

Abstract
Asthma is a chronic inflammatory disease of the airways and there are no preventions or cures. Inflammatory cells through the secretion of cytokines and pro-inflammatory molecules are thought to play a critical role in pathogenesis. Type 2 CD4+ lymphocytes (Th2 cells) and their cytokines predominate in mild to moderate allergic asthma, whereas severe steroid-resistant asthma has more of a mixed Th2/Th1 phenotype with a Th17 component. Other immune cells, particularly neutrophils, macrophages and dendritic cells, as well structural cells such as epithelial and airway smooth muscle cells also produce disease-associated cytokines in asthma. Increased levels of these immune cells and cytokines have been identified in clinical samples and their potential role in disease demonstrated in studies using mouse models of asthma. Clinical trials with inhibitors of cytokines such as interleukin (IL)-4, -5 and tumour necrosis factor-α have had success in some studies but not others. This may reflect the design of the clinical trials, including treatments regimes and the patient population included in these studies. IL-13, -9 and granulocyte-macrophage colony-stimulating factor are currently being evaluated in clinical trials or preclinically and the outcome of these studies is eagerly awaited. Roles for IL-25, -33, thymic stromal lymphopoietin, interferon-γ, IL-17 and -27 in the regulation of asthma are just emerging, identifying new ways to treat inflammation. Careful interpretation of results from mouse studies will inform the development and application of therapeutic approaches for asthma. The most effective approaches may be combination therapies that suppress multiple cytokines and a range of redundant and disconnected pathways that separately contribute to asthma pathogenesis. Astute application of these approaches may eventually lead to the development of effective asthma therapeutics. Here we review the current state of knowledge in the field.
LINKED ARTICLES
This article is part of a themed issue on Respiratory Pharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-1
Keywords: asthma, therapy, treatment, cytokine, animal models, human clinical trials
Introduction
Asthma is a prevalent and serious chronic inflammatory airway disease that affects 300 million people word wide. The mechanisms of pathogenesis are incompletely understood and there are no preventions or cures. In the mid-1980s a paradigm emerged that type 2 CD4+ lymphocytes (Th2 cells) were important in the pathogenesis of allergic asthma (Kay et al., 1991; Robinson et al., 1992) based on the observations that specific Th2 subtypes existed (Mosmann et al., 1986; Parish and Luckhurst, 1982). This paradigm has dominated asthma research for the past 30 years. However, asthma is now increasingly being recognized as a heterogeneous disorder and roles for Th1, Th2, and more recently Th17 and regulatory T cells have been identified (Figure 1) (Thorburn and Hansbro, 2010). Innate immune cells such as neutrophils, macrophages, natural killer (NK) and natural killer T (NKT) cells and dendritic cells and structural cells including epithelial and airway smooth muscle (ASM) cells release T-cell polarizing and other cytokines. Many cytokines released by T cells, innate and structural cells contribute to the different pathogenetic features of asthma. Therefore, specific anti-cytokine therapies may be effective in some subsets of asthmatics but not others. Thus, there is a need to better segregate asthma patients into subgroups that differ in the likely cause of the lung disorder. In addition, it might be necessary to block the expression and/or bioactivity of more than one cytokine to obtain significant therapeutic benefits in an asthma patient. Here we review the identification of the proposed roles of specific cytokines in asthma pathogenesis and their potential as therapeutic targets. Although we recognize their importance, due to space restraints we have not included detailed discussions of studies that have used cultured human cells to elucidate the mechanistic contribution of cytokines to asthma pathogenesis or of endogenous factors that control the half lives and catabolism of cytokines that are likely to be important in asthma. Furthermore, due to space limitations we quote a number of detailed reviews to direct the reader to the primary literature and our focus is a broad contemporary overview of the field.
Figure 1.
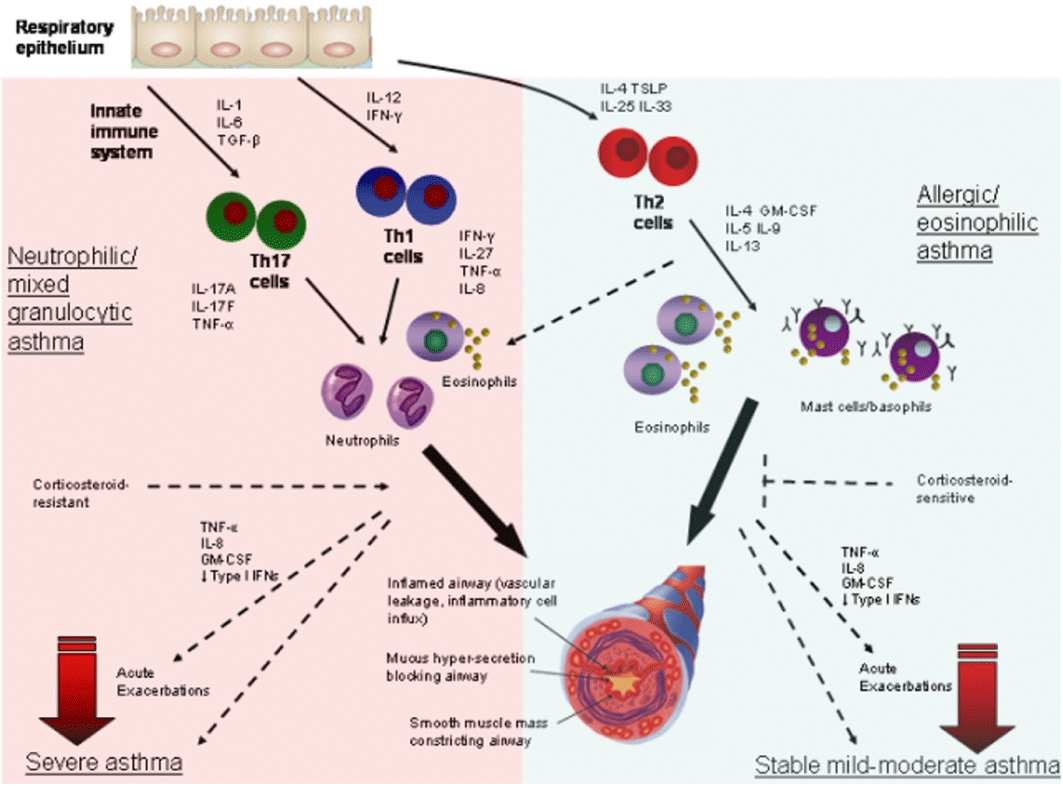
Subtypes of asthma. The respiratory epithelium can interact bi-directionally with cells from the innate immune system to release a variety of cytokines that are responsible for initiating the differentiation of the various T helper (Th) cell subsets. These innate cytokines include interleukin (IL)-1, IL-6 and TGF-β, which induce Th17 cells, IL-12 and interferon (IFN)-γ that promote Th1 cell development, and IL-4, IL-25, IL-33 and thymic stromal lymphopoietin (TSLP), which drive Th2 cell differentiation. These Th cell subsets then secrete a variety of subset specific mediators that activate unique cellular responses. These cells either directly produce and/or induce the production of IL-17A and F, tumour necrosis factor (TNF)-α (Th17), IFN-γ, IL-27 and TNF-α (Th1), or IL-4, -5, -9, -13 and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Th2). Th1 and Th17 cell responses in the lung contribute to predominantly neutrophil or mixed (neutrophils and lower proportions of eosinophils) granulocytic inflammation and neutrophilic or mixed granulocytic asthma. By contrast, Th2 cell responses induce predominately eosinophil, basophil and mast cell infiltration of the airways and promote IgE-mediated responses and allergic or eosinophilic asthma. These subtypes of asthma have the hallmark clinical features of disease including inflamed airways, mucus hypersecretion and bronchoconstriction, even though they are induced through different mechanisms. Neutrophilic asthma is largely steroid-resistant, hence this subtype often leads to severe asthma and involves TNF-α, IFN-γ, IL-17 and IL-27. Eosinophilic asthma is steroid-sensitive and can be effectively controlled by corticosteroid treatment and most patients experience stable mild-moderate disease. Patients with both subtypes of asthma can experience acute exacerbations induced by a variety of triggers, particularly infection that are associated with TNF-α, IL-8, GM-CSF and reduced type I IFNs.
Asthma pathogenesis
Mild to moderate allergic asthma is generally characterized by acute on chronic airway inflammation consisting of activated Th2 lymphocytes and eosinophil infiltrates in association with IgE production, mucus secreting cells (MSC) hyperplasia and metaplasia, remodelling of the airway wall and airway hyperresponsiveness (AHR) (Figure 1) (Bochner et al., 1994; Wills-Karp, 2004). Airway remodelling involves a thickening of the airway epithelium, MSC hyperplasia/metaplasia, subepithelial fibrosis, collagen deposition and smooth muscle hypertrophy (Temelkovski et al., 1998). The AHR is characterized by enhanced responsiveness and constriction of the airways to non-specific spasmogenic stimuli, such as methacholine. These hallmark pathological features of asthma are thought to underpin the clinical symptoms of disease such as airway obstruction, coughing, dyspnoea and wheezing. In particular, Th2 cells through the secretion of their cytokines [interleukin (IL)-3, -4, -5, -9, -13, granulocyte-macrophage colony-stimulating factor (GM-CSF), thymic stromal lymphopoietin (TSLP)] are thought contribute to various pathological features of disease (Parronchi et al., 1991; Durham et al., 1992; Hansbro et al., 2008; Kaiko et al., 2008). For example, IL-5, -9 and -13 regulate eosinophilia, mastocytosis and mucus hypersecretion respectively (Townsend et al., 2000; Kibe et al., 2003; Yang et al., 2003). Many studies have identified a central role for IL-13 in the pathogenesis of allergic inflammation and asthma. The cause of the increased numbers of Th2 cells in the airways remains unknown but may be related to the dysregulation of the activities of Th1 cells [interferon (IFN)-γ production] or regulatory T cells (IL-10 production) that can provide a counterbalance (Thorburn and Hansbro, 2010). In recent years, the innate immune cytokines IL-25, -33 and TSLP have gained prominence for their potent Th2-inducing capacity (Saenz et al., 2008). The activity of cytokines leads to the influx and activation of Th2 cells, mast cells and eosinophils that release their own mediators and promote chronic allergic airway inflammation (Flood-Page et al., 2003a).
