

Abstract
The world is running out of antibiotics. Between 1940 and 1962, more than 20 new classes of antibiotics were marketed. Since then, only two new classes have reached the market. Analogue development kept pace with the emergence of resistant bacteria until 10–20 years ago. Now, not enough analogues are reaching the market to stem the tide of antibiotic resistance, particularly among gram-negative bacteria. This review examines the existing systemic antibiotic pipeline in the public domain, and reveals that 27 compounds are in clinical development, of which two are new classes, both of which are in Phase I clinical trials. In view of the high attrition rate of drugs in early clinical development, particularly new classes and the current regulatory hurdles, it does not seem likely that new classes will be marketed soon. This paper suggests that, if the world is to return to a situation in which there are enough antibiotics to cope with the inevitable ongoing emergence of bacterial resistance, we need to recreate the prolific antibiotic discovery period between 1940 and 1962, which produced 20 classes that served the world well for 60 years. If another 20 classes and their analogues, particularly targeting gram-negatives could be produced soon, they might last us for the next 60 years. How can this be achieved? Only a huge effort by governments in the form of finance, legislation and providing industry with real incentives will reverse this. Industry needs to re-enter the market on a much larger scale, and academia should rebuild its antibiotic discovery infrastructure to support this effort. The alternative is Medicine without effective antibiotics.
LINKED ARTICLES
This article is part of a themed issue on Respiratory Pharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-1
Keywords: novel antibiotics, bacterial infections, antibiotic resistance, genomics, non-culturable bacteria, bacteriophages, non-multiplying bacteria, multiplying bacteria
Introduction
The future of modern medicine depends upon effective antibiotics (So et al., 2010). The world produced more than 20 novel classes of antibiotics between 1930 and 1962 (Coates et al., 2002; Powers, 2004). Since then, only two new classes of antibiotics have been marketed (Butler and Buss, 2006; Hair and Kean, 2007; Zappia et al., 2007). Numerous analogues of existing classes have reached the market in this time period. Meanwhile, multi-drug-resistant bacteria (superbugs) have emerged throughout the world (Levy and Marshall, 2004), and now half the deaths from clinical infection in Europe are associated with multi-drug-resistant bacteria (Watson, 2008). In the short term, according to the Infectious Diseases Society of America (IDSA, 2010) at least another 10 antibiotics, which are active against superbugs, are required to reach the market within the next 10 years. In the longer term, novel classes of antibiotics will be needed, but how many? The answer to this question is unknown. However, on the basis that most classes of antibiotics now have substantial resistance problems, at least for some species of bacteria, a new class, together with its analogues, might remain useful for 50 years. This would mean that the 20 classes which entered the market between 1930 and 1960 lasted for an average of 50 years. This assumption would lead to the conclusion that it is likely that the world will need a further 20 novel classes of antibiotics to support modern medicine during the next 50 years (see Figure 1). Some bacterial infections, such as gram-negatives, are already very difficult to treat, for example, metallo-β-lactamase producing pathogens which neutralize carbapenems (Kumarasamy et al., 2010), while others, such as gram-positive Staphylococcal infections like methicillin-resistant Staphylococcus aureus (MRSA) are still susceptible to a range of old and new antibiotics (Boucher et al., 2009; Walkey et al., 2010). Thus, the need is critical for some gram-negatives, but less so for gram-positives. However, the world's capacity for antibiotic discovery is already falling behind the rate of emergence of bacterial resistance, and so, is it likely that another 20 new classes of antibiotics will reach the market within 50 years, and perhaps, a further 20 classes between 50 and 100 years from now? Figure 1 depicts a line drawing of the number of new classes that reached the market between the 1940s and 1960s, the two new classes since 2000, and projections into the future of 20 new classes for the next 50 years, and 20 further classes required to support medicine in 50–100 years from now. Although these are only projections, they are drawn upon past performance of the pharmaceutical industry and upon the relentless emergence of antibiotic resistant bacteria. Whether or not the actual numbers of new classes required proves to be accurate, and whether the goal of 20 more classes in the next 50 years is achievable, there is clearly an urgent need for new classes of antibiotics. However, the production of only two new classes of antibiotics in the last 50 years suggests that it may be very difficult to produce enough new classes of compounds to support modern medicine during the next 50 years.
Figure 1.
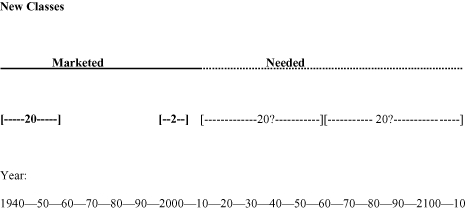
The number of new classes of antibiotics which have reached the market, and predictions of novel classes of antibiotics which are needed during the next 100 years.
The issues
Antibiotic discovery is the key to the continuance of modern medicine (So et al., 2010). Why is the pharmaceutical industry producing fewer new antibiotic classes now, when the need is increasing? The reason for the lack of new classes is not clear, but the following will be discussed later in this paper: there may be a shortage of new metabolic targets (Becker et al., 2006). Marketing of analogues is more financially feasible than developing new classes (Devasahayam et al., 2010), and it used to be thought that current antibiotics would last almost indefinitely (reviewed in Coates et al., 2002). The numbers of major pharmaceutical companies which are engaged in antibiotic discovery decreased substantially over the past two to three decades; now, only five remain active in the field (Boucher et al., 2009). Governments did not increase funding for antibiotic discovery, including education in this field, fast enough to cope with the rise in antibiotic resistance (House of Lords, 1998). This is a major concern for many countries. Meanwhile, regulatory requirements increased, which has elevated the costs of developing drugs, and the profits from competing products led pharmaceutical companies away from the antibiotic field.
Why is the scope for analogue development limited?
