

Abstract
Cystic fibrosis (CF) is a lethal, recessive, genetic disease affecting approximately 1 in 2500 live births among Caucasians. The CF gene codes for a cAMP/PKA-dependent, ATP-requiring, membrane chloride ion channel, generally found in the apical membranes of many secreting epithelia and known as CFTR (cystic fibrosis transmembrane conductance regulator). There are currently over 1700 known mutations affecting CFTR, many of which give rise to a disease phenotype. Around 75% of CF alleles contain the ΔF508 mutation in which a triplet codon has been lost, leading to a missing phenylalanine at position 508 in the protein. This altered protein fails to be trafficked to the correct location in the cell and is generally destroyed by the proteasome. The small amount that does reach the correct location functions poorly. Clearly the cohort of patients with at least one ΔF508 allele are a major target for therapeutic intervention. It is now over two decades since the CF gene was discovered and during this time the properties of CFTR have been intensely investigated. At long last there appears to be progress with the pharmaco-therapeutic approach. Ongoing clinical trials have produced fascinating results in which clinical benefit appears to have been achieved. To arrive at this point ingenious ways have been devised to screen very large chemical libraries for one of two properties: (i) agents promoting trafficking of mutant CFTR to, and insertion into the membrane, and known as correctors or (ii) agents which activate appropriately located mutant CFTR, known as potentiators. The best compounds emerging from these programmes are then used as chemical scaffolds to synthesize other compounds with appropriate pharmaceutical properties, hopefully with their pharmacological activity maintained or even enhanced. In summary, this approach attempts to make the mutant CFTR function in place of the real CFTR. A major function of CFTR in healthy airways is to maintain an adequate airway surface liquid (ASL) layer. In CF the position is further confounded since epithelial sodium channels (ENaC) are no longer regulated and transport salt and water out of the airways to exacerbate the lack of ASL. Thus an additional possibility for treatment of CF is to use agents that inhibit ENaC either alone or as adjuncts to CFTR correctors and/or potentiators. Yet a further way in which a pharmacological approach to CF can be considered is to recruit alternative chloride channels, such as calcium-activated chloride channel (CaCC), to act as surrogates for CFTR. A number of P2Y2 receptor agonists have been investigated that operate by increasing Ca2+i which in turn activates CaCC. Some of these compounds are currently in clinical trials. The knowledge base surrounding the structure and function of CFTR that has accumulated in the last 20 years is impressive. Translational research feeding from this is now yielding compounds that provide real prospects for a pharmacotherapy for this disease.
LINKED ARTICLES
This article is part of a themed issue on Respiratory Pharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-1
Keywords: epithelial chloride ion channels, epithelial sodium ion channels, airway surface liquid, CFTR, high throughput screening
Characteristics of cystic fibrosis airway disease
It is remarkable that the airways of most people are clean and sterile, even though the air we breathe contains viruses and bacteria, along with many sorts of particulate matter. Just how the airways can remain clean and sterile under these conditions is still not completely understood, but much new information has accrued in recent years as progress is made towards the complete picture of the defence mechanisms. It is also known that in cystic fibrosis (CF) airway defences are severely compromised. In CF, problems affecting the airways are the major causes of morbidity and mortality, so the major therapeutic challenge is directed to improving airway function. It is now 21 years since the CF gene was discovered (Riordan et al., 1989) and around 1700 different mutations are now known. The CF gene codes for a membrane protein, the cystic fibrosis transmembrane conductance regulator (CFTR; channel nomenclature follows Alexander et al., 2009) that acts as an anion channel, regulated by cAMP-dependent protein kinase A (PKA) and requiring ATP to cycle between open and closed states (Tsai et al., 2010). Thus in CF epithelial anion transport fails, causing functional disruption in a number of organs besides the airways, including the alimentary canal, exocrine pancreas, gall bladder, reproductive tracts and sweat ducts. This review is concerned only with airway problems and how these may be alleviated with a pharmaco-therapeutic approach.
Airway surfaces are covered with a thin layer of fluid, the airway surface liquid (ASL) consisting of a periciliary liquid or sol layer with a viscous gel layer above. Particulate matter in inhaled air is trapped by the mucus and moved towards the pharynx by ciliary action, the so-called mucociliary escalator, where it is either swallowed or expectorated. In contrast, the reduced volume hypothesis (Matsui et al., 1998) proposes that, in CF, the sol layer all but disappears and the interaction of the gel phase with the cilia renders them ineffective, leading to stasis, and providing a rich environment for infection, notably by Pseudomonas aeruginosa or Burkholderia cepacia. These organisms are notoriously difficult to eradicate, yet are innocuous in normal individuals (see Figure 1 for details).
Figure 1.
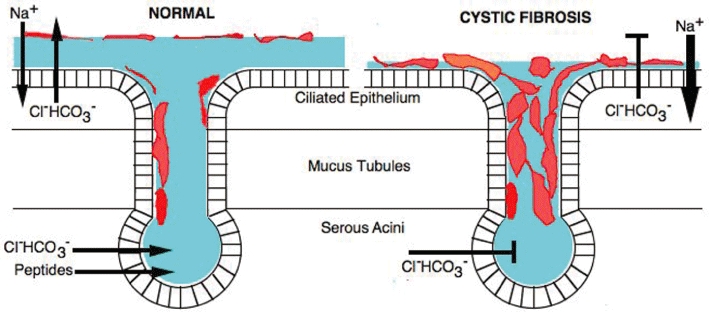
This Figure summarizes the main differences in the transport of salts (and in consequence osmotic fluid movement) in healthy and cystic fibrosis (CF) airways. The diagram represents the processes at both the surface-ciliated epithelium and in submucosal glands (containing the secretory serous acini and mucus tubules). In normal airways, chloride and bicarbonate ions are secreted into the airway surface liquid (ASL, shown in blue) by an electrogenic process, while at the same time sodium ions are absorbed from the ASL, also by an electrogenic mechanism. Of course, counterions will move passively as a result of these processes. Whether fluid (water) moves from the ASL into the submucosal space or in the opposite direction will depend on the overall osmotic balance established by the two transport processes. The acini of the submucosal glands also secrete anions, probably by a process similar to that of the surface epithelium. The secretion also contains a variety of antimicrobial peptides, concerned with defence mechanisms and mucins (shown in red) are added as the fluid moves onto the airway surface. The balance of these processes is such that they maintain an ASL with a well-defined sol layer in contact with the epithelial surface and a viscous gel layer above, an ideal way in which to trap particulate matter in mucus and move it, by ciliary action, towards the exterior. In CF neither the surface epithelium nor the submucosal glands are capable of generating anion secretion because of the lack of functioning cystic fibrosis transmembrane conductance regulator, while sodium absorption is enhanced. The osmotic balance is altered such that fluid leaves the airway surface and moves into the submucosal space. In consequence, submucosal gland ducts are blocked by mucus, which also settles down on the cilia making them unable to function and bacterial infection establishes itself, aggravated by the loss of the antimicrobial peptides.