Severe asthma has different pathological features to mild to moderate allergic asthma and is characterized by a mixed Th2/Th1 phenotype with a possible contribution from Th17 cells (Figure 1) (Cho et al., 2005; Alcorn et al., 2010). Tumour necrosis factor (TNF)-α, IFN-γ, IL-17 and -27 are elevated and may induce the influx of neutrophils (rather than eosinophils) or a mixed granulocytic airway infiltrate that is characteristic of this subtype of asthma. Notably, patients with this asthma subtype are refractory to glucocorticoid treatment and bacterial and viral infections are implicated in the induction and progression of disease (Hansbro et al., 2004; 2008; Wang et al., 2010; Horvat et al., 2010a).
Asthma exacerbations. Airway obstruction, coughing, dyspnoea and wheezing contribute to exacerbations that are precipitated by allergens and infections (Hansbro et al., 2004; 2008;). Exacerbations are mediated by eosinophils or in more common and severe cases by neutrophils, are driven by different cytokines and are steroid resistant (Figure 1). TNF-α, IL-8 and GM-CSF are implicated in more severe exacerbations that are steroid resistant and TNF-α in particular is likely to play a pivotal role (Berry et al., 2006; Ito et al., 2008). Infection-induced exacerbations may be related to reduced levels of type I IFNs (IFN-α's and -β) (Wark et al., 2005).
Steroids and combination therapies with long-acting β-agonists are the mainstay of asthma treatment and effectively suppress cytokine expression and acute inflammatory symptoms (Eklund et al., 1997). However, they do not prevent, reverse or treat the underlying causes of disease. These treatments require constant monitoring and are associated with side-effects and resistance. Therefore, there is an urgent need for new and more effective treatments and cytokines have been extensively investigated as potential therapeutic targets.
Anti-cytokine therapies
Established clinical targets
The following investigations and human trials employing inhibitors of cytokines and pathways have been performed:
Anti-IL-4/IL-4-Rα
IL-4 is produced by Th2 cells, activated mast cells and eosinophils, is required for Th2 cell differentiation and expansion, and suppresses Th1 cell development (Figure 1) (Kaiko et al., 2008). It promotes isotype switching of B cells to IgE production (Finkelman et al., 1988), the growth and development of mast cells (Madden et al., 1991) and eosinophil recruitment (Schleimer et al., 1992). IL-4 contributes to maintaining the inflammatory response to antigens, the production of eotaxins and the development of MSC and AHR (Temann et al., 1997; Hogan et al., 1997b). Inflammation is enhanced by IL-4-induced increases in vascular cell adhesion molecule (VCAM)-1 expression that promotes the migration of T cells and inflammatory cells into the lung. IL-4 also induces collagen and fibronectin synthesis and may contribute to airway remodelling (Büttner et al., 1997). Both IL-4 and -13 induce their effects by signalling through the IL-4 receptor α/IL-13Rα1 (Hart et al., 1999). An alternatively spliced transcript of IL-4 lacking exon 2 has been identified and may be a natural inhibitor of IL-4 and may have a potential as an asthma therapy (Sorg et al., 1993). Both anti-IL-4 and anti-IL-4Rα have been investigated for their capacity to suppress the induction and reverse asthma.
Mouse studies. The administration of IL-4 to mice did not induce cellular influx into the airways or AHR (Corry et al., 1996; Gavett et al., 1997). IL-4-trangenic (Tg) mice have increased serum IgE and mucus production (Tepper et al., 1990; Temann et al., 1997).
IL-4- and IL-4R-deficient (−/−) mice have been assessed in animal models of allergic airway disease (AAD) designed to recapitulate many of the hallmark features of asthma. The induction of acute AAD in IL-4−/− mice was associated with reduced eosinophil recruitment into the airways, MSC hyperplasia and IgE and IgG1 responses. However, the degree of neutrophil and lymphocyte recruitment, blood eosinophilia and airway damage were not affected (Brusselle et al., 1994; Hogan et al., 1997b; Grünig et al., 1998). AHR was suppressed in some studies but not others and Brusselle et al., showed that IL-4 may suppress AHR in the absence of inflammation (Brusselle et al., 1994). The attenuation of hallmark features of AAD was greater in IL-4R−/− mice, which results from inhibiting the activity of both IL-4 and -13.
In chronic models of asthma IL-4−/− mice had reduced airway inflammation but increased epithelial hypertrophy, subepithelial fibrosis and AHR (Foster et al., 2000) and IL-4R−/− mice had reduced epithelial hypertrophy and MSC hyperplasia but airway inflammation, fibrosis and AHR were unaffected (Kumar et al., 2002). These studies indicate that IL-4 does not play a significant role in chronic airway inflammation.
Nevertheless, models of AAD in mice have demonstrated beneficial effects of suppressing IL-4 activity. Administration of anti-IL-4 during allergen sensitization suppressed eosinophil infiltration into the airways and inhibited IL-5 release from T cells, IgE production and AHR (Coyle et al., 1995; Corry et al., 1996). However, administration before or during allergen challenge had no effect. In another study inhibition of IL-4 reduced but did not inhibit IL-13 production from lymph nodes, which may explain why AHR persisted (Webb et al., 2000). By contrast, antibody blockade of the IL-4R before antigen challenge significantly reduced eosinophil airway influx, allergen-specific IgE production, VCAM-1 expression, numbers of MSC and AHR (Renz et al., 1996; Gavett et al., 1997; Henderson et al., 2000). The effects on eosinophil recruitment may occur by down-regulation of VCAM-1 and very late antigen-4 (Nakajima et al., 1994). Blockade of the IL-4R may also inhibit receptors on B cells or mast cells. The expression or production of cytokines were either unaffected (IL-5, IFN-γ) or increased (IL-4) after treatment. This suggests that IL-4 is not required to sustain cytokine release from Th2 cells and that IL-4R blockade occurs on cells other than T cells and may prevent the uptake of IL-4 by T cells.
A mutated IL-4 that blocks IL-4 and -13 signalling through the IL-4Rα/IL-13Rα1 has also been tested. Administration during sensitization reduced IgE, eosinophil airway infiltration, IL-4 and -5 levels, MSC numbers and AHR (Hahn et al., 2003; Yang et al., 2004). However, the inhibitor was not effective in suppressing AAD after sensitization had occurred.
Taken together these studies showed that IL-4 is necessary but not sufficient for the development of AAD, while blockade of the IL-4R may suppress some pathological features of asthma.
Human studies. A humanized anti-IL-4 neutralizing antibody (pascolizumab) was generated and shown to inhibit IL-4 bioavailability to human-derived cell lines and was well tolerated in monkeys (Hart et al., 2002). However, clinical trials of IL-4-specific antagonists for asthma have failed (Wenzel et al., 2007). This has led to the concept that targeting IL-4 alone may be too selective and that alternative approaches that suppress the activity of both IL-4 and -13 may be more beneficial (O'Byrne et al., 2004).
A soluble IL-4Rα has been trialled for efficacy in inhibiting IL-4 activity and moderate persistent asthma in adults after corticosteroid withdrawal (Borish et al., 2001). Treatment was well tolerated and lacked side effects and prevented reductions in FEV1 and increases in asthma symptom scores compared to placebo.
A human monoclonal anti-IL-4Rα antibody (AMG 317) that blocks both IL-4 and -13 signalling has recently been developed and tested as an asthma therapy (Corren et al., 2010). Treatment did not have clinical efficacy with no improvements in the control of stable asthma, β-agonist use or lung function. However, treatment did suppress exacerbations, particularly in patients with higher asthma scores and reversibility. Increasing the dose, treatment duration or application to specific asthma phenotypes may be more effective.
Piktrakinra is a recombinant variant of human IL-4 that potently inhibits the binding of both IL-4 and -13 to the IL-4Rα/IL-13Rα1 complex. Therapeutic administration of piktrakinra protected allergic monkeys from airway eosinophil influx and AHR after allergen challenge (Tomkinson et al., 2010). In humans, treatment of atopic asthmatics with piktrakinra, significantly improved FEV1 upon allergen challenge and reduced spontaneous asthma attacks requiring rescue medication (Wenzel et al., 2007).
Anti-IL-4 treatments appear to be ineffective in established disease. However, targeting IL-4 during sensitization or in combination with anti-IL-5 treatment may be effective in asthma. Blocking IL-4 may have long-term benefits by suppressing Th2 development and the downstream effects of Th2 responses including IL-3, -13 and GM-CSF release, eosinophil influx, mucus hypersecretion and airway remodelling. The use of soluble IL-4R seems to be the most effective approach. Interfering with the IL-4Rα would also attenuate the effects of IL-13 and remains a therapeutic possibility for clinical benefit in asthma.
Anti-IL-5
IL-5 is increased in bronchial biopsies and is associated with increased numbers of eosinophils in asthmatics, which correlate with disease severity (Figure 1) (Azzawi et al., 1990; Hamid et al., 1991). IL-5 signals through the IL-5Rα and is critical for the growth, maturation and activation and suppresses apoptosis of eosinophils and is implicated in the induction of AHR (Hogan et al., 1997b). Indeed, inhalation of IL-5 by asthmatic patients induced eosinophil influx into the airways and AHR (Leckie et al., 2000). IL-5 also regulates its own receptor expression on eosinophils during their development. Eosinophils are considered to play an important role in asthma pathogenesis. Upon activation they degranulate and release lipid mediators cytokines, cytotoxins, leukotrienes, and platelet activating factor (PAF) and pro-fibrogenic factors such as TGF-α, TGF-β, platelet-derived growth factor (PDGF) and matrix metalloproteinase (MMP)-9 that induce airway narrowing and remodelling, and AHR (Trifilieff et al., 2001; Flood-Page et al., 2003a; Tanaka et al., 2004). These observations suggest that IL-5-induced eosinophils may contribute to mucus hypersecretion, airway remodelling and AHR.
Mouse studies. Increased IL-5 in the airways of wild-type (WT) or IL-5-Tg animals induces eosinophilic influx into the airways and structural changes in the lung (Van Oosterhout et al., 1995; Lee et al., 1997; Trifilieff et al., 2001). In IL-5−/− mice, eosinophil (but not other leukocyte) influx into the airways, numbers of MSC, airway damage, the early stages of airway remodelling and AHR were abrogated in acute models of AAD (Foster et al., 1996; Trifilieff et al., 2001). Increased numbers of eosinophils and AHR were restored by transfer of IL-5-producing vaccinia virus (Foster et al., 1996). In a model of subchronic AAD in IL-5Rα−/− mice, eosinophil influx into the airways, TGF-β levels in bronchoalveolar lavage (BAL), and airway fibrosis were also inhibited (Tanaka et al., 2004). These factors were enhanced upon the induction of AAD in IL-5-Tg mice (Tanaka et al., 2004).
