

Abstract
Recent technical advances have renewed interest in device-based therapy for the treatment of drug-resistant hypertension. Findings from recent clinical trials regarding the efficacy of electrical stimulation of the carotid sinus for the treatment of resistant hypertension are reviewed here. The main goal of this article, however, is to summarize the preclinical studies that have provided insight into the mechanisms that account for the chronic blood pressure lowering effects of carotid baroreflex activation. Some of the mechanisms identified were predictable and confirmed by experimentation. Others have been surprising and controversial and resolution will require further investigation. Although feasibility studies have been promising, firm conclusions regarding the value of this device-based therapy for the treatment of resistant hypertension awaits the results of current multicenter trials.
During the decade of the 1960’s, various medical devices for electrically activating the carotid baroreflex were designed and used with the intent of lowering arterial pressure in patients whose severe hypertension was inadequately controlled by available medication.1-3 While the findings in some early studies suggested that long-term stimulation of the carotid baroreflex might chronically lower arterial pressure, this approach for treatment of drug resistant hypertension was abandoned for two primary reasons. The technology was too crude to achieve reliable baroreflex activation and long-term blood pressure responses, particularly without adverse effects. Moreover, the advent of new, effective pharmacological therapy for the treatment of hypertension diminished interest in further developing this invasive device-based therapeutic strategy for lowering blood pressure.
Since these studies almost 50 years ago, several different classes of drugs have been developed to treat hypertension. Given the success of drug therapy in treating hypertension and reducing associated adverse cardiovascular effects, it would appear paradoxical that there are now on-going clinical trials evaluating the efficacy of this non-pharmacological approach for lowering arterial pressure. Why the recent interest in device-based therapy for the treatment of hypertension? There are two notable reasons for the resurgence of efforts directed at carotid baroreflex activation as a therapeutic strategy for the control of hypertension. First, despite the vast armamentarium of antihypertensive agents available to clinicians for treating hypertension, the percentage of patients achieving adequate blood-pressure control remains unacceptably low.4 In some cases, patients would benefit from drug therapy optimization and better compliance. However, in some patients, adequate blood pressure control is not correctable by these approaches. These patients have resistant hypertension, defined as blood pressure that remains above goal in spite of the concurrent use of 3 or more antihypertensive agents, with one being a diuretic, and all being given at an optimized dose.4-5 Despite thoughtful therapy, patients remain resistant to medical therapy and could benefit from new approaches such as baroreflex stimulation. Another major impetus for current clinical trials using baroreflex therapy has been the development of modern device technology for baroreflex activation that has overcome the technical limitations encountered in the early studies using chronic carotid baroreflex activation.
The development of device-based therapy for activation of the baroreflex requires mechanistic knowledge of the role of the baroreflex in long-term control of arterial pressure. Preclinical studies in dogs subjected to chronic electrical stimulation of the carotid sinus have provided insight into the basic physiological mechanisms that account for the sustained reductions in arterial pressure during prolonged activation of the baroreflex, and these studies will be emphasized in this review. However, it will be apparent that a complete understanding of the blood pressure lowering mechanisms still remains enigmatic. Some of the results have confirmed expected beliefs and predicted mechanisms, while others have been surprising and controversial. Finally, responses to carotid baroreflex activation in human subjects with resistant hypertension will be presented, including recently published findings from a recent feasibility trial demonstrating the safety and therapeutic efficacy of carotid sinus stimulation.