Both the surface epithelium and the submucosal glands of airways are capable of generating ASL, which not only contains ions but also glycoproteins, mucins, lactoferrin, defensins, lysozyme, IgA, antimicrobial surfactant proteins and secretory leukoprotease inhibitor, all part of the antimicrobial and immune defence mechanism.
The airway surface epithelium carries out both electrogenic sodium absorption and electrogenic, CFTR-dependent, anion secretion, in turn affecting the osmotic gradient driving the passive movement of fluid into or out of the airways.
There have been many attempts to measure depth, composition and pH of the ASL in airway tissues and cultures, with a few attempts in vivo in mice, with very variable results (see Verkman et al., 2003). In general the consensus value for depth is 5–10 µm. Nevertheless it is clear, particularly from epithelial culture models, that epithelia have the innate ability to maintain the appropriate depth of ASL by modulating ion transport (Tarran et al., 2006).
Measurements suggest that both proximal and distal airways are absorptive in the basal condition (Ballard and Taylor, 1994; Ballard et al., 1999; Inglis and Wilson, 2005) indicating that airway submucosal glands are an important source of ASL (see Figure 2). Apparently submucosal glands are not capable of sodium absorption, although the glands express the epithelial sodium channel (ENaC), possibly because of the presence of Kunitz type and non-Kunitz type serine protease inhibitors, preventing the proteolytic activation of ENaC (Joo et al., 2006). CFTR-dependent gland secretion fails in CF, yet non-CFTR-dependent signals, such as those delivered by the vagal cholinergic input (Wine, 2007) can produce some secretion but with a much increased viscosity (Jayaraman et al., 2001). It is not surprising therefore that occlusion by mucus of submucosal gland ducts is an early characteristic of CF lung disease.
Figure 2.
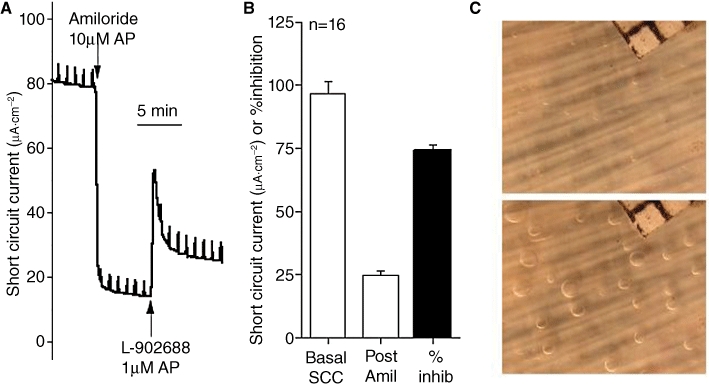
The different components of solute and fluid movement in airway tissue. All the data are derived from experiments on ovine tracheal tissue. (A) shows a recording of short circuit current (SCC), a measure of the total electrogenic transport of cations and anions across the epithelium. As cations are absorbed from the apical surface while anions are secreted in the opposite direction, these two currents will be additive. Sodium absorption can be completely eliminated by adding amiloride (Amil) to the apical surface and shows under the conditions of the experiment that it accounts for ∼75% of the total current. The remaining current, due largely to anion secretion, can be further enhanced, in this instance by the EP4 receptor agonist L-902688. Data in (B) shows that pattern seen in (A) is confirmed. These results suggest that in the basal state the airway surface epithelia are absorptive. However, it is to be remembered that the in vivo condition is very different. First, under SCC conditions, the apical surface is bathed in a solution of infinite volume, compared to the airway surface liquid (ASL), and second, the airway epithelium is removed from the humoral and neural mechanisms present in vivo. However, there is a second source of fluid that contributes to the ASL and derived from the submucosal glands. To measure secretion from individual glands the method first described by Joo et al. 2001 was used. A small area of tracheal mucosa was mounted in a special chamber and bathed on the basolateral side with Krebs solution at 37°C and superfused with 95%O2/5%CO2. The apical surface, after cleaning and drying, was covered with a thin layer of water saturated mineral oil. Secretion from individual glands collected as spherical droplets at the mouth of the glands. A sequence of photographs of the airway surface at timed intervals allowed the secretion rate to be calculated using ImageJ software. (C) shows two views of the epithelial surface (10 mm2, corner of grid with 0.5 mm squares is shown) of an ovine trachea before (upper) and a few minutes after (lower) carbachol had been added to the basolateral fluid. Note the appearance of spherical droplets at the surface opening of each gland.
While CF is a lethal genetic disease due to a mutation in a gene coding for a chloride channel, the sodium channel, ENaC, is also a key player in the condition. ENaC is normally down-regulated by CFTR, and when this regulation is lost, as in CF, electrogenic sodium absorptive processes are enhanced at the expense of the ASL. ENaC is a heteromultimeric membrane ion channel, usually composed of two α, one β and one γ subunits, but other configurations are possible sometimes with a δ subunit (Kellenberger and Schild, 2002). More recent evidence suggests ENaC may also form α, β, γ trimers.
The down-regulation of ENaC by CFTR has been confirmed in many different systems and in many laboratories, but the mechanisms are not yet clear. A decrease in channel open probability (Po) without a change in the surface expression for ENaC has been proposed (Konstas et al., 2003), while others propose that changes in intracellular chloride ion concentration are responsible (Adam et al., 2005). Other evidence suggests there is a direct interaction between ENaC and CFTR that is lost in CF (Berdiev et al., 2009). These three mechanisms are not necessarily incompatible. For further opinions on the mechanism(s) of CFTR/ENaC interactions see Berdiev et al. (2009) and Donaldson and Boucher (2007). The importance of CFTR/ENaC interactions in CF was emphasized when transgenic mice over-expressing ENaC channels were produced (Mall et al., 2004). While the CF mouse is not a particularly good model for human CF, those over-expressing ENaC showed ASL depletion, delayed mucus transport and spontaneous lung disease.
Yet another way in which ENaC can affect ASL is through the presence of local serine proteases that activate nascent (‘silent’) channels in the membrane converting them into functional, actively gating, channels. A group of membrane bound serine proteases called channel activating proteins, CAP-1 (prostasin), CAP-2 (TMPRSS4) and CAP-3 (matriptase) have been shown to activate ENaC by four- to sevenfold, apparently by an increase in channel open time, rather than any change in channel number (Vuagniaux et al., 2002). Anti-serine proteases such as α1-antitrypsin, found in plasma and lung epithelial fluid, can modify the effects of CAPs on sodium absorption so that sodium absorption depends on the protease–antiprotease balance (Lazrak et al., 2009). Further information about the regulation of ENaC by peptidases can be found in two reviews (Planes and Caughey, 2007; Rossier and Stutts, 2009).