Other studies using IL-5−/− mice and a model of chronic allergic asthma showed that IL-5 plays a pivotal role in the development of eosinophilic and chronic inflammation as well as AHR but was not required for epithelial and fibrotic changes (Foster et al., 2000). Collectively these studies confirmed the importance of IL-5 and eosinophilic inflammation in mediating inflammation and AHR in AAD of mice.
Anti-IL-5 neutralizing antibodies (TRFK-5) have potent suppressive effects on eosinophils. A single-dose inhibits eosinophil influx into the airways and their release from bone marrow when administered 8–12 weeks before allergen sensitization and challenge of mice (Garlisi et al., 1999). Pretreatment also inhibited eosinophil infiltration into the airways but did not affect AHR (Corry et al., 1996). Administration of anti-IL-5 in acute or subchronic models of AAD suppressed the development of eosinophilic inflammation (in airways, blood and bone marrow), MSC and AHR and prevented airway fibrosis (Hogan et al., 1997a; Hamelmann et al., 1999; Blyth et al., 2000; Tanaka et al., 2004) but did not affect B-cell antibody or T-cell cytokine production or mast cell function (Hamelmann et al., 1999). Delivery of anti-IL-5 to IL-4−/− mice abolished the influx of eosinophils into the airways, lung damage and AHR (Hogan et al., 1997b). This indicates that cooperative pathways are important in the pathogenesis of asthma and that combination therapies that target multiple factors may be more effective than single therapies.
In models of chronic allergic asthma, anti-IL-5 delivered once lesions had developed, suppressed airway inflammation and remodelling but had no effect on AHR (Mathur et al., 1999; Kumar et al., 2004). These studies indicate that reducing eosinophils may not suppress symptoms of chronic asthma and additional strategies to suppress AHR may be required.
Collectively, these observations suggest that IL-5 has an important role in regulating eosinophil influx into the airways, airway remodelling and AHR and indicated that anti-IL-5 may be beneficial in asthma.
Human studies. The efficacy of TRFK-5 has been tested in a non-human primate model of asthma (Mauser et al., 1995). Treatment almost completely suppressed the influx of eosinophils into the airways and AHR for up to 3 months. This led to the development of humanized anti-IL-5 monoclonal antibodies (mepolizumab, SCH55700), which block the binding of IL-5 to the IL-5Rα. Efficacy and safety of mepolizumab studies in a primate asthma model showed it had minimal adverse effects and suppressed eosinophil infiltration into the airways and blood but did not alter eosinophil precursors in the bone marrow or allergic bronchoconstriction (Hart et al., 2002).
In three small safety and efficacy clinical trials in mild and severe asthma, anti-IL-5 mAbs [mepolizumab (Leckie et al., 2000; Flood-Page et al., 2007) or SCH55700 (Kips et al., 2003)] had similar effects to those observed in chronic models of asthma in mice. Mepolizumab was safe and effectively reduced eosinophil numbers in the airways and blood, and the expression extracellular matrix (ECM) proteins, but did not suppress AHR, asthma symptoms or late phase reactions to allergen provocation or improve lung function (Leckie et al., 2000; Kips et al., 2003; Flood-Page et al., 2007). Mepolizumab suppressed late but not early eosinophil differentiation in the bone marrow and did not completely abrogate eosinophil influx (Leckie et al., 2000). Tissue infiltration of eosinophils was only reduced by 50% and there was no effect on degranulation (Flood-Page et al., 2003b). Nevertheless, even the modest reduction in tissue eosinophils correlated with significant decreases in airway TGF-β levels (which was predominantly produced by eosinophils) and deposition of some ECM (tenascin, lumincan) proteins (Flood-Page et al., 2003a).
There are several potential reasons for the lack of efficacy: (i) treatment regimes employed were short term and did not sufficiently reduce eosinophils and longer-term treatment may be required; (ii) studies were not sufficiently powered to detect clinically efficacy; (iii) patients were selected on clinical and physiological parameters rather than eosinophilic inflammation and eosinophilic outcomes were not the primary measures; (iv) eosinophils may be bystanders in the disease process or other cytokine-progenitor pathways may mediate disease; (v) anti-IL-5 suppresses IL-5Rα expression and therefore eosinophils become less responsive, which may promote eosinophil survival; and (vi) early differentiation of eosinophils may be mediated by IL-3 and GM-CSF through the IL-3/IL-5/GM-CSF βc-receptor (Asquith et al., 2008) and eosinophil influx may be induced by eotaxins and C-C chemokine receptor type 3 (CCR3) as well as IL-5 (Foster et al., 2002). Further work is needed to clarify these issues.
To address some of these issues, two more recent studies have trialled mepolizumab in asthmatics with elevated numbers of sputum eosinophils, airway symptoms and severe exacerbations that were refractory to corticosteroid treatment and over longer treatment periods (Haldar et al., 2009; Nair et al., 2009). Mepolizumab was well tolerated over 12 months, and reduced blood and sputum eosinophils, airway wall thickness and remodelling, corticosteroid use and asthma exacerbations and improved FEV1 (in one study) and quality of life in these patients. These results are supported by others that show efficacy in suppressing other eosinophil mediated diseases (Haldar et al., 2009; Nair et al., 2009) and that adjusting inhaled steroid treatment according to sputum eosinophil counts resulted in a dramatic decrease in exacerbations (Green et al., 2002; Jayaram et al., 2006). These studies confirm predictions from mouse models that IL-5 suppresses eosinophilic inflammation and airway remodelling in asthma. They demonstrate that anti-IL-5 treatment is effective in severe asthma with exacerbations where eosinophils have a pathogenetic role. Other studies show that eosinophilic inflammation is delineated from changes in lung function and AHR that may be treated with other therapies (e.g. steroids) that suppress pathogenesis through different mechanisms (Haldar et al., 2009).
Anti-TNF-α
TNF-α is increased in BAL and bronchial biopsy specimens from severe asthma patients and its expression is refractory to steroid treatment (Howarth et al., 2005). TNF-α is produced by Th1 cells and macrophages and to as lesser extent mast cells in ASM, which may induce AHR (Figure 1). TNF-α directly alters the contractility of ASM probably through alterations in calcium flux and bronchoconstrictor sensitivity (Anticevich et al., 1995).
Mouse studies. TNF-α can mediate the recruitment of neutrophils and eosinophils into the airways in antigen-driven airway inflammation in mice potentially via up-regulation of epithelial and endothelial adhesion molecules (Lukacs et al., 1995). It also directly promotes T-cell activation (Scheurich et al., 1987). TNF-α also uniquely suppresses glucocorticoid responsiveness in monocytes and up-regulates the pathways involved in chronic airway remodelling and subepithelial fibrosis (Franchimont et al., 1999; Sullivan et al., 2005).
Human studies. Several humanized anti-TNF-α neutralizing antibodies (infliximab, adalimumab and golimumab) are available (Desai and Brightling, 2009). Infliximab improved some lung function measures (diurnal variation in peak expiratory flow) but not others (morning peak expiratory flow, AHR) and reduced exacerbations in moderate asthmatics (Erin et al., 2006; Morjaria et al., 2008). The largest study used long-term treatment with golimumab for severe asthma; however, the trial was terminated early due to a large number of adverse events (Wenzel et al., 2009).
A soluble fusion protein (etanercept) that binds and neutralizes TNF-α has been developed and used with some promising results. Treatment reduced airway histamine levels and AHR and improved lung function and quality of life in patients with difficult to manage asthma. Airway eosinophil or neutrophil numbers were not altered (Howarth et al., 2005; Berry et al., 2006). Efficacy closely correlated with TNF-α mRNA expression and receptor expression on monocytes. However, the improvements were relatively modest and other studies in moderate-severe asthma have been negative. Serious concerns remain over the safety of TNF-α blockade, which may enhance susceptibility to respiratory infection (Berry et al., 2006).
Novel asthma therapies
Although there is significant experimental support for the potential targeting of the following cytokines in asthma, human trials are in their infancy.
Anti-IL-13
IL-13 expression is increased after antigen challenge of the airways of asthmatics (Huang et al., 1995). It is produced by activated Th2 cells, mast cells and dendritic cells (Figure 1) (Wills-Karp et al., 1998) and signals through the IL-4Rα/IL-13Rα1 complex, although IL-4Rα-independent signalling also occurs (Kumar et al., 2002).
Mouse studies. IL-13 induces B cells to release IgE, increases VCAM-1 expression (Wills-Karp et al., 1998) and is important in the recruitment of eosinophils into airway tissue. IL-13 can also stimulate fibroblasts to proliferate, induce MSC hyperplasia and mucus production, airway remodelling and AHR in animal models of AAD (Grünig et al., 1998; Wills-Karp et al., 1998; Kumar et al., 2004; Horvat et al., 2010b). Some of these effects may occur in IL-4Rα-dependent and T-cell independent processes; however, IL-13+ T cells alone can induce eosinophil influx, and AHR independently of the IL-4Rα (Mattes et al., 2001). The effects of IL-13 on AHR may be directly on ASM but other factors, potentially mast cells in the ASM, may also be involved (Brightling et al., 2003; Shore and Moore, 2002). Naïve IL-13-Tg mice have elevated baseline mucus production, airway remodelling and AHR (Zhu et al., 1999) and after challenge have increased IgE, mucus and susceptibility to anaphylaxis (Fallon et al., 2001).
IL-13−/− mice have suppressed MSC numbers and may or may not have AHR in acute AAD (Webb et al., 2000; Walter et al., 2001). These conflicting results may be explained by the involvement of different cells and cytokines in different models, and the development of AHR in IL-13−/− mice may result from compensatory mechanisms. These studies also show that the mechanisms of induction of mucus production and AHR may be dissociated. IL-13−/− mice show a pronounced pulmonary eosinophilic inflammation, which indicates that targeting this molecule alone will not have pronounced anti-inflammatory effects. However, depletion of IL-4 in IL-13−/− mice inhibited pulmonary eosinophil infiltration and AHR, although blood eosinophilia was still present (Webb et al., 2000). The impairment of eosinophil influx into the lung may occur by suppressing IL-4 and −13-induced adhesion molecules (e.g. VCAM-1) and chemokines (e.g. eotaxins) and eosinophil activation. Neutralization of IL-5 in IL-13−/− mice inhibited AHR (Webb et al., 2000). These studies showed that IL-13 has a modulatory role during sensitization but is pro-inflammatory during challenge.
In a model of chronic asthma IL-13−/− mice have reduced infiltration of eosinophils and inflammatory cells in the airways and decreased MSC, epithelial hypertrophy and subepithelial fibrosis although modest AHR was still present (Kumar et al., 2002). These results are in striking contrast to those produced using IL-4Rα−/− mice, which had no changes in the infiltration of eosinophils or other inflammatory cells compared to WT mice. Collectively studies indicate that IL-13 contributes to eosinophil accumulation in the airways and airway remodelling in chronic asthma but that targeting of other factors in combination may also be required.