Table 1 shows the number of analogues which have been developed in each class of antibiotics. Although there is more scope for further analogue development, it seems that such an approach is more feasible for some classes than others, cephalosporins, penicillins and quinolones being the most successful, as there are sites in the scaffolds for easy modification. However, new analogues based on one core eventually become too difficult or too expensive to make, particularly when faced with new antibiotic resistance mechanisms.
Table 1.
Main classes of marketed and withdrawn antibiotics
| Class examples |
|---|
| β-Lactams |
| Penicillins |
| Penicillin G, penicillin V, methicillin, oxacillin, cloxacillin, dicloxacillin, nafcillin, ampicillin, amoxicillin, carbenicillin, ticarcillin, mezlocillin, piperacillin, azlocillin, temocillin |
| Cephalosporins |
| First generation Cephalothin, cephapirin, cephradine, cephaloridine, cefazolin |
| Second generation Cefamandole, cefuroxime, cephalexin, cefprozil, cefaclor, loracarbef, cefoxitin, cefmetazole |
| Third generation Cefotaxime, ceftizoxime, ceftriaxone, cefoperazone, ceftazidime, cefixime, cefpodoxime, ceftibuten, cefdinir |
| Fourth generation Cefpirome, cefepime |
| Fifth generation1 Ceftaroline2, ceftobiprole3 |
| Carbapenems |
| Imipenem, meropenem, doripenem |
| Monobactams |
| Aztreonam |
| β-Lactamase inhibitors |
| Clavulanate, sulbactam, tazobactam |
| Aminoglycosides |
| Streptomycin, neomycin, kanamycin, paromomycin, gentamicin, tobramycin, amikacin, netilmicin, spectinomycin, sisomicin, dibekacin, isepamicin |
| Tetracyclines |
| Tetracycline, chlortetracycline, demeclocycline, minocycline, oxytetracycline, methacycline, doxycycline, tigecycline |
| Rifamycins |
| Rifampicin (also called rifampin), rifapentine, rifabutin, bezoxazinorifamycin, rifaximin |
| Macrolides |
| Erythromycin, azithromycin, clarithromycin |
| Ketolides |
| Telithromycin |
| Lincosamides |
| Lincomycin, clindamycin |
| Glycopeptides |
| Vancomycin, teicoplanin, telavancin |
| Lipopeptides |
| Daptomycin |
| Streptogramins |
| Quinupristin, dalfopristin, pristinamycin |
| Sulphonamides |
| Sulphanilamide, para-aminobenzoic acid, sulfadiazine, sulfisoxazole, sulfamethoxazole, sulfathalidine |
| Oxazolidinones |
| Linezolid |
| Quinolones |
| Nalidixic acid, oxolinic acid, norfloxacin, pefloxacin, enoxacin, ofloxacin/levofloxacin, ciprofloxacin, temafloxacin, lomefloxacin, fleroxacin, grepafloxacin, sparfloxacin, trovafloxacin, clinafloxacin, gatifloxacin, moxifloxacin, sitafloxacin |
| Others |
| Metronidazole, polymyxin B, colistin, trimethoprim |
Spectrum combines third generation gram-negatives with methicillin resistant Staphylococcus aureus (MRSA) and multidrug-resistant Streptococcus pneumoniae.
Pending Federal Drugs Administration review.
Marketed in Canada and Switzerland; now withdrawn.
New classes
Can we produce new classes of antibiotics many times faster than we have achieved over the past 50 years? Although only two new classes of antibiotics were marketed in the past 10 years, this shows it is still possible to discover and market new classes. It also suggests that the reason for the lack of new class development between 1962 and 2000 was that pharmaceutical companies were concentrating on analogue development, perhaps because the toxicity risks associated with analogues is lower than that for new classes. Whether it will be possible to increase antibiotic class discovery to the level it was between 1940 and 1960, is another matter. It may be difficult to do so, based upon the possibility that most of the easy-to-discover antibiotics have already been marketed. Also, the workforce in companies and in the university sector need to be re-skilled in microbiology and pharmacology, with particular emphasis on antibiotic discovery, and this will take decades to achieve and will be very expensive. Most government funding concentrates on the pathogenesis of infectious disease, and while this is important, if new antibiotics are to emerge from the university sector, a higher proportion of the funding must be allocated to antibiotic discovery and development. Thus, to generate the number of new antibiotics which are needed over the next 100 years, substantially increased funding from industry and from government will be required, and should be focused upon antibiotic discovery, with less funding for microbial pathogenesis.
The emergence of antibiotic resistance is at the heart of the gradual demise of the antibiotic era. Can we produce antibiotics which induce resistance at a lower rate than existing antibiotics? This is a key question and will be addressed further in the section on antibiotic resistance below.
The aims of this review are to discuss the advantages and disadvantages of continuing antibiotic discovery in the traditional way, namely, more of the same, meaning analogue development using the Fleming method. The role of the relentless emergence of antibiotic resistance to all antibiotics will also be discussed, as well as possible ways to design antibiotics which lead to a lower rate of emergence of resistance. The merits of trying to discover new classes of antibiotics will be discussed, including ways in which this could be achieved. Finally, the future direction of antibiotic discovery will be covered, with advantages and disadvantages of different routes.
