nearly all patients the inheritance is autosomal dominant.4 MFM mutations identified to date reside in Z-disk related proteins, namely desmin,5 alpha B-crystallin (αBC),6, 7 myotilin,8, 9 ZASP10 and filamin C11. Mutations in Bag3, another Z-disk-associated protein, present in childhood and cause progressive limb and axial muscle weakness followed by cardiomyopathy and respiratory failure in the second decade of life.12 Searching for evidence to corroborate the MFM features in the disease identified by Lacson et al.,1 we immunolocalized proteins characteristically expressed in an ectopic manner in MFM patient muscles. Although several proteins were ectopically expressed in patient muscles, αBC expression was totally absent with a monoclonal antibody raised against the entire protein as the antigen. This, in turn, provided the clue that a mutation in the corresponding gene CRYAB caused the disease.
Material and Methods
Materials studied
Paraffin sections were available for review from patients 1 to 6 described in the original publication by Lacson et al1 and from 6 more recently observed patients. DNA was available for analysis from 8 affected patients (2 isolated from blood, 4 from frozen muscle, and 2 from paraffin embedded tissue), unaffected parents of 2 patients, and an unaffected sibling of 1 patient. Table 1 shows the clinical data for patients whose DNA was analyzed.
Table 1.
Clinical data on cases with DNA analysis
| Patient | Presentation | Affected family members* |
CK (a) | Clinical Course | Death |
|---|---|---|---|---|---|
| 1 (Lacson case#1) |
Female; respiratory distress at 7 weeks |
Five siblings died <2 months age |
20-fold | Respiratory insufficiency, progressive muscle rigidity |
7 months |
| 2 (Lacson case #3) |
Male; Weak cry at 6 weeks followed by sudden death |
5 month sibling | ND | Sudden death | 6 weeks |
| 3 (Lacson case #6) |
Female; respiratory distress at 8 weeks |
None | 20-fold | Respiratory insufficiency |
3 months |
| 4 | Female; neonatal apneic spells, respiratory distress at 2 months |
2 month old brother; possible distant cousin with same surname |
15-20 fold |
Axial hypertonia, rigidity of abdominal wall, respiratory insufficiency |
3 months |
| 5 | Male; respiratory distress at 11 weeks |
One brother died at 3 months |
Truncal rigidity; alive at 4 years on mechanical ventilation |
- | |
| 6 | Female; weakness and sleep apnea since birth |
9 cousins died in early infancy; mother has mild myopathic symptoms but normal CK |
25-fold | Diffuse hypertonia and weakness; mechanical ventilation |
7.5 months |
| 7 | Female; respiratory distress |
Cousins | ND | Rigidity of abdominal wall, respiratory insufficiency |
3 months |
| 8 | Male; firm abdomen and viral pneumonia at 2 months |
Cousins | ND | Sudden death | 2 months |
Except for mother of patient 6, none of the parents reported weakness or had weakness on examination.
CK, creatine kinase, (a) reported as relative increase above the upper normal limit ND, not done
All studies were done in accordance with the ethics guidelines of the Mayo Clinic, the University of Manitoba, the University of Calgary, and the University of Western Ontario. Direct parental consent for analysis was obtained for all of the current cases. The consent documented in the cases studied retrospectively was appropriate at the time of tissue acquisition.
Histopathology and immunohistochemistry
Formalin-fixed paraffin-embedded sections of muscle were stained with hematoxylin and eosin and Masson trichrome. Routine histochemical studies were performed on frozen sections.13 Immunostains included spectrin (mouse monoclonal clone RBC2/3D5; Novocastra, Newcastle-upon-Tyne UK), myosin (mouse monoclonal clone MY-32; BioGenex, San Ramon CA), myoglobin (rabbit polyclonal; Dako, Glostrup Denmark), desmin (mouse monoclonal clone D33; Dako), myotilin (mouse monoclonal clone RS034; Novocastra), ubiquitin (rabbit polyclonal Z0458; Dako), and αBC (mouse monoclonal clone G2JF raised against amino acids 1-10; Novocastra; and mouse monoclonal clone 1B6.1-3G4 raised against the entire protein; Stressgen, Ann Arbor MI). Glutaraldehyde-fixed muscle samples were processed for plastic embedding and electron microscopy by standard methods.