Anti-IL-13 treatment during allergen challenge in acute models of AAD suppressed airway inflammation, mucus production and AHR (Grünig et al., 1998; Wang and McCusker, 2005; Wills-Karp et al., 1998). Humanized anti-IL-13 also suppressed eosinophil influx into the airways, MSC and reduced AHR that were induced my administration of human IL-13 to mice (Blanchard et al., 2005). IL-13 can also be selectively depleted using soluble IL-13Rα2-Fc fusion protein (Grünig et al., 1998). This is a naturally occurring soluble receptor that lacks signalling capabilities and silences IL-13 activity (Yasunaga et al., 2003). Treatment with sIL-13Ra2-Fc during allergen challenge of sensitized mice attenuated eosinophil (but not neutrophil) infiltration into the airways in some studies but not others, suppressed mucus hypersecretion and completely inhibited AHR (Grünig et al., 1998; Wills-Karp et al., 1998). Treatment of sensitized sheep with sIL-13Rα2-Fc or humanized anti-IL-13 before challenge abrogated bronchial constriction and AHR (Kasaian et al., 2007). There is a second IL-13 receptor (designated as IL-13Rα2) that inhibits IL-13/IL13Rα1 dependent signaling events and may have potential therapeutic use in anti-IL-13 treatment of asthma patients although this has not yet been tested.
Anti-IL-13 delivered in a model of established chronic allergic asthma suppressed cytokine/chemokine and IgE production, the accumulation of eosinophils and inflammatory cells in the airway, increases in airway MSC and remodelling but had limited effects on AHR (Blease et al., 2001; Kumar et al., 2004; Yang et al., 2004; 2005;).
Collectively these studies indicate that antagonizing IL-13 may have potential as a therapy for chronic asthma but additional suppression of eosinophilic inflammation would be required.
Human studies. Human IL-13 neutralizing antibodies (IMA-638, CAT-354 and AMG 317) have been developed that are high affinity, long lasting and are safe and well tolerated in adults with asthma (Singh et al., 2010). Further studies are eagerly awaited.
Anti-IL-9
IL-9 was originally described as a Th2 cytokine promoting T-cell growth and mastocytosis. IL-9 may control the phenotype of mast cells and other cell types in the human lung (Figure 1) (Eklund et al., 1993). IL-9-producing T cells have now been identified as being a distinct subset from Th2 cells and differentiate in response to IL-4 and TGF-β (Veldhoen et al., 2008; Staudt et al., 2010).
Mouse studies. Adoptive transfer of Th9 cells generates allergic airway inflammation in mice, which was reversed by IL-9 neutralization (Staudt et al., 2010). Tg lung-specific over-expression of IL-9 increases airway inflammation, MSC hyperplasia and AHR whereas blocking the function of IL-9 suppressed eosinophilic airway inflammation and AHR (Temann et al., 1998; Cheng et al., 2002).
Human studies. Anti-IL-9 safety trials have been completed in humans and efficacy trials for moderate-severe asthma are underway (Antoniu, 2010; Desai and Brightling, 2009).
Anti-GM-CSF
GM-CSF is produced by Th1 and Th2 cells, macrophages, eosinophils and epithelial cells (Yamashita et al., 2002) and is increased in the airways of asthmatics (Robinson et al., 1992). Early eosinophil differentiation may be mediated by IL-3 and GM-CSF through the IL-3Rα, GM-CSFRα and IL-3/IL-5/GM-CSF common β (βc)-receptors (Figure 1) (Asquith et al., 2008). GM-CSF is critical for eosinophil survival, enhances their chemotaxis, degranulation and cytokine production (Owen et al., 1987) and is essential for the activation of antigen presenting cells (Zhou and Tedder, 1996). Exposure of mature eosinophils to IL-5 or local airway allergen challenge results in a down-regulation of the IL-5 receptor and reduced responsiveness to IL-5 (Gregory et al., 2003). Therefore, eosinophil development and airway tissue eosinophils may not respond to IL-5 but may rely on IL-3 or GM-CSF, which operate through receptors unaffected by anti-IL-5 treatment.
Mouse studies. Over-expression of GM-CSF in mice enhances allergic sensitization, airway inflammation and fibrosis (Xing et al., 1996; Stämpfli et al., 1998). Anti-GM-CSF antibodies administered during allergen challenge of sensitized mice inhibited airway inflammation, mucus production and suppressed AHR (Yamashita et al., 2002).
Human studies. Phase 1 clinical safety and pharmacological trials are underway with a human neutralizing antibody for GM-CSF (MT203).
Emerging asthma targets
The following cytokines have been recently identified as new regulators of allergic inflammation and are emerging targets for clinical trials.
Anti-IL-25 (IL-17E)
IL-25, -33 and TSLP are newly discovered cytokines that are critical for the initiation of Th2 inflammatory responses and associated airway lesions (Figure 1). They are released from the airway epithelium in response to protease allergens. Therapeutic inhibition of these molecules may reduce the progression of asthma early in disease development.
IL-25 and its receptor (IL-17RB) are up-regulated in asthmatic airway biopsies. It is released by epithelial cells, basophils and eosinophils and possibly by Th2 cells. IL-25 drives allergic inflammatory responses by activating NKT cells and inducing Th2 cell differentiation (Angkasekwinai et al., 2007; Wang et al., 2007; Terashima et al., 2008; Stock et al., 2009).
Mouse studies. IL-25 is expressed during AAD in mice and promotes the development of Th2 cells (Sharkhuu et al., 2006; Angkasekwinai et al., 2007; Wang et al., 2007). IL-25 administration induces Th2 responses including IL-4, -5 and -13 expression, increased serum IgE, airway eosinophil influx, mucus production and epithelial cell hyperplasia (Fort et al., 2001). Monoclonal antibody neutralization of IL-25 reverses Th2-associated pathology in the lungs through a signal transducer and activator of transcription 6 (STAT6) and Th2 cell-dependent mechanism (Fort et al., 2001; Sharkhuu et al., 2006; Tamachi et al., 2006).
Anti-IL-33
IL-33, an IL-1 family cytokine, is increased in ASM and epithelial cells from asthmatics and this increase positively correlates with asthma severity (Prefontaine et al., 2009; 2010;). IL-33 binds to T1/ST2 receptors on Th2 cells, enhances the secretion of IL-5 and -13 and promotes chemotaxis of these cells. It may also contribute to the dendritic cell (DC)-induced development of Th2 cells and induces tryptase accumulation and cytokine release from mast cells (Schmitz et al., 2005; Komai-Koma et al., 2007; Rank et al., 2009; Kaieda et al., 2010). IL-33 can act alone or in concert with TSLP to accelerate the maturation and cytokine release of human mast cells. Similarly, IL-3 and -33 synergistically promote IL-4, -5, -8 and -13 production from human basophils and eosinophils (Pecaric-Petkovic et al., 2009). IL-33 also activates NK and NKT cells to secrete IFN-γ (Smithgall et al., 2008; Bourgeois et al., 2009) indicating that it may contribute to multiple different subtypes of asthma.
Mouse studies. IL-33 and its' soluble receptor are highly up-regulated early after allergen challenge in models of acute AAD in mice (Hayakawa et al., 2007). Treatment of the airways of mice with recombinant IL-33 induces a Th2-like inflammatory response in association with increased tissue eosinophil influx, MSC and lung epithelial cell hyperplasia and heightened IgE. Monoclonal antibody blockade of the IL-33R attenuates hallmark features of AAD (Kurowska-Stolarska et al., 2008; Kearley et al., 2009). Furthermore, the stimulatory actions of IL-33 on Th2 cells can be preventing by blocking with anti-T1/ST2 antibodies and this approach may be an alternative for inhibiting IL-33-induced inflammation.
Anti-TSLP
Like IL-33 the levels of TSLP are elevated in airways cells from asthmatics and this positively correlates with disease severity. TSLP is released by airway epithelial cells and may function early in innate immune activation to promote Th2 immune responses (Ying et al., 2005). DC-induced Th2 cell differentiation and Th2 cell chemotaxis is induced by TSLP. Furthermore TSLP can promote the release of cytokines such as IL-4, -5 and -13 from T cells and mast cells indicating that this molecules can promote the effector arm of allergic inflammation (Soumelis et al., 2002; Allakhverdi et al., 2007; Rochman et al., 2007).
Mouse studies. Lung specific TSLP-Tg mice develop severe Th2 cytokine-mediated inflammation (Zhou et al., 2005), whereas TSLP receptor−/− mice are less susceptible to the induction of acute AAD (Al-Shami et al., 2005). Notably, antibody-mediated blockade of the TSLP receptor suppresses AAD by inhibiting DC migration to the airways and CD4+ T cell priming (Shi et al.,2008).
Anti-IFN-γ
In severe asthma, which is often steroid insensitive, other cytokines such as IFN-γ, IL-17 and -27 may contribute to pathogenesis and thus be possible treatment targets (Figure 1). These cytokines are associated with Th1 and Th17 immunity rather than allergic Th2 responses, which reflects the emerging paradigm of mixed T helper cell responses promoting severe asthma.
IFN-γ is the archetypal Th1 cytokine and is implicated in both acute severe and chronic stable asthma. IFN-γ is increased in asthmatic airways and IFN-γ-producing T cells are increased in the blood (Magnan et al., 2000) and BAL (Krug et al., 1996; Brown et al., 2003), particularly in severe asthma (Cho et al., 2005). High serum levels predict deterioration in lung function in stable asthmatics (Litonjua et al., 2003). Non-specific stimulation of BAL cells leads to increased IFN-γ production (Krug et al., 1996; Brown et al., 2003), and IFN-γ also up-regulates cysteinyl leukotriene receptors on ASM cells that increases smooth muscle contractility (Amrani et al., 2001).
Mouse studies. IFN-γ has been implicated in the pathogenesis of AHR (Hessel et al., 1997). Depletion of Th1 cells during allergen sensitization and challenge in mice decreased levels of IFN-γ in BAL and suppressed the development of AHR (Fleming et al., 2001). In a model of chronic asthma IFN-γ and IFN-γ-producing CD4+ T cells were increased (Foster et al., 2003), and AHR was independent of Th2 cytokines (Foster et al., 2000; Kumar et al., 2002).