Modern Features of Device-Based Carotid Baroreflex Activation
Issues related to the technology and approach for chronic electrical activation of the carotid baroreflex have been discussed in greater detail in earlier communications.3,6 With the present system for activation of the carotid baroreflex, the stimulating electrodes are implanted in the perivascular space around the carotid sinuses and not around the carotid sinus nerve, as in some of the early studies. Although not rigorously evaluated, this virtually eliminates the possibility of damage to the carotid sinus nerve, not an uncommon occurrence in studies conducted in the 1960’s. Theoretically, stimulation of the carotid sinus also reduces the likelihood of activating carotid body chemoreceptor afferents which, along with the baroreceptor fibers, are present in the carotid sinus nerve. This is important because activation of carotid body afferent fibers increases sympathetic activity and respiratory rate, undesirable responses that would limit the therapeutic efficacy of baroreflex activation. Additionally problems reported in early studies related to extraneous nerve and muscle stimulation and pain have been solved using modern electrode technologies that permit localized electrical stimulation of carotid baroreceptors. Another important feature is that the internally implantable, miniature pulse generator is completely programmable by radiofrequency control using an external programming system similar in concept to that used for cardiac pacemakers. This provides permanent control of current delivery along the leads to the electrodes and allows time-dependent modification of stimulation parameters. However, at present, more information is needed on the selection of optimal stimulation patterns and stimulation parameters. The ability of this system to be programmed for different current delivery throughout the day and night offers the intriguing possibility of customizing therapy to the particular day-night blood pressure pattern of the individual. To date, however, this application has not been tested. Finally, the associated rapid on- and off-transient arterial pressure responses, and the potential to achieve graded and controllable long-term reductions in arterial pressure are additional desirable features of this device.
Chronic Electrical Activation of the Carotid Baroreflex Produces Sustained Reductions in Sympathetic Activity and Arterial Pressure
A potential role of baroreflexes in long-term control of arterial pressure is often conceptually dismissed because of the accepted fact that baroreflexes reset in the direction of change in arterial pressure.7-10 Resetting diminishes the ability of the baroreflex to chronically alter sympathetic activity and, thus, serve as a long-term controller of arterial pressure. Resetting of the baroreflex arc occurs as a result of adaptations manifested centrally or at the baroreceptors themselves. Unfortunately, technical limitations have precluded a quantitative evaluation of the degree of baroreflex resetting in response to long-term changes in arterial pressure and the contribution of central adaptations to the resetting process. In regard to central resetting, if this were a dominant process that counteracts changes in baroreceptor input into the brain, then chronic electrical activation of the carotid sinus would not be expected to produce appreciable, sustained reductions in sympathetic activity and arterial pressure. Clearly, this is not the case.
A unique aspect of the current technology for activation of the carotid baroreflex is that electrical stimulation of the carotid sinuses bypasses mechanotransduction at the level of the baroreceptors and allows for precise control of carotid baroreceptor input into the brain. An especially important finding from experimental studies is that prolonged baroreflex activation leads to sustained reductions in arterial pressure and heart rate.6,11-15 Furthermore, the chronic lowering of blood pressure during baroreflex activation is associated with sustained reductions in plasma NE concentration 6,11-15and whole body NE spillover to plasma,15 indicating unremitting suppression of central sympathetic outflow. These responses indicate that central resetting is relatively unimportant in diminishing the suppression of sympathetic activity and the attendant reduction of blood pressure induced by prolonged electrical activation of the carotid sinus. Additionally, despite reduced arterial pressure, it would appear that the aortic baroreflex, constrained by resetting, also fails to increase sympathetic activity appreciably and offset the sympathoinhibition induced by stimulation of the carotid sinus. It is also relevant to point out that if the reactivity of α1-adrenergic receptors in the peripheral vasculature were to increase in response to reduced sympathetic outflow, this could counteract the fall in arterial pressure.16 However, experimental studies have shown that despite the chronic suppression of sympathetic activity, increases in α1-adrenergic receptor reactivity in the peripheral circulation do not provide a significant compensatory opposition to the fall in arterial pressure induced by baroreflex activation.15 Thus, neither adaptations within the CNS nor in the peripheral circulation offset the long-term sympathoinhibitory blood pressure lowering effects of electrical stimulation of the carotid baroreflex.
The Role of the Kidneys in Mediating the Chronic Blood Pressure Lowering Effects of Baroreflex Activation
Long-term regulation of arterial pressure is closely linked to volume homeostasis through the renal body-fluid feedback mechanism.7-9,17 According to this concept, for a given level of sodium intake, long-term changes in arterial pressure cannot occur without alterations in renal excretory function. Therefore, although the baroreflex plays an important role in short-term regulation of arterial pressure by mediating changes in peripheral resistance, vascular capacity, and cardiac function, these autonomic responses, even if sustained chronically, would not be expected to produce long-term changes in arterial pressure unless accompanied by alterations in pressure natriuresis. More specifically, in the absence of an increase in renal excretory function, chronic activation of the baroreflex should lead to only a transient fall in arterial pressure. This is because the initial fall in arterial pressure would be counteracted by salt and water retention, which would persist until blood volume increased sufficiently to restore arterial pressure to normal levels. A modest degree of salt and water retention is indeed associated with the initial fall in arterial pressure and increase in vascular capacitance induced by baroreflex activation (Figure). However, because the fall in arterial pressure during carotid sinus stimulation is sustained and not just transient, baroreflex activation must have parallel effects on renal excretory function. Experimental and theoretical studies indicate that one way in which activation of the baroreflex could chronically enhance pressure natriuresis and promote sodium excretion is by suppressing RSNA.7-9,18-21
Figure.