The diagram in Figure 1 summarizes the major differences between CF and non-CF airway transport processes. In non-CF airways, anions are secreted by both the surface epithelium and the submucosal glands. While glandular secretion is shown confined to the serous acini it is possible this process continues into the mucus tubules. Note too that secretion of bicarbonate alongside chloride ions is shown; this is not to imply that HCO3- necessarily passes through the CFTR channel, but the same end result could be obtained by CFTR coexisting alongside a Cl-/HCO3- exchanger. In contrast, in CF airways, anion secretion fails and sodium absorption is enhanced resulting in a failure of mucociliary clearance and the loss of the antibacterial properties of the glandular secretions.
Strategies for treatment of CF airway disease
Following on from the discovery of the CF gene there was some expectation that gene therapy would soon provide the answer, especially for treating the airways, since topologically they are easily accessible. A variety of clinical trials using different gene transfer agents, such as adenoviral vectors or cationic liposomes, were conducted. In some of these trials ‘proof of principle’ was achieved, that is, transgene expression was detected or some biophysical measurement, such as correction of nasal potential difference (PD), was measured following the procedure. However, no clinical benefit has thus far been recorded. Access of the transfecting agents to the surface epithelial cells is hampered by the overlying mucus while transfection of the submucosal glands poses special problems. Work continues to overcome these technical difficulties, as gene therapy may yet provide the best solution, but in recent years the focus has turned towards pharmaco-therapy as likely to yield more immediate results and help reduce the burden of the disease without effecting a cure.
The emphasis on drug therapy for CF is to develop agents that normalize the CF cell's physiology. There are some obvious examples, such as reducing the excessive sodium transporting capability towards normal. Another possibility is to press into service alternative chloride channels that are present in CF cells to act as surrogates for CFTR. Yet another possibility is to make use of the mutant CFTR and provide conditions in which it can function. Mention here must be made about the most common CF mutation, namely ΔF508, the mutation found in 75% of CF alleles. Here the gene has lost a triplet codon so that the 1480 amino acid CFTR is missing phenylalanine at position 508. This protein is apparently more difficult to fold and the cellular quality control machinery directs it to destruction in the proteasome. However, a small fraction of ΔF508 CFTR is inserted into the cell membrane and shows function, but with much reduced channel open times. Humans carrying only one CF gene do not have symptoms of CF, and this together with other evidence that indicates as little as 10% normal CFTR may be sufficient, makes ΔF508 a major target for drug therapy, especially since it would benefit a majority of patients. It has been known for many years that mammalian cells expressing ΔF508 CFTR revert to wild type when the temperature is lowered (Denning et al., 1992). Obviously a chemical chaperone that could achieve the same result would be of particular interest for patients with a ΔF508 mutation. However, there is an extremely simple way to increase the depth of the ASL by drawing water onto the surface of the airways using osmosis. This can be achieved by inhalation of a small volume of hypertonic fluid or even by insufflating a small amount of water soluble powder. A treatment such as this will provide only temporary relief and patients may find it socially inconvenient; nevertheless, it is currently popular. In the following part of this review, I discuss the strategies available for treating CF, in what I suggest is the order of increasing long-term importance.
Hypertonic saline and mannitol
Inhaled hypertonic saline has become a popular treatment for CF and apparently is well tolerated and effective provided treatment is prolonged. Water is osmotically drawn onto the apical airway surface, thus maintaining the ASL layer and improving mucociliary clearance. There have been a number of clinical trials where improvements in lung function in CF patients have been recorded, for example following inhalation of 4 mL of 7% saline, twice daily for 48 weeks (Elkins et al., 2006). In an attempt to produce a greater osmotic gradient that dissipates more slowly attention turned to mannitol. Inhalation of dry mannitol powder (420 mg), twice daily for 2 weeks improved lung function in CF patients and was well tolerated (Jaques et al., 2008).
The route taken by water molecules moving into the apical space is unclear. In model systems, evidence was found that hypertonic saline increases tight junction permeability in airway epithelium (Hogman et al., 2002). In airway submucosal glands, the apical membranes of the gland epithelial cells are the rate-limiting barrier to water movement, largely governed by the density of aquaporin-5 channels present (Song and Verkman, 2001).
Reducing ENaC activity in CF
The highly selective ENaC blocking drug amiloride was used in a clinical trial 25 years ago in an attempt to prevent volume depletion of the ASL in CF patients (Kohler et al., 1986; Knowles et al., 1990). Amiloride sprayed into the airways of patients was intended to block the apical ENaC channels and improve mucociliary clearance. A number of further trials were carried out over the next decade but no therapeutic improvement was established. The reason appeared to be the low potency and the rapid clearance of amiloride from the lung. The benzyl derivative of amiloride (benzamil) was shown to be an order of magnitude more effective than the parent compound (Cuthbert, 1976; Cuthbert and Fanelli, 1978) and had a longer duration of effect in two clinical trials using nasal PD measurements as the response (Hofmann et al., 1998; Rodgers and Knox, 1999). It was suggested that benzamil might be useful in the long-term treatment of the defect in the lungs of CF patients. In experiments in sheep, benzamil increased mucociliary clearance and raised tracheal mucus velocity (Hirsh et al., 2004) but not significantly more than with amiloride. Apparently more rapid absorption of benzamil offset its greater potency. More recently even more potent amiloride-like drugs have been synthesized and evaluated in model systems (Hirsh et al., 2006; 2008;). The lead compound, 552-02, is two orders of magnitude more effective than amiloride and with a much slower off rate. Four hours after aerosol administration to sheep, 552-02 gave a persistent increase in mucociliary clearance. Results from clinical trials with this new compound are awaited.
As short duration of action has been a problem with the pyrazine carboxamides, such as amiloride, attention turned to other ways of inhibiting ENaC. BAY 39-9437 is a 170 amino acid recombinant bikunin-like serine protease inhibitor with an EC50 of 25 nM for inhibition of ENaC function in cultured monolayers of both wild-type and CF human bronchial epithelia (Bridges et al., 2001). Camostat is a low MW inhibitor of channel activating proteases and gave a potent and prolonged attenuation of ENaC function (EC50 of 50 nM). When aerosolized into the airways of conscious sheep it increased mucociliary clearance for at least 5 h (Coote et al., 2009). Clinical trials with channel protease inhibitors are progressing.