Anti-IFN-γ attenuates lymphocyte accumulation that may suppress inflammatory mediators of AHR (Issekutz et al., 1988). Anti-IFN-γ treatment during repeated Ova challenges in acute AAD prevented the development of AHR, although eosinophilic airway inflammation was not affected (Hessel et al., 1997). Anti-IFN-γ administered during established disease substantially reduced the influx of chronic inflammatory cells into the airways and IFN-γ-producing CD4+ T cells and effectively inhibited AHR but did not alter eosinophil influx or airway remodelling (Kumar et al., 2004).
Taken together these studies show that IFN-γ may be an important mediator of AHR in chronic asthma. However, anti-IFN-γ has not yet been assessed in human clinical trials for asthma.
Anti-IL-17
IL-17 is released by alveolar macrophages, Th17 and γδT cells, is associated with Th1 and Th17 immunity and may play a critical role in the mixed T-cell response and pathogenesis of severe asthma. Serum and airway IL-17 levels are elevated in severe compared to mild asthma (Bullens et al., 2006; Agache et al., 2010). IL-17 plays a crucial role in driving neutrophil influx into the airways and is implicated in fibrosis and airway remodelling (Chakir et al., 2003). Bacterial infections may induce IL-17 responses that promote the development of neutrophilic AAD (Horvat et al., 2010a).
Mouse studies. The precise role of IL-17 in AAD in mice remains controversial (Laan et al., 1999). IL-17 release by γδT cells may be important in the resolution of inflammation in AAD in mice (Murdoch and Lloyd, 2010). Administration of recombinant IL-17A during allergen challenge had a suppressive effect (Schnyder-Candrian et al., 2006), whereas delivery after challenge exacerbated inflammation and AHR (Wilson et al., 2009). Pulmonary over-expression of IL-17F enhanced AHR in both the presence and absence of airway inflammation (Oda et al., 2005). These results suggest that the temporal expression of the IL-17 inflammatory response will determine its' effects on disease. Notably, IL-17−/− mice develop equivalent airway inflammation and AHR as WT mice during acute AAD (Nakae et al., 2002).
Anti-IL-27
Increased levels of IL-27, a member of the IL-6 family of cytokines, are present in the induced sputum of steroid-refractory asthmatic patients (Li et al., 2010).
Mouse studies. Recently a unique role for IL-27 in cooperating with IFN-γ to induce steroid-resistant AHR has been demonstrated in a mouse model of severe asthma (Li et al., 2010). Induction of AHR was dependent on MyD88-signalling in alveolar macrophages but was not associated with airway neutrophil influx. Notably, treatment with IL-27 and IFN-γ inhibited dexamethasone-induced translocation of the glucocorticoid receptor into the nucleus of pulmonary macrophages.
Cytokine therapies
Administration of several cytokines (IFN-γ, -α and -β and IL-12), have been trialled in attempts to suppress Th2 responses in asthma but these have been largely disappointing with lack of efficacy and adverse effects. Treatment of mild allergic asthmatics with IL-12 reduced blood and airway eosinophil numbers but had no significant effect on AHR or the late asthmatic response (Bryan et al., 2000). Two small trials of long-term IFN-α (IFN-αcon-1 or IFN-α 2a) administration suppressed Th2 cytokine production from peripheral blood mononuclear cells in severe asthmatics and improved lung function, medication requirements, asthma symptoms and hospitalizations (Simon et al., 2003; Kroegel et al., 2006).
New anti-inflammatory approaches and combination therapies
The concept of treating asthma by targeting a single molecule has had limited success. Indeed, steroid therapy, which is currently the mainstay therapy, is thought to act by suppressing a range of pro-inflammatory pathways. The emergence of subtypes of asthma and the complexity of the diseases process where many different inflammatory mediators, cytokines, chemokines and cells are dysregulated further highlights that new ways to treat disease are required. Molecular redundancy and the existence of pathways that operate in parallel are also significant therapeutic hurdles for the treatments of inflammatory disease such as asthma. Many studies show that airway inflammation, remodelling and AHR are dissociated and may be mediated by different mechanisms under different conditions. New anti-inflammatory approaches that suppress critical factors that promote the activation of inflammatory networks such as microRNA may be beneficial (Mattes et al., 2009). Alternatively, combination anti-cytokine therapy may be a more effective therapeutic approach for asthma.
Contributions of studies using mouse models to the field
Collectively mouse models of asthma have demonstrated the regulatory complexity of allergic inflammation and that targeting single-cytokines/inflammatory molecules will not be effective for the resolution of allergic inflammation. For example, many studies have demonstrated inflammation and AHR persist in the absence of IL-4, -5, IL-4Rα, IL-13, -9, eotaxin or IgE. Thus, blocking a single-cytokine pathway may be insufficient to reverse features of human asthma. Depletion of a single cytokine may also induce compensatory effects that may induce features of asthma through other pathways. In some studies, anti-IL-5 treatment suppressed eosinophil influx into the airways but increased neutrophil and lymphocyte recruitment (Mathur et al., 1999) and in the absence of both IL-4 and IL-13, levels of IL-5 and blood eosinophilia were enhanced (Webb et al., 2000). The pro-inflammatory properties of IL-13 during allergen challenge are independent of IL-4, which suggests that common targeting may be needed for therapeutic efficacy. Indeed, cytokine deficient mice have been used to demonstrate the coordinated activity of IL-4, -5 and -13 will need to be targeted to suppress airway inflammation and AHR (Webb et al., 2000). Targeting receptors such as the IL-4Rα/IL-13Rα1 or the IL-3/IL-5/GM-CSF βc-receptor (Asquith et al., 2008) may attenuate the activity of multiple cytokines and cells (e.g. Th2, eosinophils and neutrophils) and may be more effective than blocking a single pathway. Th2/Th1 responses and IFN-γ are important in AHR in severe and chronic asthma and so targeting combinations of these cytokines (e.g. IL-5 and IFN-γ) may be more effective in these asthma subtypes (Kumar et al., 2004). Although, information from animal models is highly valuable it needs to be interpreted within the context of the experimental system and the complexity of the diseased state.
Future studies
Improved understanding of the role of cytokines in asthma would facilitate the development and application of anti-cytokine therapies. Asthma is a heterogeneous syndrome and future studies of tailored application of anti-cytokine therapies need to be trialled in individuals with subtypes of asthma that would be responsive to these specific treatments. Mild to moderate allergic asthma that is underpinned by aberrant Th2 and eosinophilic responses may benefit from therapies such as combinations of anti-IL-5/-13 and/or common receptors such as the IL-4Rα/IL-13Rα1, or the IL-3/IL-5/GM-CSF βc-receptor that target multiple arms of the pathways involved. Severe asthma that may be mediated by TNF-α, IFN-γ, IL-17 and -27 may respond to therapies that target these cytokines. Severe asthma exacerbations that are driven by TNF-α may be most responsive to anti-TNF-α treatment and infection-induced exacerbations that result from deficiencies in type I IFNs may potentially be treated with these cytokines. The optimization of dose, route of delivery and treatment duration may also be critical and combination and/or anti-receptor therapies may be more effective that single-mediator treatments. However, these approaches need to be carefully tested in well-designed and tailored trials in appropriate cohorts. These cytokines have specific immune functions and care must be taken not to induce susceptibility to infections or cancer, and the challenge may be to reduce effector cytokine to normal levels. Genetic polymorphisms in cytokine genes may have important biological significance and studies are required to determine the effects of point mutations on cytokine bioactivity and/or turnover. For example, an inspection of the single nucleotide polymorphism (SNP) database at GenBank for the human IL-4 gene (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=3565) revealed missense mutations that alter amino acids 27, 53, 59, 101 and 120 in the translated protein. An important future area of investigation will be to compare the bioactivities of recombinant IL-4 isoforms that differ at these five residues.
Conclusions
Clinical and basic research has identified many possible therapeutic cytokine targets for asthma and the need to fully elucidate the contributions of these factors to disease. Soluble IL-4R and anti-IL-5 have inhibited asthma progression and reduced exacerbations. It is likely that earlier and longer-term interventions may be more effective and have sustained effects on asthma pathogenesis particularly persistent airway inflammation and remodelling that may moderate other features of disease. The soluble TNF-α receptor has produced some promising results in patients with difficult to manage asthma and may be important in severe steroid-resistant asthma. Recent studies on IL-25, -33 and TSLP show that they are important in the initial priming of Th2 responses that may promote the development of asthma. Therefore, we may be able to better identify infants who are susceptible to the development of Th2 responses and asthma and utilize targeted anti-cytokine treatments against IL-25, -33 and TSLP to intervene in disease progression early in life. Combination therapies hold great potential for the future. Results of human studies highlight the absolute need for clinical studies to use appropriate patient groups and disease subtypes, which may otherwise confound the field. If these issues are taken into account there is substantial promise for the use of anti-cytokine therapies for the effective treatment of asthma.
Acknowledgments
Work in the authors' laboratories is supported by the National Health and Medical Research Council and the Australian Research Council.
Glossary
Abbreviations
- βc
common β
- −/−
deficient
- AAD
allergic airway disease
- AHR
airway hyperresponsiveness
- ASM
airway smooth muscle
- BAL
bronchoalveolar lavage
- ECM
extracellular matrix
- MSC
mucus secreting cells
- Tg
transgenic
- Th2
cells, type 2 CD4+ lymphocytes
- VCAM-1
vascular cell adhesion molecule-1
- WT
wild-type
Conflict of interest
The authors have no declared conflicts of interest.
Supporting Information
Teaching Materials; Fig 1 as PowerPoint slide.