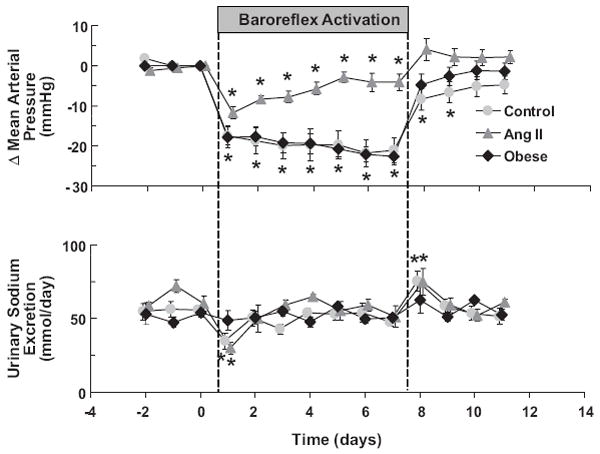
Mean arterial pressure and sodium excretory responses to prolonged baroreflex activation in dogs before (control) and after induction of ANG II hypertension. Responses to baroreflex activation in an additional group of dogs with obesity hypertension are also illustrated. Values are mean ±SEM (n=6). P<0.05 vs control. Values for MAP before baroreflex activation: control=93±1 mmHg, ANG II hypertension=129±3 mmHg, and obese hypertension=110±3 mmHg. (From Lohmeier et al).13
Angiotensin II Hypertension is Relatively Resistant to the Blood Pressure Lowering Effects of Baroreflex Activation
A noteworthy observation during chronic baroreflex activation is the failure of renin secretion to increase, despite substantial reductions in arterial pressure.6,11-15 Quantitative studies in chronically instrumented dogs have demonstrated that reductions in renal perfusion pressure ≥15 mmHg lead to marked increases in renin secretion.22-23 Therefore, baroreflex activation must have an inhibitory influence on renin secretion as sustained reductions in mean arterial pressure (MAP) of 20-25 mmHg during carotid sinus stimulation do not increase plasma renin activity (PRA). As alterations in renal adrenergic activity have both acute and chronic effects on renin secretion, it is likely that sustained reductions in RSNA account for the long-term inhibitory effects of baroreflex activation on renin secretion, an effect that counteracts pressure-dependent renin release. Based upon this impressive inhibitory influence on renin secretion, we hypothesized that were it not for the sustained renal sympathoinhibitory effects on renin secretion, the long-term blood-pressure lowering response to baroreflex activation would be greatly diminished.
This hypothesis was tested during equivalent degrees of carotid sinus stimulation before and during continuous infusion of angiotensin II. Tests were performed in the same dogs at an infusion rate producing high physiological levels of the potent sodium retaining hormone.11 Under control conditions prior to ANG II infusion, acute and chronic reductions in MAP (~20mmHg) in response to baroreflex activation were comparable. Next, after induction of ANG II hypertension, the acute fall in MAP in response to baroreflex activation was found to be similar to that observed under control conditions. However, this robust hypotensive response did not persist during chronic baroreflex activation and concurrent infusion of ANG II (Figure). Compared to the response under control conditions, the chronic blood pressure lowering effect of baroreflex activation was diminished by 75-80% in ANG II hypertension. This study clearly demonstrates that the long-term antihypertensive effects of baroreflex activation are markedly diminished in the presence of modest and persistent increases in circulating levels of ANG II and, therefore, emphasizes the importance of reflex-mediated suppression of renin secretion in counteracting pressure-dependent renin release and permitting lowering of arterial pressure.