Recruiting alternate chloride channels to act as surrogates for CFTR
Another method of preventing volume depletion is to use alternate chloride channels to act as surrogates for CFTR, provided they are appropriately situated in the epithelial cells of the airways. Preferably the channels should be located apically so that treatment by aerosol is possible, but it is essential that the signal transduction pathways are independent of cAMP metabolism. It was established that both purine (ATP) and pyrimidine (UTP) triphosphate nucleotides met the second criterion and were able to activate P2Y2 receptors located on the apical membranes of airway epithelial cells. When applied to either normal or CF airway epithelia, these agents increase Cl- secretion via increases in Ca2+i that secondarily activate calcium-activated chloride channels (CaCC) present in the apical membrane (Knowles et al., 1991). Although both ATP and UTP were equipotent, UTP analogues were chosen for development as metabolism of ATP by ectonucleotidases give rise to ADP, AMP and adenosine, all of which can cause bronchoconstriction in some circumstances. A further bonus of developing P2Y2 agonists for potential clinical use was the realization that they also inhibit electrogenic sodium absorption in both wild-type and ΔF508 CFTR expressing human bronchial epithelia (Devor and Pilewski, 1999). A second generation dinucleotide P2Y2 receptor agonist that is highly resistant to breakdown by ectonucleotidases, due to the replacement of one of the two uridine groups with deoxycytidine, has been developed, INS37217 (Yerxa et al., 2002). In various model systems, as well as increasing chloride and fluid secretion, INS37217 increased cilia beat frequency and mucin release and transport. In a 28-day Phase 2 clinical trial, Denufusol tetrasodium (INS37217) significantly improved lung function in patients with mild CF lung disease when applied three times daily by nebulizer (Deterding et al., 2007). Results from Phase 3 clinical trials are expected in early 2011.
The nonapeptide antibiotic duramycin (Cloutier et al., 1990) (Moli-1901) is in phase 2 clinical trial for CF in Europe. It is claimed that it causes calcium release from intracellular stores and stimulates calcium influx (Cloutier et al., 1993) thus activating CaCC. However, others have shown that duramycin formed non-specific ion channels in both artificial and biological membranes (Roberts et al., 1991; Sheth et al., 1992). Coalescence of initially small channels into larger ones with increasing duramycin concentration is thought responsible for the loss of cellular integrity at all but low duramycin concentrations.
The chloride channel type 2 (ClC-2) is widely distributed in epithelia and can contribute to native chloride secretion (Mohammad-Panah et al., 2001). Lubiprostone, a putative ClC-2 channel opener, apparently caused non-PKA-dependent chloride secretion in T84 monolayers (Cuppoletti et al., 2004). Lubiprostone also increased chloride secretion in airways from sheep, pigs and humans (Joo et al., 2009) but recent studies have shown that in epithelial systems from both the airways and intestine a major target for lubiprostone is the prostanoid EP4 receptor (Bijvelds et al., 2009; Cuthbert, 2010) coupled to Gs and cAMP generation and CFTR activation.
Recruiting mutant CFTR into normal function
Types of mutation
There are now over 1700 mutations known for the CF gene, so it is no surprise that attempts are made to classify them on a simple basis in relation to the fate of the CFTR protein. Six different classes are described. Class 1 mutations cause total loss of the protein, due either to nonsense mutations or premature stop codons, an example beingW1282X. This type of mutation is found in 5% of CF chromosomes. Aminoglycoside antibiotics, such as G-418 and gentamicin, allow translational read through allowing it to proceed to the end of the transcript (Howard et al., 1996), and correction of nasal PDs with gentamicin has been shown in CF patients with stop mutations (Wilschanski et al., 2003).
Class 2 mutations affect maturation and transport of CFTR, so that little mutated protein reaches the membrane. ΔF508 CFTR is an important and common example of this type, 75% of CF chromosomes carrying this mutation. Class 3 mutations produce CFTR in which the regulation of the ion channel is disturbed, as in G551D, a relatively common mutation accounting for 3% of CF mutations. Class 4 mutations allow the CFTR protein to be correctly translocated to the cell membrane but with reduced ion fluxes and altered selectivity because of mutations affecting the pore region of the channel, as in R117H. In addition, there are mutations that affect mRNA stability (Class 5) and CFTR stability (Class 6). Sometimes it is difficult to make a clear distinction between two or more of the classes of mutation, for example ΔF508 CFTR is poorly transported to the membrane and functions poorly compared to CFTR.
Correctors and potentiators
In recent times a new terminology has been introduced that is relevant, especially to ΔF508 CFTR, namely ‘correctors’ and ‘potentiators’. Correctors are compounds that specifically promote trafficking of the mutant CFTR to the membrane, whereas potentiators promote activity of mutant CFTR once delivered to the appropriate location in the cell. In earlier literature, the term ‘opener’ was usually applied to potentiators. However, potentiator is the more precise term since generally the channels need to be ‘primed’ before the potentiator can activate the channel. In the experimental situation this might be achieved by adding forskolin, while in the clinical situation priming would result from the normal physiological inputs, arising from neuronal and humoral mechanisms. Indeed an opener per se may not be useful, leading to excessive activity.
With mutations such as ΔF508 CFTR both trafficking and activity are poor so that both a corrector and potentiator would be required, unless both properties can be incorporated in a single molecule.
High throughput screening
In the last decade literally many thousands of compounds and chemical libraries have been interrogated. This work could not have been achieved without the development of automated screening instrumentation. High throughput screens looking for compounds of interest were developed, particularly in the laboratories of Alan Verkman in San Francisco and Luis Galietta in Genoa (Ma et al., 2002). The basic principles are shown in Figure 3. Epithelial cells (usually Fischer rat thyroid cells) expressing ΔF508 CFTR (or CFTR or G551D CFTR) together with the yellow fluorescent protein YP-H148Q that fluoresces in the presence of Cl- ions but not with I- ions, are grown on 100-well plates. Culture at low temperature (27°C) traffics ΔF808 CFTR to the membrane, where test compounds are added automatically, which may or may not lead to increases of channel opening. Rapidly changing the apical Cl- containing solution to one substituted with I- allows rapid changes in fluorescence to be measured, following Cl-/I- exchange. This example tests specifically for CFTR potentiators. Clearly the methodology can be adapted to other purposes, such as finding correctors by omitting the culture at low temperature stage. Figure 3 indicates the rapid changes in fluorescence that occur when I- replaces Cl- inside the cells. Using a weakly acting standard compound, such as genistein, allows more potent compounds to be selected. It is surprising just how many active compounds have been discovered and a few are currently in clinical trials.
Figure 3.
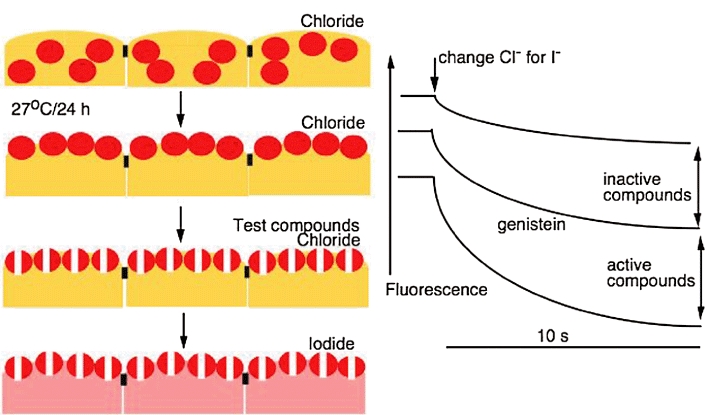
The principles of high throughput screening for cystic fibrosis transmembrane conductance regulator (CFTR) potentiators. Epithelial cells expressing ΔF508 CFTR (shown in red) and also a halide sensitive yellow fluorescent protein (yellow cells), are grown in 100-well plates. As the screen is for potentiator molecules, the CFTR has first to be assisted to the membrane. By incubation at 27°C for 24 h transfer is achieved. Test compounds are added to the wells and here are shown to open channels. This is measured by changing chloride for iodide in the bathing solution. A rapid loss of fluorescence is indicative of a strong potentiator. Note in the absence of compounds there will be some loss of fluorescence. Also by including a standard compound, such as genistein, weaker compounds can be eliminated. The method described is capable of adaptation. For example, if the search is for CFTR correctors, then the 27°C stage would be eliminated. Also a variety of different cell lines can be used and in some instances a primer may be required, such as a low concentration of forskolin.