More of the same
The traditional, and the most successful way of making antibiotics has been to find a natural product (Singh and Barrett, 2006) or, in a few cases, a chemically synthesized one (Fernandes, 2006) by screening for activity against a bacterial culture which is in log phase growth. For example, penicillin (Fleming, 1929) was discovered by Alexander Fleming by observing the antibacterial effect of the Penicillium fungus. This discovery led to the creation of numerous analogues, many of which were marketed (see Table 1). The pharmaceutical industry was able to build the analogues in such a way that they were active against penicillin-resistant bacteria. The advantage of this route is that analogues tend to have a similar solubility, protein binding and toxicity as the parent compound. A further advance that was made was to add an anti-bacterial resistance factor to the antibiotic, which rendered it active in the patient. An example of this approach is the addition of clavulanic acid, a beta-lactamase inhibitor, to amoxicillin, which is marketed as Augmentin. The disadvantage of these routes of development is that there is a limit to the number of analogues which can be made from a single chemical core, or ones which can counteract bacterial resistance mechanisms, and eventually, bacteria can evolve resistance beyond the scope of even the most ingenious medicinal chemist. Other discoveries followed that of penicillin, the most notable of which, in terms of analogue development, was cephalosporins (see Table 1). Even today, cephalosporin analogues are being developed with anti-superbug activity, one of which, ceftaroline, is in pre-registration (see Table 2). Clearly, there is some scope for more analogue development, and a new trimethoprim derivative, iclaprim, was recently rejected by the Federal Drugs Administration (FDA) for failure to demonstrate non-inferiority. However, it is also clear that some antibiotic families are more amenable to analogue development than others. For instance (Table 1), the most prolific number of analogues has been made using the cephalosporin and penicillin cores, but the quinolone and aminoglycoside cores have also been very productive. Unfortunately, resistance arises to all these compounds, and thus analogue development merely ‘buys time’ until the discovery of the next novel class. However, in our view, more analogues are likely to be marketed in the next 20 years. Table 2 shows the antibiotics which are in development and which are accompanied by information in the public domain. It can be seen that although few new classes are represented, there are some significant improvements. Concern has to be noted that pleuromutilins, which are new to human therapy but have been used in veterinary medicine for about 30 years, are susceptible to the cfr plasmid-mediated resistance which affects virtually all antibiotics that target the 50S ribosomal subunit. In this review, we do not count pleuromutilins as a new class because they have been widely used for decades. Another potential new class, PDF inhibitors, has been abandoned by a number of companies over the past decade because of resistance emergence.
Table 2.
Antibacterial compounds in development
| Class | Product | Spectrum | Iv/oral | Indications | Phase | Company (Licensor) |
|---|---|---|---|---|---|---|
| Glycopeptide | Dalbavancin | Gram-positive (excluding VRE) | IV; once-weekly | cSSTI | Phase III | Durata Therapeutics (Pfizer) |
| Glycopeptide | Telavancin | Gram-positive (excluding VRE) | IV | cSSTI and HAP | Marketed for SSTI in USA; HAP approval stalled at FDA | Astellas (Theravance) |
| Glycopeptide | Oritavancin | Gram-positive (including VRE) | IV; single dose treatment | cSSTI | Phase III | The Medicines Company (Lilly) |
| Cephalosporin | Ceftaroline | Gram-positive and gram-negative excluding ESBLs etc. and non-fermenters | IV | SSTI, CAP | Pre-registration | Forest (Cerexa) |
| Glycopeptide-cephalosporin hybrid | TD-1792 | Gram-positive | IV | SSTI, HAP | Phase IIa | Theravance |
| Ketolide | Cethromycin | Gram-positive and respiratory tract infection pathogens | Oral and IV | Community-acquired RTIs biothreat pathogens | Phase III | Advanced Life Sciences (Abbott) |
| Ketolide | EDP-420 | Gram-positive and respiratory tract infection pathogens | Oral | Community-acquired RTIs | Phase II/III | Enanta (Shionogi) |
| Fluoroketolide | Solithromycin | Gram-positive and respiratory tract infection pathogens | IV/oral | Community-acquired RTIs biothreat pathogens | Phase I/II | Cempra (Optimer) |
| Pleuromutilin | BC-3781 | Gram-positive and respiratory tract infection pathogens | Oral IV | Community-acquired RTIs and SSTIs | Phase IIPreclinical | Nabriva |
| Peptide deformylase inhibitor (new class) | GSK1322322 | Gram-positive and Respiratory tract pathogens | Oral | Community-acquired RTIs and SSTIs | Phase I | GlaxoSmithKline |
| Oxazolidinone | Torezolid | Gram-positive (including linezolid- and daptomycin resistant strains) | IV/oral | cSSTI | Phase IIa | Trius (DongA) |
| Oxazolidinone | Radezolid | Gram-positive (including linezolid- and daptomycin resistant strains) | IV/oral | SSTI | Phase IIa | Rib-X |
| Oxazolidinone | PNU-100480 | TB | Oral | TB | Phase I | Pfizer |
| FabI Inhibitor (new class) | AFN-1252 | Staphylococci | Oral | Staph infections | Phase I | Affinium |
| FabI Inhibitor (new class) | MUT056399 | Staphylococci | IV | Staph infections | Phase I | FAB Pharma |
| Aminomethylcycline (tetracycline) | PTK0796 | Gram-positive, RTI and SSTI pathogens | IV/oral | cSSTI, CAP | Phase III | Novartis (Paratek) |
| Fluoroquinolone | Delafloxacin | Broad-spectrum including fluoroquinolone-resistant MRSA | IV/oral | cSSTI, | Phase III | Rib-X |
| Fluoroquinolone | Finafloxacin | Broad-spectrum; enhanced activity at acid pH | Merlion | |||