The pre-existing level of RSNA may be another determinant of the long-term arterial pressure response to baroreflex activation and a contributing factor to the diminished antihypertensive response to electrical stimulation of the carotid sinus in ANG II hypertension. Experimental studies in chronically instrumented animals have clearly demonstrated that naturally-induced, baroreflex-mediated suppression of RSNA is a sustained response to ANG II hypertension.18-20 Thus, if RSNA is already suppressed in ANG II hypertension as a result of the natural engagement of the baroreflex, it would be reasonable to expect diminished inhibition of RSNA and arterial pressure during electrical activation of the baroreflex. If this rationale is correct, the antihypertensive effects of baroreflex activation would be expected to be substantial in forms of hypertension associated with increased RSNA, such as obesity hypertension.
Obesity Hypertension is Totally Abolished by Baroreflex Activation
There is considerable evidence that activation of the sympathetic nervous system plays an important role in the pathogenesis of obesity hypertension.24-26 Furthermore, increased RSNA is clearly implicated in the hypertensive process. Therefore, because chronic activation of the baroreflex has sustained effects to suppress RSNA, we hypothesized that electrical stimulation of the baroreflex would greatly diminish the severity of hypertension in obese dogs fed a high-fat diet. After 4 weeks of feeding dogs a high-fat diet, MAP increased ~15 mmHg in association with a 50% increase in weight gain.13 Tachycardia and increased plasma levels of NE were prominent features associated with the hypertension, consistent with activation of the sympathetic nervous system. Baroreflex activation during week 5 of the high-fat diet greatly attenuated the tachycardia and totally abolished the increments in plasma NE concentration and arterial pressure associated with weight gain. There were no changes in PRA during prolonged stimulation of the carotid baroreflex. Thus, in contrast to the ANG II model of hypertension, the antihypertensive effects of baroreflex activation were pronounced and sustained in obesity hypertension (Figure). These findings suggest greater degrees of baroreflex-induced renal sympathoinhibition in obesity than in ANG II hypertension, with pre-existing levels of RSNA being high in obesity and low in ANG II hypertension. Furthermore, as in normotensive dogs, the absence of an increase in PRA, despite the large fall in MAP, supports the hypothesis that reductions in RSNA counteract pressure-dependent renin secretion during baroreflex activation. Once again, had renal sympathoinhibition not offset pressure-dependent renin release, it is likely that the antinatriuretic effects of increased circulating ANG II would have diminished the antihypertensive response to baroreflex activation.
The Presence of the Renal Nerves is not an Obligate Requirement for Long-Term Reductions in Arterial Pressure During Baroreflex Activation
The above studies, along with observations in hypertensive animals during natural activation of the baroreflex, indicate that the renal nerves provide a logical link between reductions in central sympathetic outflow and increased renal excretory function that leads to chronic lowering of blood pressure during prolonged activation of the baroreflex. Therefore, we directly evaluated the importance of the renal nerves in mediating long-term baroreflex-mediated reductions in arterial pressure by chronically activating the carotid baroreflex in the same normotensive dogs before and, following a recovery period, after bilateral renal denervation.12 We hypothesized that if the renal nerves are critical mediators of the long-term blood pressure-lowering effects of the baroreflex, then chronic electrical stimulation of the carotid sinus would not lead to sustained reductions in arterial pressure after renal denervation. To our great surprise, the lowering of arterial pressure in response to activation of the baroreflex after renal denervation was not significantly different from when the renal nerves were intact, although there was a tendency for greater sodium retention and a smaller fall in arterial pressure in the absence than in the presence of the renal innervation. Therefore, we speculated that after renal denervation the sustained fall in arterial pressure during baroreflex activation was dependent upon a greater degree of fluid retention and attendant, exaggerated activation of redundant natriuretic mechanisms. While not tested experimentally, theoretical support for this hypothesis was provided from computer simulations using an established integrative mathematical model of human physiology (Quantitative Human Physiology, QHP2008).21 This model predicted that after renal denervation, there is excessive fluid retention during baroreflex activation and that increased plasma levels of atrial natriuretic peptide and renal interstitial pressure become increasingly important in promoting sodium excretion and lowering arterial pressure.