A brief survey of the search for compounds that increase the trafficking of ΔF508 CFTR to the membrane (correctors), or activate Δ508 CFTR (potentiators) once it is correctly located, or both of these is given below.
The chance discovery of the benzimidazolone, NS004 (Gribkoff et al., 1994) and the tyrosine kinase inhibitor, genistein (Illek et al., 1995) showed that it was possible to directly activate CFTR with chemical agents. Gribkoff et al. (1994) claimed that NS004 was the first ‘activator’ of CFTR and ΔF508 CFTR provided prior exposure to forskolin had been made. They suggested that the PKA phosphorylated form of the CFTR Cl channel was the target.
Screening of a small library of benzoquinolizinium compounds showed that some of these not only activated CFTR, ΔF508 CFTR and G551D but also redirected the mutant forms to the correct cell location; that is, they were both correctors and potentiators (Becq et al., 1999; Dormer et al., 2001). Two hundred and twenty three compounds based largely on genistein were examined and one, UCCF-029, was found to be the most potent with a Kd of 5 µM for activating CFTR (Galietta et al., 2001). Using UCCF-029 as a structural guide other similar compounds were investigated resulting in UCCF-339 with a Kd of 1.7 µM (Springsteel et al., 2003). A further 60 000 compounds were examined by Ma et al. 2002 that showed potentiators with Kd values of 200 nM for activation of CFTR were possible. Focusing upon potentiators of ΔF508 CFTR, Yang et al. (2003) found six novel chemical classes of high affinity potentiators amongst 100 000 diverse small molecules.
Another useful approach is to screen compounds already approved for medicinal use as this reduces the time normally required to study toxicity, bioavailability and the like and make them available to CF patients. For example, many of the antihypertensive 1,4 dihydropyridines, like nifedipine, nicardipine and nimodipine activated CFTR, ΔF508 CFTR and G551D (Pedemonte et al., 2005a). Using epithelial cells expressing ΔF508 CFTR and the yellow fluorescent protein and not subjecting them to the low temperature regime means the ΔF protein remains mainly in the cell. Using 150 000 chemically diverse compounds a study was made to examine how many compounds produced a greater apical chloride current than low-temperature rescue. A group of bisaminomethylbithiazoles corrected ΔF508/ΔF508 expressed in a human bronchial epithelial (HBE) cell line but not the temperature-sensitive mutant P547H-CFTR (Pedemonte et al., 2005b).
Those who might wish to learn more about what has been achieved through high throughput screening for CF drugs and how, surprisingly, academia rather than industry led the way may wish to consult Conese et al. (2009) and Verkman (2004).
It must be remembered that compounds emerging from these screening programmes, some with very high affinities, have generally not been examined for other pharmacological properties, some of which may be synergistic or antagonistic to the aim of the screening procedure. For example, an excellent ΔF508 corrector with CFTR channel blocking activity is of no use. Chloro-benzo[F]isoquinoline, a potentiator of CFTR and ΔF508 CFTR, also activates KCNN4 channels in human airway epithelial cells, possibly a useful asset since activating basolateral K+ channels will increase the potential gradient for Cl flux through potentiated ΔF508 CFTR channels (Szkotak et al., 2004; Murthy et al., 2005).
From laboratory to clinic
At this stage the reader may feel that if all these thousands of miscellaneous chemicals have been screened for useful activity in CF why some are not progressing into development for clinical use, or at least into clinical trials?
At least two molecules are now in development, VX-770 (Figure 4) and VX-809. The former is now entering Phase 3, while the latter is in Phase 2. Both compounds were produced by a consortium of the Vertex Company with the American Cystic Fibrosis Foundation. Using high throughput screening the consortium examined 228 000 compounds from which a lead scaffold was selected for medicinal chemistry optimization. VX-770 was chosen on its ability to potentiate several forms of CFTR, including CFTR, G551D and R117H. While VX-770 is a potentiator VX-809 is a corrector.
Figure 4.
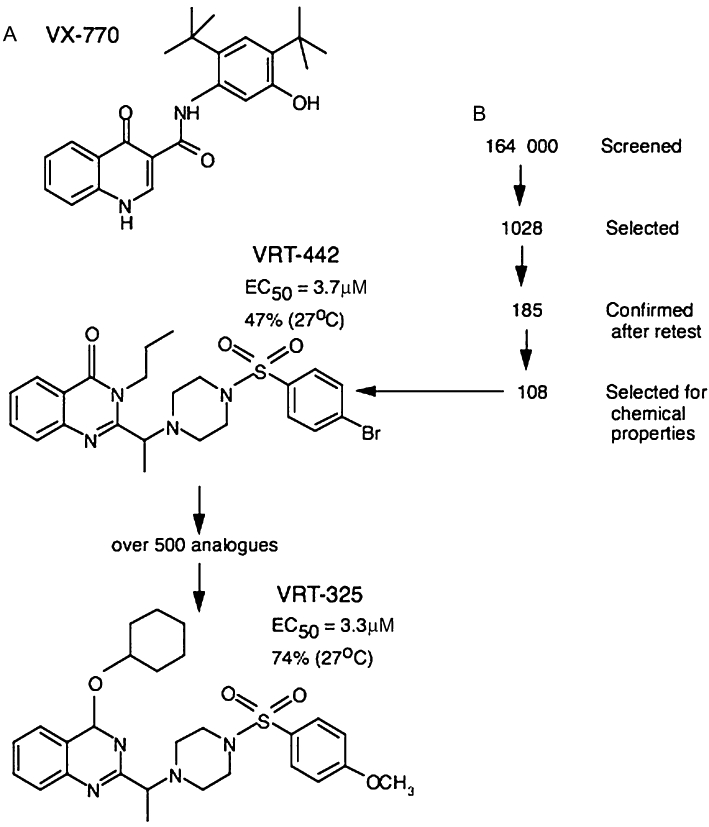
(A) shows the chemical structure of the cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770, currently in Phase 3 clinical trial. (B) illustrates the path followed to obtain the CFTR corrector compound VRT-325. Note the original library had 164 000 compounds of which 1028 were selected through high throughput screening, but only 18% survived retesting. One of the better compounds, VRT-442 had an EC50 in the µM range and gave a correction equivalent to 47% of that achievable by temperature lowering to27°C. A further 500 analogues using this scaffold were then prepared and VRT-325 selected. Note that in VRT-325, the EC50 is barely changed, yet it can achieve a correction of 74% of that of the temperature shift.