DNA mutation analysis
Mutation analysis was done in 8 patients using DNA. Polymerase-chain-reaction (PCR) primers were designed to analyze all exons and their flanking noncoding regions of CRYAB.11 PCR-amplified fragments were sequenced using fluorescently labeled dideoxy terminators. CRYAB nucleotides were numbered according to the mRNA sequence (GenBank reference no: NM001885.1).
Results
Clinical features
All 8 affected children were of Canadian Aboriginal descent from First Nations in central and western Canada ranging from northern Ontario to Alberta. Seven patients had a family history of similarly affected individuals, but not all families were found to be related. Clinical features of the 8 patients whose DNA was analyzed, including 3 previously reported patients,1 are shown in Table 1. All but one affected child presented with truncal hypertonia evident soon after birth, and all had progressive respiratory distress. None had dysmorphic features. All patients had normal age appropriate cognitive function, and patient 4 had a normal brain MRI. Among those for which serum creatine kinase (CK) was measured, all had levels elevated 15-20 fold above normal. Treatment with muscle relaxants or neuromuscular blockage, failed to abolish hypertonia, even when the patients were no longer capable of spontaneous movements. Thus, the hypertonia resides in muscle. All but one patient died between 6 weeks to 7 months of age; one remains alive at age 4 years on mechanical ventilation.
Muscle Pathology
All available patient muscle samples showed similar abnormalities, with truncal muscles more severely affected than limb muscles. The muscle fiber diameter varied pathologically from 10 to >100μm. Some fibers were necrotic and a comparable number of fibers were regenerating. Scattered fibers displayed multiple vacuoles. Numerous fibers harbored granular or larger hyaline deposits (Figure 1A), or amorphous material that stained blue or blue-red in trichrome sections (Figure 1G). The hyaline masses were devoid of oxidative enzyme activity. The larger inclusions in numerous abnormal fibers displayed mild to moderate congophilia in Congo red stained sections viewed under rhodamine optics. Increases of acid phosphatase occurred in relation to many cytoplasmic inclusions (Figure 1B). There was a moderate to marked increase in endomysial fibrous and perimysial fibrous and fatty connective tissue (Figure 1G). Notably, in one patient a quadriceps muscle sample showed only minimal fiber size variation and hypercontracted fibers, but a rectus abdominis specimen obtained 4 weeks later displayed typical MFM pathology. Scattered lymphocytes were present in three autopsy and one surgical biopsy specimen, and large collections of lymphocytes were identified in an autopsy specimen.
Figure 1.

Histopathological features in rectus abdominis muscle specimen obtained from a boy at 14 weeks age. A. Hematoxylin and eosin stained paraffin section shows considerable fiber irregularity and dense eosinophilic deposits. Fiber in center harbors large vesicular nuclei suggesting attempt at regeneration (arrow). B. Acid phosphatase reacted frozen muscle specimen showing reactivity (red) in abnormal fibers and in endomysial inflammatory cells. C. Immunostain for αBC on paraffin section using the antibody that recognizes amino acids 1-10. Labeling is detected in some regions (arrows). D. Immunostain for desmin on immediately adjacent paraffin section shows patchy labeling and reduced expression in αBC immunoreactive regions (arrows). E. Immunostain for αBC on frozen section using the antibody that recognizes the entire protein is comparable to background staining. F. Immunostain for spectrin on frozen section. Note fiber size variation and focal internal labeling (arrow). G. Modified Gomori trichrome stained frozen section shows fiber size variation, endomysial fibrosis, and dark staining inclusions (arrow). H. Electron micrograph showing fiber with sinuous inclusion material (arrow) arising from Z disk surrounded by a region of myofibrillar disorganization. I. Higher magnification electron micrograph shows dense granular deposits amidst the sarcomeres. Bar = 16 μm for A, C, D, G. Bar = 25 μm for B, E, F. Bar = 4 μm for H. Bar = 0.5 μm for I.
The abnormal fibers displayed strong immunoreactivity for desmin (Figure 1D), myotilin, and NCAM. Desmin and myotilin immunoreactivities were concentrated at the periphery of the fibers, surrounding abnormal inclusions that reacted for myosin and myoglobin. Spectrin and dystrophin were normally expressed at the sarcolemma and at multiple loci within the fibers, sometimes surrounding small vacuoles (Figure 1F). Small granular deposits in rimmed vacuoles reacted for ubiquitin but the large inclusions did not. αBC immunoreactivity was entirely absent using the monoclonal antibody that was raised against the entire protein (Figure 1E) but was moderately strongly expressed in the abnormal inclusions and faintly in other fiber regions with the monoclonal antibody raised against the first 10 residues of αBC (Figure 1C) both in frozen and paraffin sections. Resin sections of muscle clearly displayed the marked endomysial fibrosis and pleomorphic intrafiber inclusions. On electron microscopy, even the least affected fibers exhibited sarcomeric disarray with focal Z-disk streaming. In many fibers the Z-disks were replaced by, or merged with, coarse granular deposits (Figure 1I). In some areas the granular deposits formed large sinuous inclusions (Figure 1H). Some fibers also contained vacuoles harboring cytoplasmic degradation products.