| Fluoroquinolone | JNJ-Q2 | Enhanced gram-positive activity including fluoroquinolone-resistance-resistant MRSA | IV/oral | Phase I | J&J | |
| Fluorocycline (tetracycline) | TP-434 | Broad gram-positive, -negative and anaerobic activity including Acinetobacter but not P. aeruginosa, less active versus Proteae and some K. pneumoniae | IV/oral | cSSTI, cIAI | Phase I | Tetraphase |
| Aminoglycoside | ACHN-490 | MDR enterobacteriaceae and S. aureus, including aminoglycoside-resistant and metallo-ß-lactamase producers | IV | cUTI, cIAI | Phase I | Achaogen |
| leucyl-tRNA synthase inhibitor (new class) | GSK2251052 | MDR enterobacteriaceae and P. aeruginosa | IV | Anacor/GSK | ||
| Penicillin | CXA-101 | MDR P. aeruginosa and susceptible enterobacteriaceae | IV | cUTI | Phase II | Cubist |
| Penicillin/β-lactamase-inhibitor | CXA-101 /tazobactam (CXA-201) | MDR P. aeruginosa and enterobacteriaceae, excluding metallo-ß-lactamases | IV | cUTI, cIAIVAP | Phase I | Cubist |
| Non-β-lactam β-lactamase-inhibitor (new class) | NXL104 | Class A, C and some class D β-lactamases, including OXA-48 | AstraZeneca (Novexel) | |||
| Cephalosporin/β-lactamase-inhibitor | Ceftaroline/ NXL104 | MRSA and MDR enterobacteriaceae, excluding metallo-ß-lactamases | IV | cUTI, SSTI, CAP | Phase II ready | AstraZeneca/Forest |
| Cephalosporin/β-lactamase-inhibitor | Ceftazidime/ NXL104 | MDR P. aeruginosa and enterobacteriaceae, excluding metallo-ß-lactamases | IV | cUTI, SSTI, VAP | Phase III ready | AstraZeneca/Forest |
| Non-β-lactam β-lactamase-inhibitor (new class) | MK-7655 | Class A and C β-lactamases | Merck & Co | |||
| Carbapenem/β-lactamase-inhibitor | Imipenem/ MK-7655 | MDR P. aeruginosa and enterobacteriaceae, excluding metallo-ß-lactamases; Acinetobacter | IV | cUTI, cIAI, HAP/VAP | Phase II ready | Merck & Co |
| Sulfactam (siderophore monobactam) | BAL30072 | MDR P. aeruginosa Acinetobacter including metallo-ß-lactamases and enterobacteriaceae | IV | Preclinical | Basilea | |
| Monobactam/ carbapenem | BAL30072/ meropenem | Most MDR gram-negatives including resistant Enterobacteriaceae and anaerobes | IV | cUTI, Hospital-acquired cIAI, VAP | Preclinical | Basilea |
| Monocarbam (siderophore monobactam) | MC-1 | MDR P. aeruginosa including metallo-ß-lactamases S. maltophilia and enterobacteriaceae | IV | cUTI, Hospital-acquired cIAI, VAP | Preclinical | Pfizer |
These data are based upon information in the public domain.
Products shaded blue = primarily gram-positive; unshaded = broad(ish)-spectrum; shaded red = MDR gram-negatives.
MDR, multidrug resistant; MRSA, methicillin-resistant Staphylococcus aureus.
For gram-negatives, the non-β-lactamase β-lactamase-inhibitor from Novexel has broad coverage against class A, C and some class D, such as OXA-48, but not those of Acinetobacter; it has no activity against metallo-β-lactamases. Merck & Co. also have a similar analogue, but there is less information available in public domain about this molecule.
The siderophore monobactams from Basilea and Pfizer have similar structures but seem to have different activities, especially against Acinetobacter spp. The combination of BAL30072 with meropenem seems to cope with many of the β-lactam-resistance mechanisms in gram-negatives: this is an example of more than just inhibiting resistance mechanisms, but of broadening the spectrum. However, none of the developments is a panacea. Even the new class from Anacor/GlaxoSmithKline has gaps, especially Acinetobacter spp. The neoglycoside from Achaogen is immune to nearly, but not all, the aminoglycoside modifying enzymes found in Enterobacteriaceae but is not active against Pseudomonas aeruginosa.
It should be noted that in Table 2, the indications for products at preclinical, Phase I and Phase II are largely guesswork
Resistance – the antibiotic slayer
The Achilles heel of antibiotics is resistance development of target pathogenic bacteria. Within 2 years of marketing, resistance is usually observed, even to new classes of compounds (Bax et al., 1998). When the proportion of bacteria, which cause specific types of clinical infection, rises to 20% or more, the antibiotic becomes poorly effective for that indication and may be withdrawn. This is a unique feature of anti-microbial drugs, namely that they become ineffective over time, something which does not affect other drug groups such as cardiovascular, central nervous system and anti-inflammatories. However, some bacteria mutate slowly, while others do so faster or acquire resistance elements from other species. For example, some strains of Neisseria meningitidis (du Plessis et al., 2008) still show only a reduced susceptibility to penicillin, while for S. aureus (Chambers and Deleo, 2009) resistance to penicillin appeared in the early 1940s, shortly after its introduction into the market, and resistance to methicillin was recorded just 1 year after its introduction. In 2005 in the USA, it has been estimated that S. aureus caused 10 800 deaths, of which 5500 were due to MRSA (Klein et al., 2007). The majority of hospital infections in USA are caused by the so-called ESKAPE pathogens (Rice, 2010), for which new antibiotics are urgently needed. These are Enterococcus faecium, S. aureus, Acinetobacter baumanii, Klebsiella pneumoniae, P. aeruginosa and Enterobacter species. Gram-negative bacteria are particularly troublesome in this regard, and have become progressively resistant to each antibiotic class. Now Enterobacteriaceae (Kumarasamy et al., 2010), which are resistant to carbapenem conferred by New Delhi metallo-β-lactamase 1, are being isolated from patients in several countries. Major efforts are being adopted by many countries to prevent such infections (Coates and Hu, 2008) by improvements in infection control in hospitals, withdrawal of vulnerable classes of antibiotics, restrictions in the use of antibiotics and the introduction of new vaccines.