Thus far there has been only one published in vitro study (Van Goor et al., 2009) on the mechanisms of VX-770 (N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide). In HBE cultures carrying both the G551D and the ΔF508 mutations VX-770 increased chloride secretion by ∼10-fold, to ∼50% of that observed in wild-type HBE epithelia. At the same time excessive Na+ and fluid absorption was reduced while cilia beat frequency was increased. The initial clinical trial with VX770 was for patients with G551D mutations (∼3% of the CF population) as the mutant protein is delivered to the membrane but cannot be activated. Patch clamping studies showed that when expressed in FRT cells, the Po of G551D was increased from 0.1 to 0.45 by VX-770, while with ΔF508 CFTR, Po increased from 0.08 to 0.52 (Van Goor et al., 2009).
Both the VX compounds are bio-available when taken orally. With VX770, 150 mg was given twice daily at 12 h intervals. After 28 days, FEV was increased by 11.6% (P < 0.01), sweat chloride concentration was reduced by 52.8 mM (P < 0.01) and nasal PD fell by 4.3 mV (P < 0.05) compared to the placebo group (Accurso et al., 2008). VX-770 has now entered into Phase 3 (48-week trial), which will include patients homozygous for ΔF508 CFTR mutations, since these too might benefit from potentiating the low level mutant CFTR protein that reaches the correct destination. These are precisely the changes thought to be needed for improving the human condition in CF.
Currently it is known that the other VX compound, VX-809 (100/200 mg twice daily) in homozygous ΔF508 CFTR patients caused a significant fall in sweat chloride. A trial involving both VX-770 and VX-809 is anticipated to start sometime within 2010. This is a particularly exciting prospect as dual therapy may tackle the disease present in the largest cohort of patients. If a significant portion of the non-trafficked ΔF508 CFTR can be moved to the apical membranes of airway cells by VX-809 and be potentiated by VX-770, when appropriate physiological signals are present, then a major step forward will have been achieved.
Medicinal chemistry is the key?
The sudden availability of several compounds that have been shown to make a clinical difference, are well tolerated by patients and available in a pharmaceutically acceptable form, prompts the question ‘what has made the difference?’ Some insight into this question can be gained by studying how the corrector compound VRT-325 was developed (Van Goor et al., 2006). The test system was NIH 3T3 cells expressing ΔF508 CFTR and were used to screen 164 000 compounds at 10 µM. The method was the same as described in Figure 3, except that no cooling to 27°C was performed as the search was for agents that trafficked the mutant protein to the membrane. The first screen gave 1028 compounds that made the criteria for further investigation, that is, gave a value that was >2.5 standard deviations from the mean, corresponding to 32% of 27°C control. These compounds were subjected to further retests from which only 185 survived. More than a third of these compounds were then rejected for reasons of presence of impurities, high molecular weight, high cLogP, toxic or reactive functionalities, limited protein interaction sites and lack of structures amenable to rapid analogue synthesis. The remaining 108 compounds representing 13 distinct scaffolds were subject to functional assays measuring short circuit currents, from which the quinazolinone derivative VRT-422 was chosen as one of the most potent. Over 500 analogues of the compound were then generated and tested by high throughput screening, eventually yielding VRT-325 in which the magnitude of the correction that could be achieved was much greater than with VFT-422, even though their potencies were similar (Figure 4). Further functional studies have been made with VRT-325. In one of these, VRT-325 rescued CFTR mutants R258G, S945L and H949Y as well as ΔF508 CFTR (Loo et al., 2005). In another study, a test was made to examine whether VRT-325 interacted directly with ΔF508 CFTR (Chiaw et al., 2010). After ΔF508 CFTR had been trafficked to the membrane by lowering the temperature, VRT-325 caused a small, but significant, inhibition of halide flux and decreased ATP affinity, both results suggesting that VRT-325 not only promoted trafficking but also compromised activity of CFTR. What I believe this example shows is that not only is it necessary to have a high throughput screening system, but also a preparedness to indulge in extensive medicinal chemistry to make a product fit for purpose.
Concluding remarks
Since the discovery of the CF gene in 1989, the mean life expectancy of CF patients has approximately doubled, and now stands at 37 years. Virtually all this improvement has resulted from better treatment of the consequences of the disease itself rather than the basic cause. Among these can be listed the appropriate use of antibiotics to combat infection, mucolytics and bronchodilators to clear airway blockages of mucus, coupled with improved physiotherapy, oral pancreatic enzymes to help fat digestion and better insulin therapy for CF related diabetes, not forgetting drugs to deal with intestinal blockages. However, we may now be on the cusp of discoveries that will begin to treat the molecular lesion of the disease itself. I refer, of course, to the encouraging clinical data emerging with the VX compounds. It would be wrong to think the battle is won. Almost certainly we will need compounds better than those already found, which raises the question of how to find them. Do we go on screening more and more compounds and making more and more analogues of the best ones? Alternatively, will it be more important to discover at the crystal structure level just how the correctors and potentiators interact with, say ΔF508 CFTR and then design the model drug? It is as yet unknown whether compounds like these, taken over many years, could be tolerated by patients. Whatever the answers may be to these questions, it would seem that pharmaco-therapy for CF has reached a hopeful stage.
Acknowledgments
The author is grateful to the Cystic Fibrosis Trust for support.
Glossary
Abbreviations
- ASL
airway surface liquid
- CaCC
calcium-activated chloride channel
- CAP
channel activating protease
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- ClC-2
chloride channel type 2
- ENaC
epithelial sodium channel
- PD
potential difference
- PKA
protein kinase A
- Po
channel open probability
- SCC
short circuit current
- UTP
uridine triphosphate
Conflict of interest
There is no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–4 as PowerPoint slide.