The heart was examined from autopsies of 5 patients. It was grossly normal in all and microscopically normal in 4. In one patient, rare fibers in left ventricle showed segmental eosinophilia. None of the patients had developmental brain abnormalities in autopsy. Two showed scattered neuronal changes suggestive of mild hypoxic damage.
Mutation Analysis
All 8 tested patients were homozygous for the c.60C deletion mutation that predicts a Ser to Ala change at codon 21 and a stop codon after 23 missense residues (p.Ser21AlafsX24) (Fig. 2). DNA samples available for 2 sets of unaffected parents were heterozygous for the mutation. An unaffected sibling did not carry the mutation.
Figure 2.
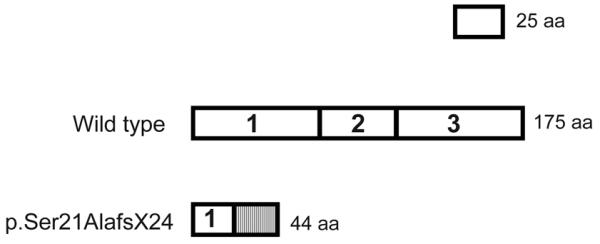
p.Ser21AlafsX24 in exon 1 is followed by 23 missense amino acids and a stop codon. Shaded box represents the missense residues.
Discussion
The fatal autosomal recessive infantile hypertonic muscular dystrophy observed in Canadian Aboriginals shows the pathologic features of MFM. The clue to a defect in αBC came from the absence of αBC immunoreactivity using a monoclonal antibody raised against the entire αBC protein. This finding differed sharply from those in previously reported MFM patients with autosomal dominant CRYAB mutations whose muscles showed markedly increased and ectopic expression of αBC.6, 7, 14 The most likely explanation was a biallelic null mutation that behaves in a genetic recessive manner. However, a monoclonal antibody raised against the first 10 residues of αBC did reveal some immunoreactivity in some muscle fibers. These findings are readily reconciled by the position of the identified mutation that may allow expression of the first 20 αBC residues, though expression is likely attenuated by nonsense mediated mRNA decay. Presence of the same mutation in affected children in different regions of Canada points to a founder mutation.
Interestingly, CRYAB knockout is not embryonic or neonatal lethal in mice. After 40 weeks of age, the mice lose weight and body fat, and develop kyphosis and a myopathy involving the posterior tongue and axial muscles with lesser involvement of the extremity muscles. Our patients show a similar selective involvement of neck and truncal muscles, but are more severely affected than the knockout mice.15 Tongue, examined at autopsy in one patient, showed advanced pathologic changes. The predilection for truncal muscles may be related to the greater expression of αBC in type 1 fibers which are enriched in large postural muscles.16 As in our patients, the heart of the knockout mice is unaffected.15
αBC is a small heat shock protein that inhibits aggregation and precipitation of denatured proteins.17, 18 The myofibrils, and especially the Z-disks, are constantly stressed by physical activity but are protected from unfolding and denaturation by molecular chaperones.19 The close association of αBC with the Z-disk implies that αBC is particularly important in protecting the Z-disk from disintegration that results in myofibrillar breakdown. Specifically, αBC appears to have a high affinity for desmin20 and titin21 under situations of cellular stress. We postulate that the interaction of αBC with titin is relevant in this novel myopathy as titin is largely responsible for muscle elasticity. Thus the lack of αBC could derange titin in such a way to contribute to reduced muscle elasticity. There is some indirect evidence for this pathophysiology in the αBC/HSPB2 deficient mouse model wherein cardiac ischemia induces more rapid and sustained resting muscle tension compared to controls.22 Hypertonia is not typically seen in myopathic conditions and will need to be adequately addressed in any proposed model of this muscle disease. In addition, αBC protects myosin from thermal denaturation.23 An additional pathogenetic factor in this disease is the marked and early endomysial fibrosis that impairs chest wall compliance and contributes to early respiratory failure. Similar early and marked endomysial fibrosis has not been documented in other types of MFM, suggesting the αBC may have an additional role in regulating connective tissue proliferation in muscle as has been suggested in skin wound healing.24
Although some pathogenetic aspects of the fatal αB-crystallinopathy of the Canadian Aboriginals remain to be elucidated, deciphering the gene defect has important implications for diagnosis, genetic counseling, carrier detection, preconceptional and prenatal testing, and eventual prevention of the disease.