However, superbugs which are resistant to key classes of antibiotics continue to emerge. Some antibiotics induce resistance readily, for example, rifampicin (Lambert, 2005), while others, such as those which target the cell membrane, may do so more slowly (Zhanel et al., 2008). The key mechanisms of genetic resistance are (Coates et al., 2002): (i) bacteria can inactivate the antibiotic by producing, for example, β-lactamase which degrades the β-lactam ring which is a key part of penicillins and cephalosporins; (ii) reduce membrane permeability to the antibiotic; (iii) increase the efflux of antibiotic from the cell; (iv) overproduce the target enzyme; (v) bypass the inhibited step; and (vi) alter the site of action of the antibiotic. Some antibiotics, notably fluoroquinolones, induce the SOS response, which increases the error rate of DNA replication and speeds the development of resistance (Da Re et al., 2009)
In addition, before the onset of genetic resistance, bacteria can survive antibiotic treatment by entering into a slow or non-multiplying state (Coates et al., 2002). It is thought that about 60% of all clinical infections contain bacteria in this state (Coates and Hu, 2008). Commensal bacteria, for example, those which naturally live on the skin, in the mouth, nose and intestines contain large numbers of antibiotic-resistant organisms, and these may be a source of antibiotic-resistance markers for pathogenic bacteria (Gillings et al., 2008). Also, about half of all antibiotics which are used each year in the world are consumed by animals. This is also thought to be a source of antibiotic resistance in humans, although this is disputed by some (Soulsby, 2008). Recently (van Cleef et al., 2010), MRSA has been found in half of the pig farms in Europe and has spread to pig farmers and their families, but has not, as of yet, spread into the general population.
These data suggest that antibiotic resistance should not be considered, particularly in gram-negatives, to be isolated to a small number of superbugs. Rather, it is part of a much larger picture, namely the whole of the bacterial kingdom which seems to operate cooperatively, horizontally transferring antibiotic resistance containing DNA between different species. Also, the resistant bacteria are fit to be able to survive and persist for a long period of time, even though no antibiotic selective pressure is present (Andersson, 2006)
Novel classes of antibiotics
Novel classes of antibiotics are urgently needed for the future. The two new classes of antibiotics which have been introduced into the market, oxazolidinone (linezolid by Pfizer) and cyclic lipopeptide (daptomycin by Cubist) are active against gram-positive bacteria, such as MRSA, but there are no new classes in Phase II or III clinical trials, and none in the pre-registration stage (see Figure 2). There are two new classes in Phase I and a small number in the pre-clinical and discovery phases, for example, a new class which targets type IIA topoisomerases with a new mechanism of action (Bax et al., 2010). It is difficult to estimate the number of drugs in preclinical development, because most are not published, so no attempt has been made to do so in this review. It should be noted that the antibiotics in clinical development, which appear in Table 2 and Figure 2, are based upon information which is in the public domain. However, this may be an underestimate, because many companies do not publish data in this area. Figure 2 shows that there are at least 27 anti-bacterial compounds which are in clinical development, of which 11 are in Phase I clinical trials, 8 in Phase II, but only 6 in Phase III and 2 at the pre-registration stage. Of the antibiotics in clinical development, two belong to new classes. There are no antibiotics against the major gram-negative pathogens K. pneumoniae, P. aeruginosa and A. baumanii in Phase IIb and III. Furthermore, there are only a few compounds against these pathogens in earlier stages of development. Most of these compounds are analogues of existing marketed antibiotics. Because there are no new class in the later stages of development, Phase II, III and pre-registration (Figure 2), there may be no new classes in the market in the short term. Although there are two potential new class compounds in early clinical Phase I development, the high attrition rate, particularly for new classes, means that the odds are against these compounds reaching the market, and it is possible that no new classes will reach the market within 10 years. In the longer term, during the next 20 years, the likelihood of discovering 20 novel classes including many broad spectrum antibiotics, similar to the achievements in the 1940–1960s, seems to be remote, particularly for multi-drug-resistant gram-negatives.
Figure 2.
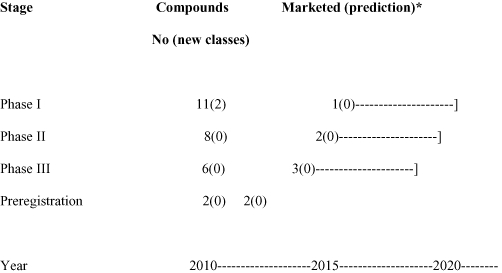
The number of systemic compounds and classes in development and predictions of the number which will reach the market. Based on estimates of drugs in development which are published in the public domain. New classes are in brackets. *Assumptions: percent of compounds which reach the market (CMR International 2009) – 6.25% in Phase I, 25% in Phase II, 50% in Phase III and 75% in Pre-registration. Phase II includes drugs which are in Phase I/II, II ready, II and IIa. Phase III includes drugs in II/III, III ready and III. Market predictions are shown with – which indicates a range of years due to current uncertainty of the length of time – which is required to complete Phase III trials, for example for hospital-acquired pneumonia/ventilator-associated pneumonia.
Why is there a shortage of new classes of antibiotics in development?
The risk and cost of developing a new class is considerably greater than that of an analogue. For example, the starting point for an analogue is likely to give rise to compounds which are soluble and are not toxic, on the grounds that the parent molecule has these characteristics. Toxic side effects are a common cause of failure of a compound to proceed past Phase I clinical trials. In contrast, a new class of compound has unknown toxic potential, and may have chemico-physical features which make them unsuitable for drug development.
However, there may be a more fundamental reason why so few new classes have reached the market during the past 50 years. Simply, it may be more difficult to find new classes which are broad spectrum, than it was in the golden age of antibiotic discovery. It has been suggested (Becker et al., 2006) that anti-bacterial classes have been developed and marketed already against all the main metabolic pathways, and that there are no more available. In other words, it is possible that all the classes of broad-spectrum antibacterials have been discovered. If this were to be the case, the antibiotic era is likely to fade away, sooner rather than later.
Another blow to the antibiotic discovery field is the considerable investment which the pharmaceutical industry invested in the genomics approach (Brotz-Oesterhelt and Sass, 2010) that failed to deliver a new class into the market. This approach uses bacterial DNA sequence data to predict enzymatic pathways which can be inhibited by novel classes of compounds.
The recent difficulties with registration of antibiotics are also likely to suppress the enthusiasm of large pharmaceutical companies to enter the field. For example, the FDA in the USA, having agreed to the protocol for telavancin hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) studies with clinical response as the primary endpoint, the agency requested additional data and analyses to support an evaluation of all-cause mortality as the primary efficacy endpoint. As a result, the company may have to conduct new studies. Initial applications for dalbavancin and iclaprim have also been rejected by the FDA.