References
- Accurso FJ, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick DB, et al. Interim results of a phase 2a study of VX-770 to evaluate safety, pharmacokinetics and biomarkers of CFTR activity in cystic fibrosis subjects with G551D. Pediatr Pulmonol Suppl. 2008;31:295. [Google Scholar]
- Adam G, Ousingsawat J, Schreiber R, Kunzelmann K. Increase in intracellular Cl- concentration by cAMP and Ca2+-dependent stimulation of M1 collecting duct cells. Eur J Physiol. 2005;449:470–478. doi: 10.1007/s00424-004-1356-4. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard ST, Taylor AE. Bioelectric properties of proximal bronchiolar epithelium. Am J Physiol. 1994;267:79–84. doi: 10.1152/ajplung.1994.267.1.L79. [DOI] [PubMed] [Google Scholar]
- Ballard ST, Trout L, Bebok Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am J Physiol. 1999;277:L694–L699. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- Becq F, Mettey E, Gray MA, Galietta LJV, Dormer RL, Merton M, et al. Development of substituted benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem. 1999;274:27415–27425. doi: 10.1074/jbc.274.39.27415. [DOI] [PubMed] [Google Scholar]
- Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5:123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijvelds MJC, Bot AGM, Escher JC, De Jonge HR. Activation of intestinal chloride secreion by lubprostone requires the cystic fibrosis transmembrane conductance regulator. Gastroenterology. 2009;137:976–985. doi: 10.1053/j.gastro.2009.05.037. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Newton BB, Pilewski JM, Devor DD, Poll CT, Hall RL. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39-9437. Am J Physiol. 2001;281:L16–L23. doi: 10.1152/ajplung.2001.281.1.L16. [DOI] [PubMed] [Google Scholar]
- Chiaw PK, Wellhauser L, Huan LJ, Ramjeesingh M, Bear CE. A chemical corrector modifies the channel function of F508del-CFTR. Mol Pharmacol. 2010;78:411–418. doi: 10.1124/mol.110.065862. [DOI] [PubMed] [Google Scholar]
- Cloutier MM, Guernsey L, Mattes P, Koeppen B. Duramycin enhances chloride secretion in airway epithelium. Am J Physiol. 1990;259:C450–C454. doi: 10.1152/ajpcell.1990.259.3.C450. [DOI] [PubMed] [Google Scholar]
- Cloutier MM, Guernsey L, Sha'afi RI. Duramycin increases intracellular calcium in airway epithelium. Membr Biochem. 1993;10:107–118. doi: 10.3109/09687689309150258. [DOI] [PubMed] [Google Scholar]
- Conese M, Romano M, Furnari ML, Copreni E, Fino ID, Pardo F, et al. New genetic and pharmacological treatments for cystic fibrosis. Curr Ped Rev. 2009;5:8–27. [Google Scholar]
- Coote K, Atherton-Watson HC, Sugar R, Young A, MacKenzie-Beevor A, Gosling M, et al. Camostat attenuates airway epithelial sodium channel function in vivo through inhibition of a channel-activating protease. J Pharmacol Exp Ther. 2009;329:764–774. doi: 10.1124/jpet.108.148155. [DOI] [PubMed] [Google Scholar]
- Cuppoletti J, Malinowska DH, Tewari KP, Li Q-j, Sherry AM, Patchen ML, et al. SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol. 2004;287:C1173–C1183. doi: 10.1152/ajpcell.00528.2003. [DOI] [PubMed] [Google Scholar]
- Cuthbert AW. Importance of guanidinium groups for blocking sodium channels in epithelia. Mol Pharmacol. 1976;12:945–957. [PubMed] [Google Scholar]
- Cuthbert AW. Lubiprostone targets prostanoid EP4 receptors in ovine airways. Br J Pharmacol. 2010;162:508–520. doi: 10.1111/j.1476-5381.2010.01058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW, Fanelli GM. Effects of some pyrazine carboxamides on sodium transport in frog skin. Br J Pharmacol. 1978;63:139–149. doi: 10.1111/j.1476-5381.1978.tb07783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Deterding RR, LaVange LM, Engels JM, Mathews DW, Coquillette SJ, Brody AS, et al. Phase 2 randomised safety and efficacy tial of nebulized Denufosol tetrasodium in cystic fibrosis. Am J Respir Crit Care Med. 2007;176:362–369. doi: 10.1164/rccm.200608-1238OC. [DOI] [PubMed] [Google Scholar]
- Devor DC, Pilewski JM. UTP inhibits Na+ absorption in wild-type and ΔF508 CFTR-expressing human bronchial epithelia. Am J Physiol. 1999;276:C827–C837. doi: 10.1152/ajpcell.1999.276.4.C827. [DOI] [PubMed] [Google Scholar]
- Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631–1636. doi: 10.1378/chest.07-0288. [DOI] [PubMed] [Google Scholar]
- Dormer RL, Derand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Metaye T, et al. Correction of delF508-CFTR activity with benzo(c) quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci. 2001;114:4073–4081. doi: 10.1242/jcs.114.22.4073. [DOI] [PubMed] [Google Scholar]
- Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]
- Galietta LJV, Sprinsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, et al. Novel CFTR channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Champigny G, Babry P, Dworetzky SI, Meanwell NA, Lazdunski M. The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J Biol Chem. 1994;269:10983–10986. [PubMed] [Google Scholar]
- Hirsh AJ, Sabater JR, Zamurs A, Smith RT, Paradiso AM, Hopkins S, et al. Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2004;311:929–938. doi: 10.1124/jpet.104.071886. [DOI] [PubMed] [Google Scholar]
- Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ, et al. Design,synthesis, and structure-activity relationships of novel 2-substituted pyrazinoylguanidine epithelial sodium channel blockers: Drugs for cystic fibrosis and chronic bronchitis. J Med Chem. 2006;49:4098–4115. doi: 10.1021/jm051134w. [DOI] [PubMed] [Google Scholar]
- Hirsh AJ, Zhang J, Zamurs A, Fleegle J, Thelin WR, Caldwell RA, et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)N'-4-[4(2,3-dihydroxypropoxy)phenyl]butyl-guanidine methanesulphonate (552-02), a novel epithelial sodium channel blocker with a potential clinical efficacy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2008;325:77–88. doi: 10.1124/jpet.107.130443. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Stutts MJ, Ziersch A, Ruckes C, Weber WM, Knowles MR, et al. Effects of topically delivered benzamil and amiloride on nasal potential difference in cystic fibrosis. Am J Respir Crit Care Med. 1998;157:1844–1849. doi: 10.1164/ajrccm.157.6.9709043. [DOI] [PubMed] [Google Scholar]
- Hogman M, Mork A-C, Roomans GM. Hypertonic saline increases tight junction permeability in airway epithelium. Eur Respir J. 2002;20:1444–1448. doi: 10.1183/09031936.02.00017202. [DOI] [PubMed] [Google Scholar]
- Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- Illek B, Fischer H, Santos GF, Widdicombe JH, Machen TE, Reenstra WW. cAMP independent inactivation of CFTR chloride channels by the tyrosine kinase inhibitor genistein. Am J Physiol. 1995;268:886–893. doi: 10.1152/ajpcell.1995.268.4.C886. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Wilson SM. Cystic fibrosis and airway submucosal glands. Ped Pulmonol. 2005;40:279–284. doi: 10.1002/ppul.20183. [DOI] [PubMed] [Google Scholar]