References
- Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. 2010;104:1131–1137. doi: 10.1016/j.rmed.2010.02.018. [DOI] [PubMed] [Google Scholar]
- Alcorn J, Crowe C, Kolls J. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, Chartier S, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829–839. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, Moore P, Hoffman R, Shore S, Panettieri RJ. Interferon-gamma modulates cysteinyl leukotriene receptor-1 expression and function in human airway myocytes. Am J Respir Crit Care Med. 2001;1164:2098–2101. doi: 10.1164/ajrccm.164.11.2108005. [DOI] [PubMed] [Google Scholar]
- Angkasekwinai P, Park H, Wang YH, Chang SH, Corry DB, Liu YJ, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevich SZ, Hughes JM, Black JL, Armour CL. Induction of human airway hyperresponsiveness by tumour necrosis factor-alpha. Eur J Pharmacol. 1995;284:221–225. doi: 10.1016/0014-2999(95)00463-u. [DOI] [PubMed] [Google Scholar]
- Antoniu SA. MEDI-528, an anti-IL-9 humanized antibody for the treatment of asthma. Curr Opin Mol Ther. 2010;12:233–239. [PubMed] [Google Scholar]
- Asquith K, Ramshaw H, Hansbro P, Beagley K, Lopez A, Foster P. The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J Immunol. 2008;180:1199–1206. doi: 10.4049/jimmunol.180.2.1199. [DOI] [PubMed] [Google Scholar]
- Azzawi M, Bradley B, Jeffery P, Frew A, Wardlaw A, Knowles G, et al. Identification of activated T lymphocytes and eosinophils in bronchial biopsies in stable atopic asthma. Am Rev Respir Dis. 1990;142:1407–1413. doi: 10.1164/ajrccm/142.6_Pt_1.1407. [DOI] [PubMed] [Google Scholar]
- Berry M, Hargadon B, Shelley M, Parker D, Shaw D, Green R, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354) Clin Exp Allergy. 2005;35:1096–1103. doi: 10.1111/j.1365-2222.2005.02299.x. [DOI] [PubMed] [Google Scholar]
- Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel S, Hogaboam C. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol. 2001;166:5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- Blyth DI, Wharton TF, Pedrick MS, Savage TJ, Sanjar S. Airway subepithelial fibrosis in a murine model of atopic asthma: suppression by dexamethasone or anti-interleukin-5 antibody. Am J Respir Cell Mol Biol. 2000;23:241–246. doi: 10.1165/ajrcmb.23.2.3999. [DOI] [PubMed] [Google Scholar]
- Bochner B, Undem B, Lichtenstein L. Immunological aspects of allergic asthma. Annu Rev Immunol. 1994;12:295–335. doi: 10.1146/annurev.iy.12.040194.001455. [DOI] [PubMed] [Google Scholar]
- Borish L, Nelson H, Corren J, Bensch G, Busse W, Whitmore J, et al. Efficacy of soluble IL-4 receptor for the treatment of adults with asthma. J Allergy Clin Immunol. 2001;107:963–970. doi: 10.1067/mai.2001.115624. [DOI] [PubMed] [Google Scholar]
- Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol. 2009;39:1046–1055. doi: 10.1002/eji.200838575. [DOI] [PubMed] [Google Scholar]
- Brightling C, Symon F, Holgate S, Wardlaw A, Pavord I, Bradding P. Interleukin-4 and -13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. 2003;33:1711–1716. doi: 10.1111/j.1365-2222.2003.01827.x. [DOI] [PubMed] [Google Scholar]
- Brown V, Warke T, Shields M, Ennis M. T cell cytokine profiles in childhood asthma. Thorax. 2003;58:311–316. doi: 10.1136/thorax.58.4.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusselle G, Kips J, Tavernier J, van der Heyden J, Cuvelier C, Pauwels R, et al. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. 1994;24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- Bryan S, O'Connor B, Matti S, Leckie M, Kanabar V, Khan J, et al. Effects of recombinant human interleukin-12 on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2149–2153. doi: 10.1016/S0140-6736(00)03497-8. [DOI] [PubMed] [Google Scholar]
- Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner C, Skupin A, Reimann T, Rieber E, Unteregger G, Geyer P, et al. Local production of interleukin-4 during radiation-induced pneumonitis and pulmonary fibrosis in rats: macrophages as a prominent source of interleukin-4. Am J Respir Cell Mol Biol. 1997;17:315–325. doi: 10.1165/ajrcmb.17.3.2279. [DOI] [PubMed] [Google Scholar]
- Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- Cheng G, Arima M, Honda K, Hirata H, Eda F, Yoshida N, et al. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am J Respir Crit Care Med. 2002;166:409–416. doi: 10.1164/rccm.2105079. [DOI] [PubMed] [Google Scholar]
- Cho S, Stanciu L, Holgate S, Johnston S. Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am J Respir Crit Care Med. 2005;171:224–230. doi: 10.1164/rccm.200310-1416OC. [DOI] [PubMed] [Google Scholar]
- Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med. 2010;181:788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- Corry D, Folkesson H, Warnock M, Erle D, Matthay M, Wiener-Kronish J, et al. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle A, Le Gros G, Bertrand C, Tsuyuki S, Heusser C, Kopf M, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- Desai D, Brightling C. Cytokine and anti-cytokine therapy in asthma: ready for the clinic? Clin Exp Immunol. 2009;158:10–19. doi: 10.1111/j.1365-2249.2009.03998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham S, Ying S, Varney V, Jacobson M, Sudderick R, Mackay I, et al. Cytokine messenger RNA expression for IL-3, IL-4, IL-5, and granulocyte/macrophage-colony-stimulating factor in the nasal mucosa after local allergen provocation: relationship to tissue eosinophilia. J Immunol. 1992;148:2390–2394. [PubMed] [Google Scholar]
- Eklund K, Ghildyal N, Austen K, Stevens R. Induction by IL-9 and suppression by IL-3 and IL-4 of the levels of chromosome 14-derived transcripts that encode late-expressed mouse mast cell proteases. J Immunol. 1993;151:4266–4273. [PubMed] [Google Scholar]
- Eklund K, Humphries D, Xia Z, Ghildyal N, Friend D, Gross V, et al. Glucocorticoids inhibit the cytokine-induced proliferation of mast cells, the high affinity IgE receptor-mediated expression of TNF-alpha, and the IL-10-induced expression of chymases. J Immunol. 1997;158:4373–4380. [PubMed] [Google Scholar]
- Erin E, Leaker B, Nicholson G, Tan A, Green L, Neighbour H, et al. The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthma. Am J Respir Crit Care Med. 2006;174:753–762. doi: 10.1164/rccm.200601-072OC. [DOI] [PubMed] [Google Scholar]
- Fallon P, Emson C, Smith P, McKenzie A. IL-13 overexpression predisposes to anaphylaxis following antigen sensitization. J Immunol. 2001;166:2712–2716. doi: 10.4049/jimmunol.166.4.2712. [DOI] [PubMed] [Google Scholar]
- Finkelman F, Katona I, Urban JJ, Holmes J, Ohara J, Tung A, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol. 1988;141:2335–2341. [PubMed] [Google Scholar]
- Fleming C, He H, Ciota A, Perkins D, Finn P. Administration of pentoxifylline during allergen sensitization dissociates pulmonary allergic inflammation from airway hyperresponsiveness. J Immunol. 2001;167:1703–1711. doi: 10.4049/jimmunol.167.3.1703. [DOI] [PubMed] [Google Scholar]
- Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig M, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003a;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil's role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003b;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–1071. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- Foster P, Hogan S, Ramsay A, Matthaei K, Young I. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P, Ming Y, Matthei K, Young I, Temelkovski J, Kumar R. Dissociation of inflammatory and epithelial responses in a murine model of chronic asthma. Lab Invest. 2000;80:655–662. doi: 10.1038/labinvest.3780068. [DOI] [PubMed] [Google Scholar]
- Foster P, Hogan S, Yang M, Mattes J, Young I, Matthaei K, et al. Interleukin-5 and eosinophils as therapeutic targets for asthma. Trends Mol Med. 2002;8:162–167. doi: 10.1016/s1471-4914(02)02302-x. [DOI] [PubMed] [Google Scholar]
- Foster P, Webb D, Yang M, Herbert C, Kumar R. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signalling in experimental chronic asthma. Clin Exp Allergy. 2003;33:688–695. doi: 10.1046/j.1365-2222.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- Franchimont D, Martens H, Hagelstein MT, Louis E, Dewe W, Chrousos GP, et al. Tumor necrosis factor alpha decreases, and interleukin-10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptor. J Clin Endocrinol Metab. 1999;84:2834–2839. doi: 10.1210/jcem.84.8.5931. [DOI] [PubMed] [Google Scholar]
- Garlisi CG, Kung TT, Wang P, Minnicozzi M, Umland SP, Chapman RW, et al. Effects of chronic anti-interleukin-5 monoclonal antibody treatment in a murine model of pulmonary inflammation. Am J Respir Cell Mol Biol. 1999;20:248–255. doi: 10.1165/ajrcmb.20.2.3327. [DOI] [PubMed] [Google Scholar]
- Gavett S, O'Hearn D, Karp C, Patel E, Schofield B, Finkelman F, et al. Interleukin-4 receptor blockade prevents airway responses induced by antigen challenge in mice. Am J Physiol. 1997;272:L253–L261. doi: 10.1152/ajplung.1997.272.2.L253. [DOI] [PubMed] [Google Scholar]
- Green R, Brightling C, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- Gregory B, Kirchem A, Phipps S, Gevaert P, Pridgeon C, Rankin S, et al. Differential regulation of human eosinophil IL-3, IL-5, and GM-CSF receptor alpha-chain expression by cytokines: IL-3, IL-5, and GM-CSF down-regulate IL-5 receptor alpha expression with loss of IL-5 responsiveness, but up-regulate IL-3 receptor alpha expression. J Immunol. 2003;170:5359–5366. doi: 10.4049/jimmunol.170.11.5359. [DOI] [PubMed] [Google Scholar]
- Grünig G, Warnock M, Wakil A, Venkayya R, Brombacher F, Rennick D, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn C, Teufel M, Herz U, Renz H, Erb K, Wohlleben G, et al. Inhibition of the IL-4/IL-13 receptor system prevents allergic sensitization without affecting established allergy in a mouse model for allergic asthma. J Allergy Clin Immunol. 2003;111:1361–1369. doi: 10.1067/mai.2003.1527. [DOI] [PubMed] [Google Scholar]
- Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelmann E, Cieslewicz G, Schwarze J, Ishizuka T, Joetham A, Heusser C, et al. Anti-interleukin 5 but not anti-IgE prevents airway inflammation and airway hyperresponsiveness. Am J Respir Crit Care Med. 1999;160:934–941. doi: 10.1164/ajrccm.160.3.9806029. [DOI] [PubMed] [Google Scholar]