The requirement of the FDA for a smaller non-inferiority margin and to restrict nosocomial pneumonia trials to VAP is unlikely to result in new agents for multidrug resistant (MDR) gram-negative pneumonia. For a non-inferiority margin of 7%, the cost of a HAP/VAP study would total $600 million and take 8–10 years for enrolment, even using hundreds of centres. This would double if the trial were to be VAP only. Further regulatory difficulties are envisaged that will discourage the development of MDR-gram-negative antibiotics. With no standard therapy, non-inferiority studies are not possible; historical controls are considered irrelevant and superiority studies are not ethical as patients cannot be randomized to ineffective therapy for infections with high mortality.
Most large pharmaceutical companies have now left the antibiotic discovery field. This has led to de-skilling of the workforce in the industry over the past 30 years. In parallel, many universities have closed academic departments which underpin antibiotic discovery and development. The net effect of the de-skilling in antibiotic discovery worldwide, compared with the 1940–1960s, is that it will take decades to rebuild the teaching and training which is needed to support the discovery of numerous new classes of antibiotics.
Future direction
If bacteria continue to develop new resistance mechanisms, and if the pharmaceutical industry produces novel classes of antibiotics at the existing rate, the future for medicine is bleak. In our view, it is unlikely that the industry will dramatically increase the rate of production of new classes of antibiotics. This means that the present global antibiotic discovery process is probably unsustainable in the long term.
How can we improve this situation?
Reduce the rate of emergence of resistance
The most important step would be to break the cycle of marketing a new antibiotic, followed by resistance arising within a few years. Tuberculosis (TB) chemotherapy (Mitchison, 2005) has addressed this issue by using combinations of antibiotics which reduce the rate of emergence of resistance in the target pathogen. However, multi-drug-resistant TB is on the increase, and so, while this approach can slow the emergence of resistance, it cannot stop it. However, combination therapy is an approach which could be applied more widely for the treatment of pyogenic bacteria. A potential disadvantage of this approach is that it could increase the rate of resistance emergence in normal bacterial flora, and these bacteria might then transfer resistance to pathogens. Included in this combination approach is the use of inhibitors such as clavulanic acid which block the action of bacterial β-lactamase (Brogden et al., 1981). Another potential way to deal with this issue is to develop antibiotics which induce resistance at a lower rate than existing antibiotics. For example, some compounds which target complex bacterial systems such as the membrane (Tenover, 2006; Hurdle et al., 2011), may induce resistance less readily than those such as rifampicin (Falagas et al., 2007), which inhibit single enzymes like RNA polymerase. Presumably, because synthesis of the membrane requires so many enzymes, bacteria cannot easily produce mutants with altered membranes. While this is a plausible hypothesis, resistance to daptomycin, a drug which targets bacterial membranes, is already occurring (Sakoulas et al., 2006).
Increase the rate of production of new classes of antibiotics
Historically, natural compounds have been the source of most new classes of antibiotics. The search for new classes needs to be intensified in both natural and chemical potential sources and broadened to include novel approaches. For example, an actinomyte from the deep ocean has recently been found which produces a novel antibiotic called abyssomycin (Riedlinger et al., 2004). Other sources such as plants (Mitscher et al., 1987), reptiles (Wang et al., 2008) and mammals (Flores-Villasenor et al., 2010) are also being searched. Another possible source of new classes is non-cultivable bacteria (Daniel, 2004), which constitute the majority of bacterial species. It has been suggested that the antibiotic-producing genes of these bacteria could be expressed in cultivable bacteria, and so new antibiotic classes might be produced. Chemical libraries are also being screened with the hope of finding new classes of anti-bacterials. In addition, the genomics approach (Payne et al., 2007) in which essential enzyme pathways in bacteria are targeted by inhibitors, may provide new classes, although none have been marketed so far. A further new method which targets non-multiplying bacteria (Hu et al., 2010) may also have potential, and a new class topical compound derived in this way is now in Phase III clinical trials, but no new compounds have been marketed using this approach thus far.
A crisis of this magnitude clearly needs government incentives, although money alone will be insufficient. There needs to be a political appreciation globally that infections (including those of the lower respiratory tract, human immunodeficiency virus/acquired immunodeficiency syndrome, diarrhoea, TB, malaria), of which antibiotic-resistant microbes are an important part, kill more people than heart attacks or strokes (Lopez and Mathers, 2006). The current effects of climate change on health are more difficult to judge, but if predictions are accurate, deaths due to infections such as diarrhoeal disease may increase as a result of climate*induced changes (IPCC, 2007). In addition, it needs to be understood that antibiotics are not like other groups of drugs such as anti-inflammatory agents as they become redundant in a few decades and so have to be continually replaced. Many governments already provide financial incentives, such as grants to academia and industry in the field of infectious disease. However, thus far, an insufficient proportion of these grants have been allocated to academia and industry to stimulate the generation of enough new classes of antibiotics with which to counteract the tide of antibiotic resistance.
The pharmaceutical industry needs to be encouraged to return to the antibiotic discovery arena. Only the prospect of profit from a marketed antibiotic product will persuade them to do this. The simplest way forward would be for governments to agree to a cost per unit for novel antibiotics which is comparable to anti-cancer drugs. Patent protection should be specifically extended for antibiotics on the grounds so that this will encourage companies to discover novel antibiotics, and the enhanced price should reduce the usage of the product and should extend the life of the antibiotic.
Universities should be encouraged to rebuild their antibiotic discovery sectors and to replace lost skills in this field. Clearly this will take decades, but antibiotic discovery is something that will need to be continued into the foreseeable future.
The considerable resources of the charitable sector should be channelled in part into antibiotic discovery for new classes of drugs against gram-negative bacteria, in a similar way to that for the TB and malaria programmes.