Acknowledgements
This work was supported by a K08 NS050106 grant from NIH to Dr. Selcen. Dr. Del Bigio holds the Canada Research Chair in Developmental Neuropathology.
References
- 1.Lacson AG, Seshia SS, Sarnat HB, et al. Autosomal recessive, fatal infantile hypertonic muscular dystrophy among Canadian Natives. Can J Neurol Sci. 1994;21:203–212. doi: 10.1017/s0317167100041172. [DOI] [PubMed] [Google Scholar]
- 2.De Bleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol. 1996;55:563–577. doi: 10.1097/00005072-199605000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Nakano S, Engel AG, Waclawik AJ, et al. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol. 1996;55:549–562. doi: 10.1097/00005072-199605000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain. 2004;127:439–451. doi: 10.1093/brain/awh052. [DOI] [PubMed] [Google Scholar]
- 5.Goldfarb LG, Park KY, Cervenakova L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- 6.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 7.Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54:804–810. doi: 10.1002/ana.10767. [DOI] [PubMed] [Google Scholar]
- 8.Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004;62:1363–1371. doi: 10.1212/01.wnl.0000123576.74801.75. [DOI] [PubMed] [Google Scholar]
- 9.Olive M, Goldfarb LG, Shatunov A, et al. Myotilinopathy: refining the clinical and myopathological phenotype. Brain. 2005;128:2315–2326. doi: 10.1093/brain/awh576. [DOI] [PubMed] [Google Scholar]
- 10.Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005;57:269–276. doi: 10.1002/ana.20376. [DOI] [PubMed] [Google Scholar]
- 11.Vorgerd M, van der Ven PF, Bruchertseifer V, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304. doi: 10.1086/431959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel AG. The muscle biopsy. In: Engel AG, Franzini-Armstrong C, editors. Myology. 3rd ed. Vol. 1. McGraw-Hill; New York: 2004. pp. 681–690. [Google Scholar]
- 14.Reilich P, Schoser B, Schramm N, et al. The p.G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul Disord. 2010;20:255–259. doi: 10.1016/j.nmd.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Brady JP, Garland DL, Green DE, et al. AlphaB-crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- 16.Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem Cell Biol. 2004;122:415–425. doi: 10.1007/s00418-004-0711-z. [DOI] [PubMed] [Google Scholar]
- 17.Markossian KA, Yudin IK, Kurganov BI. Mechanism of suppression of protein aggregation by alpha-crystallin. Int J Mol Sci. 2009;10:1314–1345. doi: 10.3390/ijms10031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Augusteyn RC. alpha-crystallin: a review of its structure and function. Clin Exp Optom. 2004;87:356–366. doi: 10.1111/j.1444-0938.2004.tb03095.x. [DOI] [PubMed] [Google Scholar]
- 19.Phillips SM, Glover EI, Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol. 2009;107:645–654. doi: 10.1152/japplphysiol.00452.2009. [DOI] [PubMed] [Google Scholar]
- 20.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 21.Bullard B, Ferguson C, Minajeva A, et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 22.Golenhofen N, Redel A, Wawrousek EF, Drenckhahn D. Ischemia-induced increase of stiffness of alphaB-crystallin/HSPB2-deficient myocardium. Pflugers Arch. 2006;451:518–525. doi: 10.1007/s00424-005-1488-1. [DOI] [PubMed] [Google Scholar]
- 23.Melkani GC, Cammarato A, Bernstein SI. alphaB-crystallin maintains skeletal muscle myosin enzymatic activity and prevents its aggregation under heat-shock stress. J Mol Biol. 2006;358:635–645. doi: 10.1016/j.jmb.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed MR, Gopinath D, Gomathi K, et al. Alpha-crystallin-incorporated collagen matrices as an aid for dermal wound healing. J Biomed Mater Res B Appl Biomater. 2004;69:241–248. doi: 10.1002/jbm.b.30003. [DOI] [PubMed] [Google Scholar]