- Jaques A, Daviskas E, Turton JA, McKay K, Cooper P, Stirling RG, et al. Inhaled mannitol improves lung function in cystic fibrosis. Chest. 2008;133:1388–1396. doi: 10.1378/chest.07-2294. [DOI] [PubMed] [Google Scholar]
- Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na+] and pH but elevated viscosity. Proc Natl Acad Sci USA. 2001;98:8119–8123. doi: 10.1073/pnas.131087598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo NS, Wu JV, Krouse ME, Senz Y, Wine JJ. An optical method for quantifying rates of mucus secretion from single submucosal glands. Am J Physiol. 2001;281:L458–L468. doi: 10.1152/ajplung.2001.281.2.L458. [DOI] [PubMed] [Google Scholar]
- Joo NS, Irokawa T, Robbins RC, Wine JJ. Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J Biol Chem. 2006;281:7392–7398. doi: 10.1074/jbc.M512766200. [DOI] [PubMed] [Google Scholar]
- Joo NS, Wine JJ, Cuthbert AW. Lubiprostone stimulates secretion from tracheal submucosal glands of sheep,pigs and humans. Am J Physiol. 2009;296:L811–L824. doi: 10.1152/ajplung.90636.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Church NL, Waltner NE, Yankaskas JR, Gilligan P, King M, et al. A pilot study of aerosolised amiloride for the treatment of lung disease in cystic fibrosis. N Engl J Med. 1990;322:1189–1194. doi: 10.1056/NEJM199004263221704. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Clarke LL, Boucher RC. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N Engl J Med. 1991;325:533–538. doi: 10.1056/NEJM199108223250802. [DOI] [PubMed] [Google Scholar]
- Kohler D, App E, Schmitz-Schumann M, Wurtemberger G, Matthys H. Inhalation of amiloride improves the mucociliary clearance and the cough clearance in patients with cystic fibroses. Eur J Respir Dis. 1986;146:319–326. [PubMed] [Google Scholar]
- Konstas AA, Koch J-P, Korbmacher C. cAMP-dependent activation of CFTR inhibits epithelial sodium channel (ENaC) without affecting its surface expression. Eur J Physiol. 2003;445:513–521. doi: 10.1007/s00424-002-0957-z. [DOI] [PubMed] [Google Scholar]
- Lazrak A, Nita I, Subramaniyam D, Wei S, Song W, Ji H-L, et al. Alpha 1-antitrypsin inhibits epithelial Na+ transport in vitro and in vivo. Am J Respir Cell Mol Biol. 2009;41:261–270. doi: 10.1165/rcmb.2008-0384OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TW, Bartlett C, Clarke DM. Rescue of ΔF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol Pharmaceut. 2005;2:407–413. doi: 10.1021/mp0500521. [DOI] [PubMed] [Google Scholar]
- Ma T, Vetrivel L, Yang H, Pedemonte N, Zegarra-Moran O, Galietta LJV, et al. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem. 2002;277:37235–37241. doi: 10.1074/jbc.M205932200. [DOI] [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nature Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airway disease. Cell. 1998;95:1005–1010. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Mohammad-Panah R, Gyomorey K, Rommens J, Choudhury M, Li C, Wang Y, et al. ClC-2 contributes to native chloride secretion by a human intestinal cell line Caco-2. J Biol Chem. 2001;276:8306–8313. doi: 10.1074/jbc.M006764200. [DOI] [PubMed] [Google Scholar]
- Murthy M, Pedemonte N, MacVinish L, Galietta L, Cuthbert AW. Chlorobenzo[F]isoquinoline (CBIQ) a novel activator of CFTR and ΔF508 CFTR. Eur J Pharmacol. 2005;516:118–124. doi: 10.1016/j.ejphar.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Diena T, Caci E, Nieddu E, Mazzei M, Ravazzolo R, et al. Antihypertensive 1,4-dihydropyridines as correctors of the cystic fibrosis transmembrane conductance regulator channel gating defect caused by cystic fibrosis mutations. Mol Pharmacol. 2005a;68:1736–1746. doi: 10.1124/mol.105.015149. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJV, et al. Small molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005b;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planes C, Caughey GH. Regulation of the epithelial Na+ channel by peptidases. Curr Top Dev Biol. 2007;78:23–44. doi: 10.1016/S0070-2153(06)78002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Roberts M, Hladky SB Pickles RJ, Cuthbert AW. Stimulation of sodium transport by duramycin in cultured human colonic epithelia. J Pharmacol Exp Ther. 1991;259:1050–1058. [PubMed] [Google Scholar]
- Rodgers HC, Knox AJ. The effect of topical benzamil and amiloride on nasal potential difference in cystic fibrosis. Eur Respir J. 1999;14:693–696. doi: 10.1034/j.1399-3003.1999.14c32.x. [DOI] [PubMed] [Google Scholar]
- Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Ann Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- Sheth TR, Henderson RM, Hladky SB, Cuthbert AW. Ion channel formation by duramycin. Biochim Biophys Acta. 1992;1107:179–185. doi: 10.1016/0005-2736(92)90345-m. [DOI] [PubMed] [Google Scholar]
- Song Y, Verkman AS. Aquaporin-5 dependent fluid secretion in airway submucosal glands. J Biol Chem. 2001;276:41288–41292. doi: 10.1074/jbc.M107257200. [DOI] [PubMed] [Google Scholar]
- Springsteel MF, Galietta LJ, Ma T, By K, Berger GO, Yang H, et al. Benzoflavone activators ot the cystic fibrosis transmembrane conductance regulator: towards a pharmacophore model for the nucleotide-binding domain. Bioorg Med Chem. 2003;11:4113–4120. doi: 10.1016/s0968-0896(03)00435-8. [DOI] [PubMed] [Google Scholar]
- Szkotak M, Murthy M, MacVinish LJ, Duszyk M, Cuthbert AW. 4-chloro-benzo{F]isoquinoline (CBIQ) activates CFTR chloride channels and KCNN4 potassium channels in human airway epithelial cells. Br J Pharmacol. 2004;142:531–542. doi: 10.1038/sj.bjp.0705846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J Gen Physiol. 2006;127:591–604. doi: 10.1085/jgp.200509468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M-F, Li M, Hwang T-C. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J Gen Physiol. 2010;135:5399–5414. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Strayley KS, Cao D, Gonzalez G, Hadida S, Hazlewood A, et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, et al. Rescue of a CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Nat Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Drug discovery in academe. Am J Physiol. 2004;286:C465–C474. doi: 10.1152/ajpcell.00397.2003. [DOI] [PubMed] [Google Scholar]
- Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol. 2003;284:C2–C15. doi: 10.1152/ajpcell.00417.2002. [DOI] [PubMed] [Google Scholar]
- Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum and glucocorticoid-regulated kinase(Sgk1) in Xenopus oocytes. J Gen Physiol. 2002;120:191–201. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- Wine JJ. Parasympathetic control of airway submucosal glands: Central reflexes and the airway intrinsic nervous system. Autonom Neurosci. 2007;133:35–54. doi: 10.1016/j.autneu.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, et al. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, et al. Pharmacology of INS37217 [P1-(uridine 5′)-P4-92-deoxycytidine 5′) tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.