Infection control also has a role, as have better diagnostics and new vaccines.
A major change is now needed in the regulation of antibiotics. The rules are too restrictive and need to be relaxed for this group of drugs because the world is running out of antibiotics. For example, in pivotal clinical trials, specific surrogate endpoints could be allowed, such as a microbiological endpoint, rather than a clinical endpoint.
Conclusion
The existing antibiotic pipeline and the infrastructure which underpins it, is not sufficient to cope with the emergence of resistance worldwide, particularly among gram-negative bacteria. If the antibiotic discovery process continues at the same rate as at present ‘More of the same’, the antibiotic era will end within a few decades, at least for many gram-negative infections.
Potential ways forward include the use of combinations of antibiotics to reduce the rate of emergence of resistance. Concentrating on the discovery of compounds that target complex bacterial systems, such as membranes, could also reduce resistance emergence.
In the long term, many more new classes of antibiotics are needed. This will require the intervention of governments worldwide, with increased grants, subsidies, tax incentives and an increase in the unit price of new classes of antibiotics. Universities, charities and regulatory bodies will also need to change in order to encourage antibiotic discovery.
Acknowledgments
AC and YH would like to acknowledge financial support from the Medical Research Council, European Commission and the Burton Trust.
Glossary
Abbreviations
- DNA
deoxyribonucleic acid
- HAP
Hospital acquired pneumonia
- HIV/AIDS
human immunodeficiency/ acquired immunodeficiency syndrome
- MRSA
methicillin resistant Staphylococcus aureus
- RNA
ribonucleic acide
- spp
species
- TB
tuberculosis
- VAP
Ventilator associated pneumonia
Conflicts of interests
AC is founder, director and shareholder of Helperby Therapeutics Group Ltd which makes new antibiotics targeted at non-multiplying bacteria. AC and YH receive grants from Helperby Therapeutics.
Supporting Information
Teaching Materials; Figs 1–2 as PowerPoint slide.
References
- Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol. 2006;9:461–465. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Bax RP, Anderson R, Crew J, Fletcher P, Johnson T, Kaplan E, et al. Antibiotic resistance – what can we do? Nat Med. 1998;4:545–546. doi: 10.1038/nm0598-545. [DOI] [PubMed] [Google Scholar]
- Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, et al. Type 11A topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;19:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, et al. Robust salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;16:303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Carmine A, Heel RC, Morley PA, Speight TM, Avery GS. Combination clavulanic acid. Drugs. 1981;22:337–362. doi: 10.2165/00003495-198122050-00001. [DOI] [PubMed] [Google Scholar]
- Brotz-Oesterhelt H, Sass P. Postgenomic stategies in antibacterial drug discovery. Future Microbiol. 2010;5:1553–1579. doi: 10.2217/fmb.10.119. [DOI] [PubMed] [Google Scholar]
- Butler MS, Buss AD. Natural products – the future scaffolds for novel antibiotics? Biochem Pharmacol. 2006;71:919–929. doi: 10.1016/j.bcp.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Chambers HF, Deleo FR. Resistance Staph aureus to penicillin and methicillin. Nat Rev Microbiol. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Cleef BA, Verkade EJ, Wulf MW, Buiting AG, Voss A, Huijsdens XW, et al. Prevalence of livestock-associated MRSA in communities with high pig-densities in the Netherlands. PLoS ONE. 2010;5:e9385. doi: 10.1371/journal.pone.0009385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates AR, Hu Y. Targeting non-multiplying organisms as a way to develop novel antimicrobials. Trends Pharmacol Sci. 2008;29:143–150. doi: 10.1016/j.tips.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Coates A, Hu Y, Bax R, Page C. The future challenges facing the development of new antimicrobial drugs. Nature Rev Drug Discovery. 2002;1:895–910. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- CMR International. 2009. Available at http://cmr.thomsonreuters.com/pdf/cmrfb-brochure.pdf (accessed February 2011)
- Da Re S, Garnier F, Guerin E, Campoy S, Denis F, Ploy MC. The SOS response promotes qnrB quinolone-resistance determinant expression. EMBO Rep. 2009;10:929–933. doi: 10.1038/embor.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R. The soil metagenome – a rich resource for the discovery of novel natural products. Curr Opin Biotechnol. 2004;15:199–204. doi: 10.1016/j.copbio.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Devasahayam G, Scheld WM, Hoffman PS. Newer antibacterial drugs for a new century. Expert Opin Investig Drugs. 2010;19:215–234. doi: 10.1517/13543780903505092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas ME, Bliziotis IA, Fragoulis KN. Oral rifampin for eradication of Staphylococcus aureus carriage from healthy and sick populations: a systematic review of the evidence from comparative trials. Am J Infect Control. 2007;35:106–114. doi: 10.1016/j.ajic.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Fernandes P. Antibacterial discovery and development – the failure of success? Nat Biotechnol. 2006;24:1497–1503. doi: 10.1038/nbt1206-1497. [DOI] [PubMed] [Google Scholar]
- Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B.influenza. Br J Exp Path. 1929;10:226–236. [Google Scholar]
- Flores-Villasenor H, Canizalez-Roman A, Reyes-Lopez M, Nazmi K, de la Garza M, Zazueta-Beltran J, et al. Bactericidal effect of bovine lactoferrin, Lfcin, Lfampin and Lfchimera on antibiotic-resistant Staphylococcus aureus and Escherichia coli. Biometals. 2010;23:569–578. doi: 10.1007/s10534-010-9306-4. [DOI] [PubMed] [Google Scholar]