- Hamid Q, Azzawi M, Ying S, Moqbel R, Wardlaw A, Corrigan C, et al. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991;87:1541–1546. doi: 10.1172/JCI115166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansbro P, Beagley K, Horvat J, Gibson P. Role of atypical bacterial infection of the lung in predisposition/protection of asthma. Pharmacol Ther. 2004;101:193–210. doi: 10.1016/j.pharmthera.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Hansbro N, Horvat J, Wark P, Hansbro P. Understanding the mechanisms of viral induced asthma: new therapeutic directions. Pharmacol Ther. 2008;117:313–353. doi: 10.1016/j.pharmthera.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Blackburn M, Brigham-Burke M, Dede K, Al-Mahdi N, Zia-Amirhosseini P, et al. Preclinical efficacy and safety of pascolizumab (SB 240683): a humanized anti-interleukin-4 antibody with therapeutic potential in asthma. Clin Exp Immunol. 2002;130:93–100. doi: 10.1046/j.1365-2249.2002.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart P, Bonder C, Balogh J, Dickensheets H, Donnelly R, Finlay-Jones J. Differential responses of human monocytes and macrophages to IL-4 and IL-13. J Leukoc Biol. 1999;66:575–578. [PubMed] [Google Scholar]
- Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282:26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- Henderson WJ, Chi E, Maliszewski C. Soluble IL-4 receptor inhibits airway inflammation following allergen challenge in a mouse model of asthma. J Immunol. 2000;164:1086–1095. doi: 10.4049/jimmunol.164.2.1086. [DOI] [PubMed] [Google Scholar]
- Hessel E, Van Oosterhout A, Van Ark I, Van Esch B, Hofman G, Van Loveren H, et al. Development of airway hyperresponsiveness is dependent on interferon-gamma and independent of eosinophil infiltration. Am J Respir Cell Mol Biol. 1997;16:325–334. doi: 10.1165/ajrcmb.16.3.9070618. [DOI] [PubMed] [Google Scholar]
- Hogan S, Koskinen A, Foster P. Interleukin-5 and eosinophils induce airway damage and bronchial hyperreactivity during allergic airway inflammation in BALB/c mice. Immunol Cell Biol. 1997a;75:284–288. doi: 10.1038/icb.1997.43. [DOI] [PubMed] [Google Scholar]
- Hogan S, Mould A, Kikutani H, Ramsay A, Foster P. Aeroallergen-induced eosinophilic inflammation, lung damage, and airways hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest. 1997b;99:1329–1339. doi: 10.1172/JCI119292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat J, Starkey M, Kim R, Beagley K, Preston J, Gibson P, et al. Chlamydial respiratory infection during allergen sensitization drives neutrophilic allergic airways disease. J Immunol. 2010a;184:4159–4169. doi: 10.4049/jimmunol.0902287. [DOI] [PubMed] [Google Scholar]
- Horvat J, Starkey M, Kim R, Phipps S, Gibson P, Beagley K, et al. Early-life chlamydial lung infection enhances allergic airways disease through age-dependent differences in immunopathology. J Allergy Clin Immunol. 2010b;125:671–625. doi: 10.1016/j.jaci.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Howarth P, Babu K, Arshad H, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Xiao H, Kleine-Tebbe J, Paciotti G, Marsh D, Lichtenstein L, et al. IL-13 expression at the sites of allergen challenge in patients with asthma. J Immunol. 1995;155:2688–2694. [PubMed] [Google Scholar]
- Issekutz T, Stoltz J, vd Meide P. Lymphocyte recruitment in delayed-type hypersensitivity. The role of IFN-gamma. J Immunol. 1988;140:2989–2993. [PubMed] [Google Scholar]
- Ito K, Herbert C, Siegle J, Vuppusetty C, Hansbro N, Thomas P, et al. Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am J Respir Cell Mol Biol. 2008;39:543–550. doi: 10.1165/rcmb.2008-0028OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram L, Pizzichini M, Cook R, Boulet L, Lemière C, Pizzichini E, et al. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J. 2006;27:483–494. doi: 10.1183/09031936.06.00137704. [DOI] [PubMed] [Google Scholar]
- Kaieda S, Shin K, Nigrovic P, Seki K, Lee R, Stevens R, et al. Synovial fibroblasts promote the expression and granule accumulation of tryptase via interleukin-33 and its receptor ST-2 (IL1RL1) J Biol Chem. 2010;285:21478–21486. doi: 10.1074/jbc.M110.114991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko G, Horvat J, Beagley K, Hansbro P. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasaian MT, Donaldson DD, Tchistiakova L, Marquette K, Tan XY, Ahmed A, et al. Efficacy of IL-13 neutralization in a sheep model of experimental asthma. Am J Respir Cell Mol Biol. 2007;36:368–376. doi: 10.1165/rcmb.2006-0244OC. [DOI] [PubMed] [Google Scholar]
- Kay A, Ying S, Varney V, Gaga M, Durham S, Moqbel R, et al. Messenger RNA expression of the cytokine gene cluster, interleukin 3 (IL-3), IL-4, IL-5, and granulocyte/macrophage colony-stimulating factor, in allergen-induced late-phase cutaneous reactions in atopic subjects. J Exp Med. 1991;173:775–778. doi: 10.1084/jem.173.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med. 2009;179:772–781. doi: 10.1164/rccm.200805-666OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibe A, Inoue H, Fukuyama S, Machida K, Matsumoto K, Koto H, et al. Differential regulation by glucocorticoid of interleukin-13-induced eosinophilia, hyperresponsiveness, and goblet cell hyperplasia in mouse airways. Am J Respir Crit Care Med. 2003;167:50–56. doi: 10.1164/rccm.2110084. [DOI] [PubMed] [Google Scholar]
- Kips J, O'Connor B, Langley S, Woodcock A, Kerstjens H, Postma D, et al. Effect of SCH55700, a humanized anti-human interleukin-5 antibody, in severe persistent asthma: a pilot study. Am J Respir Crit Care Med. 2003;167:1655–1659. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37:2779–2786. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- Kroegel C, Bergmann N, Foerster M, Workalemahu G, Machnik A, Mock B, et al. Interferon-alphacon-1 treatment of three patients with severe glucocorticoid-dependent asthma. Effect on disease control and systemic glucocorticosteroid dose. Respiration. 2006;73:566–570. doi: 10.1159/000088660. [DOI] [PubMed] [Google Scholar]
- Krug N, Madden J, Redington A, Lackie P, Djukanovic R, Schauer U, et al. T-cell cytokine profile evaluated at the single cell level in BAL and blood in allergic asthma. Am J Respir Cell Mol Biol. 1996;14:319–326. doi: 10.1165/ajrcmb.14.4.8600935. [DOI] [PubMed] [Google Scholar]
- Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;32:1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- Kumar R, Herbert C, Webb D, Li L, Foster P. Effects of anticytokine therapy in a mouse model of chronic asthma. Am J Respir Crit Care Med. 2004;170:1043–1048. doi: 10.1164/rccm.200405-681OC. [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- Leckie M, ten Brinke A, Khan J, Diamant Z, O'Connor B, Walls C, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- Lee J, McGarry M, Farmer S, Denzler K, Larson K, Carrigan P, et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Wang W, Baines KJ, Bowden NA, Hansbro PM, Gibson PG, et al. IL-27/IFN-gamma induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J Immunol. 2010;185:4401–4409. doi: 10.4049/jimmunol.1001039. [DOI] [PubMed] [Google Scholar]
- Litonjua A, Sparrow D, Guevarra L, O'Connor G, Weiss S, Tollerud D. Serum interferon-gamma is associated with longitudinal decline in lung function among asthmatic patients: the Normative Aging Study. Ann Allergy Asthma Immunol. 2003;90:422–428. doi: 10.1016/S1081-1206(10)61827-3. [DOI] [PubMed] [Google Scholar]
- Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J Immunol. 1995;154:5411–5417. [PubMed] [Google Scholar]
- Madden K, Urban JJ, Ziltener H, Schrader J, Finkelman F, Katona I. Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J Immunol. 1991;147:1387–1391. [PubMed] [Google Scholar]
- Magnan A, Mély L, Camilla C, Badier M, Montero-Julian F, Guillot C, et al. Assessment of the Th1/Th2 paradigm in whole blood in atopy and asthma. Increased IFN-gamma-producing CD8(+) T cells in asthma. Am J Respir Crit Care Med. 2000;161:1790–1796. doi: 10.1164/ajrccm.161.6.9906130. [DOI] [PubMed] [Google Scholar]
- Mathur M, Herrmann K, Li X, Qin Y, Weinstock J, Elliott D, et al. TRFK-5 reverses established airway eosinophilia but not established hyperresponsiveness in a murine model of chronic asthma. Am J Respir Crit Care Med. 1999;159:580–587. doi: 10.1164/ajrccm.159.2.9712018. [DOI] [PubMed] [Google Scholar]
- Mattes J, Yang M, Siqueira A, Clark K, MacKenzie J, McKenzie A, et al. IL-13 induces airways hyperreactivity independently of the IL-4R alpha chain in the allergic lung. J Immunol. 2001;167:1683–1692. doi: 10.4049/jimmunol.167.3.1683. [DOI] [PubMed] [Google Scholar]
- Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci U S A. 2009;106:18704–18709. doi: 10.1073/pnas.0905063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauser P, Pitman A, Fernandez X, Foran S, Adams G, Kreutner W, et al. Effects of an antibody to interleukin-5 in a monkey model of asthma. Am J Respir Crit Care Med. 1995;152:467–472. doi: 10.1164/ajrccm.152.2.7633694. [DOI] [PubMed] [Google Scholar]
- Morjaria J, Chauhan A, Babu K, Polosa R, Davies D, Holgate S. The role of a soluble TNFalpha receptor fusion protein (etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax. 2008;63:584–591. doi: 10.1136/thx.2007.086314. [DOI] [PubMed] [Google Scholar]
- Mosmann T, Cherwinski H, Bond M, Giedlin M, Coffman R. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- Murdoch J, Lloyd C. Resolution of allergic airway inflammation and airway hyperreactivity is mediated by IL-17-producing {gamma}{delta}T cells. Am J Respir Crit Care Med. 2010;182:464–476. doi: 10.1164/rccm.200911-1775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Sano H, Nishimura T, Yoshida S, Iwamoto I. Role of vascular cell adhesion molecule 1/very late activation antigen 4 and intercellular adhesion molecule 1/lymphocyte function-associated antigen 1 interactions in antigen-induced eosinophil and T cell recruitment into the tissue. J Exp Med. 1994;179:1145–1154. doi: 10.1084/jem.179.4.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Byrne P, Inman M, Adelroth E. Reassessing the Th2 cytokine basis of asthma. Trends Pharmacol Sci. 2004;25:244–248. doi: 10.1016/j.tips.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Oda N, Canelos PB, Essayan DM, Plunkett BA, Myers AC, Huang SK. Interleukin-17F induces pulmonary neutrophilia and amplifies antigen-induced allergic response. Am J Respir Crit Care Med. 2005;171:12–18. doi: 10.1164/rccm.200406-778OC. [DOI] [PubMed] [Google Scholar]
- Owen WJ, Rothenberg M, Silberstein D, Gasson J, Stevens R, Austen K, et al. Regulation of human eosinophil viability, density, and function by granulocyte/macrophage colony-stimulating factor in the presence of 3T3 fibroblasts. J Exp Med. 1987;166:129–141. doi: 10.1084/jem.166.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish W, Luckhurst E. Eosinophilia VI, spontaneous synthesis of chemokinetics, chemotactic, complement receptor-inducing activities for eosinophils by bronchial t lymphocytes of asthmatic-bronchitic patients. Clin Allergy. 1982;12:475–488. doi: 10.1111/j.1365-2222.1982.tb01646.x. [DOI] [PubMed] [Google Scholar]