- Gillings M, Boucher Y, Labbate M, Holmes A, Krishnan S, Holley M, et al. The evolution of class 1 integrons and the rise of antibiotic resistance. J Bacteriol. 2008;190:5095–5100. doi: 10.1128/JB.00152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hair PI, Kean SJ. Daptomycin: a review of its use in the management of complicated skin and soft-tissue infections and Staphylococcus aureus bacteraemia. Drugs. 2007;67:1483–1512. doi: 10.2165/00003495-200767100-00008. [DOI] [PubMed] [Google Scholar]
- House of Lords. Inquiry into antimicrobial resistance. Commun Dis Rep CRD Wkly. 1998;24:8. [PubMed] [Google Scholar]
- Hu Y, Shamaei-Tousi A, Liu Y, Coates A. A new approach for the discovery of antibiotics by targeting non-multiplying bacteria: a novel topical antibiotic for staphylococcal infections. PLoS ONE. 2010;5:e11818. doi: 10.1371/journal.pone.0011818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle JG, O'Neill AJ, Chopra I, Lee RE. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDSA. ID Physicians call for 10 new antibiotics by 2020. 2010. Available at http://www.journals.uchicago.edu/doi/full/10.1086/652237 (accessed 19 February 2010)
- IPCC. Climate change 2007: synthesis report. Contribution of working groups I, II and III to the fourth assessment report of the intergovernmental panel on clinate change. In: Pachauri RK, Reisinger A, editors. Core Writing Team. Geneva: IPCC; 2007. p. 104. http://www.ipcc.ch/publicationsanddata/publicationsipccfourthassessmentreportsynthesisreport.htm. [Google Scholar]
- Klein E, Smith DL, Laxminarayan R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis. 2007;13:1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological and epidemiological study. Lancet Infect Dis. 2010;10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PA. Bacterial resistance to antibiotics: modified target sites. Adv Drug Deliv Rev. 2005;57:1471–1485. doi: 10.1016/j.addr.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Lopez AD, Mathers CD. Measuring the global burden of disease and epidemiological transitions: 2002–2030. Ann Trop Med Parasitol. 2006;100:481–499. doi: 10.1179/136485906X97417. [DOI] [PubMed] [Google Scholar]
- Mitchison DA. The diagnosis and therapy of tuberculosis during the past 100 years. Am J Respir Crit Care Med. 2005;171:699–706. doi: 10.1164/rccm.200411-1603OE. [DOI] [PubMed] [Google Scholar]
- Mitscher LA, Drake S, Gollapudi SR, Okwute SK. A modern look at folkloric use of anti-infective agents. J Nat Prod. 1987;50:1025–1040. doi: 10.1021/np50054a003. [DOI] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- du Plessis M, von Gottberg A, Cohen C, de Gouveia L, Klugman KP. Neisseria meningitidis intermediately resistant to penicillin and causing invasive disease in South Africa in 2001 to 2005. J Clin Microbiol. 2008;46:3208–3214. doi: 10.1128/JCM.00221-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JH. Antimicrobial drug development – the past, the present, and the future. Clin Microbiol Infect. 2004;4:23–31. doi: 10.1111/j.1465-0691.2004.1007.x. [DOI] [PubMed] [Google Scholar]
- Rice LB. Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol. 2010;1:S7–S10. doi: 10.1086/655995. [DOI] [PubMed] [Google Scholar]
- Riedlinger J, Reicke A, Zahner H, Krismer B, Bull AT, Maldonado LA, et al. Abyssomicins, inhibitors of the para-aminobenzoic acid pathway produced by the marin Verrucosispora strain AB- 18-032. J Antibiot (Tokyo) 2004;57:271–279. doi: 10.7164/antibiotics.57.271. [DOI] [PubMed] [Google Scholar]
- Sakoulas G, Alder J, Thauvin-Eliopoulos C, Moellering RC, Eliopoulos GM. Induction to daptomycin heterogeneous susceptibility in Staphylococcus aureus by exposure to vancomycin. Antimicrob Agents Chemother. 2006;50:1581–1585. doi: 10.1128/AAC.50.4.1581-1585.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Barrett JF. Empirical antibacterial drug discovery – foundation in natural products. Biochem Pharmacol. 2006;30:1996–1015. doi: 10.1016/j.bcp.2005.12.016. [DOI] [PubMed] [Google Scholar]
- So AD, Gupta N, Cars O. Tackling antibiotic resistance. BMJ Editorial. 2010;340:c2071. doi: 10.1136/bmj.c2071. [DOI] [PubMed] [Google Scholar]
- Soulsby L. The 2008 Garrod Lecture: antimicrobial resistance-animals and the environment. J Antimicrob Chemother. 2008;62:229–233. doi: 10.1093/jac/dkn183. [DOI] [PubMed] [Google Scholar]
- Tenover FC. Mechanisms of antimicrobial resistance in bacteria. Am J Infect Control. 2006;34:S3–10. S64–73. doi: 10.1016/j.ajic.2006.05.219. [DOI] [PubMed] [Google Scholar]
- Walkey AJ, O'Donnell MR, Wiener RS. Linezolid versus glycopeptide antibiotics for the treatment of suspected methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a meta-analysis of randomized controlled trials. Chest. 2010;10:1556. doi: 10.1378/chest.10-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hong J, Liu X, Yang H, Liu R, Wu J, et al. Snake cathelicidin from Bungarus fasciatus is a potent peptide antibiotics. PLoS ONE. 2008;B3:e3217. doi: 10.1371/journal.pone.0003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R. Multidrug resistance responsible for half of deaths from healthcare associated infections in Europe. BMJ. 2008;336:1266. doi: 10.1136/bmj.39601.623808.4E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia G, Menendez P, Monache GD, Misiti D, Nevola L, Botta B. The contribution of oxazolidinone frame to the biological activity of pharmaceutical drugs and nature products. Mini Rev Med Chem. 2007;7:389–409. doi: 10.2174/138955707780363783. [DOI] [PubMed] [Google Scholar]
- Zhanel GG, Trapp S, Gin AS, DeCorby M, Lagace-Wiens PR, Rubinstein E, et al. Dalbavancin and telavancin: novel lipoglycopeptides for the treatment of Gram-positive infections. Expert Rev Anti Infect Ther. 2008;6:67–81. doi: 10.1586/14787210.6.1.67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.