- Parronchi P, Macchia D, Piccinni M, Biswas P, Simonelli C, Maggi E, et al. Allergen- and bacterial antigen-specific T-cell clones established from atopic donors show a different profile of cytokine production. Proc Natl Acad Sci U S A. 1991;88:4538–4542. doi: 10.1073/pnas.88.10.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–1534. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–5103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol. 2009;123:1047–1054. doi: 10.1016/j.jaci.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz H, Bradley K, Enssle K, Loader J, Larsen G, Gelfand E. Prevention of the development of immediate hypersensitivity and airway hyperresponsiveness following in vivo treatment with soluble IL-4 receptor. Int Arch Allergy Immunol. 1996;109:167–176. doi: 10.1159/000237216. [DOI] [PubMed] [Google Scholar]
- Robinson D, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley A, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- Rochman I, Watanabe N, Arima K, Liu YJ, Leonard WJ. Cutting edge: direct action of thymic stromal lymphopoietin on activated human CD4+ T cells. J Immunol. 2007;178:6720–6724. doi: 10.4049/jimmunol.178.11.6720. [DOI] [PubMed] [Google Scholar]
- Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurich P, Thoma B, Ucer U, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: induction of TNF receptors on human T cells and TNF-alpha-mediated enhancement of T cell responses. J Immunol. 1987;138:1786–1790. [PubMed] [Google Scholar]
- Schleimer R, Sterbinsky S, Kaiser J, Bickel C, Klunk D, Tomioka K, et al. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J Immunol. 1992;148:1086–1092. [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkhuu T, Matthaei KI, Forbes E, Mahalingam S, Hogan SP, Hansbro PM, et al. Mechanism of interleukin-25 (IL-17E)-induced pulmonary inflammation and airways hyper-reactivity. Clin Exp Allergy. 2006;36:1575–1583. doi: 10.1111/j.1365-2222.2006.02595.x. [DOI] [PubMed] [Google Scholar]
- Shi L, Leu SW, Xu F, Zhou X, Yin H, Cai L, et al. Local blockade of TSLP receptor alleviated allergic disease by regulating airway dendritic cells. Clin Immunol. 2008;129:202–210. doi: 10.1016/j.clim.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Shore S, Moore P. Effects of cytokines on contractile and dilator responses of airway smooth muscle. Clin Exp Pharmacol Physiol. 2002;29:859–866. doi: 10.1046/j.1440-1681.2002.03756.x. [DOI] [PubMed] [Google Scholar]
- Simon H, Seelbach H, Ehmann R, Schmitz M. Clinical and immunological effects of low-dose IFN-alpha treatment in patients with corticosteroid-resistant asthma. Allergy. 2003;58:1250–1255. doi: 10.1046/j.1398-9995.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- Singh D, Kane B, Molfino NA, Faggioni R, Roskos L, Woodcock A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10:3. doi: 10.1186/1471-2466-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- Sorg R, Enczmann J, Sorg U, Schneider E, Wernet P. Identification of an alternatively spliced transcript of human interleukin-4 lacking the sequence encoded by exon 2. Exp Hematol. 1993;21:560–563. [PubMed] [Google Scholar]
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- Stämpfli M, Wiley R, Neigh G, Gajewska B, Lei X, Snider D, et al. GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J Clin Invest. 1998;102:1704–1714. doi: 10.1172/JCI4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt V, Bothur E, Klein M, Lingnau K, Reuter S, Grebe N, et al. Interferon-Regulatory Factor 4 Is Essential for the Developmental Program of T Helper 9 Cells. Immunity. 2010;33:192–202. doi: 10.1016/j.immuni.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Stock P, Lombardi V, Kohlrautz V, Akbari O. Induction of airway hyperreactivity by IL-25 is dependent on a subset of invariant NKT cells expressing IL-17RB. J Immunol. 2009;182:5116–5122. doi: 10.4049/jimmunol.0804213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DE, Ferris M, Pociask D, Brody AR. Tumor necrosis factor-alpha induces transforming growth factor-beta1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am J Respir Cell Mol Biol. 2005;32:342–349. doi: 10.1165/rcmb.2004-0288OC. [DOI] [PubMed] [Google Scholar]
- Tamachi T, Maezawa Y, Ikeda K, Kagami S, Hatano M, Seto Y, et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J Allergy Clin Immunol. 2006;118:606–614. doi: 10.1016/j.jaci.2006.04.051. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Komai M, Nagao K, Ishizaki M, Kajiwara D, Takatsu K, et al. Role of interleukin-5 and eosinophils in allergen-induced airway remodeling in mice. Am J Respir Cell Mol Biol. 2004;31:62–68. doi: 10.1165/rcmb.2003-0305OC. [DOI] [PubMed] [Google Scholar]
- Temann U, Prasad B, Gallup M, Basbaum C, Ho S, Flavell R, et al. A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion. Am J Respir Cell Mol Biol. 1997;16:471–478. doi: 10.1165/ajrcmb.16.4.9115759. [DOI] [PubMed] [Google Scholar]
- Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–1320. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temelkovski J, Hogan S, Shepherd D, Foster P, Kumar R. An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–856. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper R, Levinson D, Stanger B, Campos-Torres J, Abbas A, Leder P. IL-4 induces allergic-like inflammatory disease and alters T cell development in transgenic mice. Cell. 1990;62:457–467. doi: 10.1016/0092-8674(90)90011-3. [DOI] [PubMed] [Google Scholar]
- Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, et al. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med. 2008;205:2727–2733. doi: 10.1084/jem.20080698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn A, Hansbro P. Harnessing regulatory T cells to suppress asthma: from potential to therapy. Am J Respir Cell Mol Biol. 2010;43:511–519. doi: 10.1165/rcmb.2009-0342TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkinson A, Tepper J, Morton M, Bowden A, Stevens L, Harris P, et al. Inhaled vs subcutaneous effects of a dual IL-4/IL-13 antagonist in a monkey model of asthma. Allergy. 2010;65:69–77. doi: 10.1111/j.1398-9995.2009.02156.x. [DOI] [PubMed] [Google Scholar]
- Townsend J, Fallon G, Matthews J, Smith P, Jolin E, McKenzie N. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000;13:573–583. doi: 10.1016/s1074-7613(00)00056-x. [DOI] [PubMed] [Google Scholar]
- Trifilieff A, Fujitani Y, Coyle AJ, Kopf M, Bertrand C. IL-5 deficiency abolishes aspects of airway remodelling in a murine model of lung inflammation. Clin Exp Allergy. 2001;31:934–942. doi: 10.1046/j.1365-2222.2001.01084.x. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout A, Fattah D, Van Ark I, Hofman G, Buckley T, Nijkamp F. Eosinophil infiltration precedes development of airway hyperreactivity and mucosal exudation after intranasal administration of interleukin-5 to mice. J Allergy Clin Immunol. 1995;96:104–112. doi: 10.1016/s0091-6749(95)70039-0. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- Walter D, McIntire J, Berry G, McKenzie A, Donaldson D, DeKruyff R, et al. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J Immunol. 2001;167:4668–4675. doi: 10.4049/jimmunol.167.8.4668. [DOI] [PubMed] [Google Scholar]
- Wang Y, McCusker CT. Interleukin-13-dependent bronchial hyper-responsiveness following isolated upper-airway allergen challenge in a murine model of allergic rhinitis and asthma. Clin Exp Allergy. 2005;35:1104–1111. doi: 10.1111/j.1365-2222.2005.02301.x. [DOI] [PubMed] [Google Scholar]
- Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li J, Foster P, Hansbro P, Yang M. Potential therapeutic targets for steroid-resistant asthma. Curr Drug Targets. 2010;11:957–970. doi: 10.2174/138945010791591412. [DOI] [PubMed] [Google Scholar]
- Wark P, Johnston S, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DC, McKenzie AN, Koskinen AM, Yang M, Mattes J, Foster PS. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J Immunol. 2000;165:108–113. doi: 10.4049/jimmunol.165.1.108. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Wilbraham D, Fuller R, Getz E, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1396–1398. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Barnes P, Bleecker E, Bousquet J, Busse W, Dahlén S, et al. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179:549–558. doi: 10.1164/rccm.200809-1512OC. [DOI] [PubMed] [Google Scholar]
- Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:43–71. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben T, Karp C, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Braciak T, Ohkawara Y, Sallenave J, Foley R, Sime P, et al. Gene transfer for cytokine functional studies in the lung: the multifunctional role of GM-CSF in pulmonary inflammation. J Leukoc Biol. 1996;59:481–488. doi: 10.1002/jlb.59.4.481. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Tashimo H, Ishida H, Kaneko F, Nakano J, Kato H, et al. Attenuation of airway hyperresponsiveness in a murine asthma model by neutralization of granulocyte-macrophage colony-stimulating factor (GM-CSF) Cell Immunol. 2002;219:92–97. doi: 10.1016/s0008-8749(02)00565-8. [DOI] [PubMed] [Google Scholar]
- Yang M, Hogan S, Mahalingam S, Pope S, Zimmermann N, Fulkerson P, et al. Eotaxin-2 and IL-5 cooperate in the lung to regulate IL-13 production and airway eosinophilia and hyperreactivity. J Allergy Clin Immunol. 2003;112:935–943. doi: 10.1016/j.jaci.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, et al. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther. 2005;313:8–15. doi: 10.1124/jpet.104.076133. [DOI] [PubMed] [Google Scholar]
- Yang G, Volk A, Petley T, Emmell E, Giles-Komar J, Shang X, et al. Anti-IL-13 monoclonal antibody inhibits airway hyperresponsiveness, inflammation and airway remodeling. Cytokine. 2004;28:224–232. doi: 10.1016/j.cyto.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Yasunaga S, Yuyama N, Arima K, Tanaka H, Toda S, Maeda M, et al. The negative-feedback regulation of the IL-13 signal by the IL-13 receptor alpha2 chain in bronchial epithelial cells. Cytokine. 2003;24:293–303. doi: 10.1016/j.cyto.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- Zhou L, Tedder T. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci U S A. 1996;93:2588–2592. doi: 10.1073/pnas.93.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Homer R, Wang Z, Chen Q, Geba G, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.