Baroreflex Activation Enhances Drug-Induced Lowering of Arterial Pressure in Experimental Studies
As indicated above, an impetus for the development of technology for electrical stimulation of the carotid sinus has been the need for new treatments for blood pressure control in patients with resistant hypertension. Because subjects with resistant hypertension have inadequate blood pressure control despite therapy involving multiple drug regimens, we evaluated whether baroreflex activation might potentiate the blood pressure lowering effects of different classes of antihypertensive drugs. Clinical studies have shown that the chronic lowering of blood pressure associated with some drugs, including diuretics and calcium channel blockers, leads to chronic increases in sympathetic activity, which might be expected to attenuate antihypertensive efficacy.27-29
Our previous studies in dogs suggested that prolonged baroreflex activation had the capacity to reduce arterial pressure considerably more than chronic blockade of α1- and β1,2-adrenergic receptors.30 Therefore, we determined whether central inhibition of sympathetic outflow by baroreflex activation would lower arterial pressure by mechanisms distinct from diminished activation of the adrenergic receptors believed to be most important in mediating the actions of the sympathetic nervous system.14 During chronic administration of prazosin and propranolol, at doses that completely blocked the normal agonist stimulation of α1- and β1,2-adrenergic receptors, MAP decreased ~20 mmHg. However, this lowering of blood pressure during chronic adrenergic blockade was associated with sustained activation of the sympathetic nervous system, as reflected by a 3-fold increase in plasma NE concentration. The additional advantage of reflex-induced sympathoinhibition in lowering arterial pressure was apparent during the subsequent week of baroreflex activation and continued adrenergic blockade. During the 7 days of baroreflex activation, MAP fell by an additional 10 mmHg, concomitant with reductions in plasma NE concentration to control levels. Although possibly hormonally mediated, the abrupt changes in arterial pressure immediately after baroreflex activation suggested that this additional long-term fall in arterial pressure was due to reduced release of NE and/or cotransmitters from adrenergic nerve terminals. Based on the pronounced vasoactive responses to localized arterial administration of α2-adrenergic receptor antagonists in dogs and human subjects,31-32 we focused on the possibility that diminished NE release and attendant activation of postjunctional vasoconstrictor α2-adrenergic receptors contributed importantly to the additional blood pressure-lowering effects of baroreflex activation during α1-and β1-adrenergic blockade. Indeed, in response to acute administration of a peripherally acting α2-antagonist, there was a considerably smaller fall in MAP during adrenergic blockade + baroreflex activation than during adrenergic blockade alone.
A similar scenario occurred during chronic administration of another commonly used antihypertensive agent, the calcium channel blocker amlodipine.33 As with adrenergic blockade, the chronic lowering of blood pressure during amlodipine administration (MAP decreased ~ 15 mmHg) was associated with a sustained increase in sympathetic activity (plasma NE concentration). In parallel with activation of the sympathetic nervous system, PRA increased ~ 6-fold. Moreover, with continued amlodipine administration, MAP continued to decrease by more than 10 mmHg during the subsequent 7 days of baroreflex activation. This additional reduction in arterial pressure during baroreflex-mediated suppression of central sympathetic outflow occurred concomitantly with the return of plasma NE concentration and PRA to or almost to control levels.
Taken together, these findings suggest that the fall in arterial pressure with some classes of antihypertensive drugs is associated with sustained activation of the sympathetic nervous system, presumably due to the natural unloading of arterial baroreceptors. Furthermore, they indicate that this reflex-induced sympathoexcitation attenuates the lowering of blood pressure and that concomitant inhibition of central sympathetic outflow by baroreflex activation lowers arterial pressure further. Thus, persistent baroreflex-mediated sympathetic activation during chronic antihypertensive therapy may be a mechanism contributing to resistant hypertension. Furthermore, the ability of carotid sinus stimulation to counteract drug-induced sympathetic activation by suppressing central sympathetic outflow may partially account for the lowering of arterial pressure in patients with resistant hypertension.34-36
Baroreflex Activation Therapy in Patients with Resistant Hypertension
The first comprehensive evaluation of baroreflex activation therapy for resistant hypertension is now completed (please see Online Data Supplement at http://hyper.ahajournals.org for a listing of all CVRx sponsored trials registered at www.clinicaltrials.gov). The DEBuT-HT (Device-Based Therapy in Hypertension Trial, identifier NCT00710190) was a multicenter, prospective, nonrandomized feasibility trial evaluating the safety and efficacy of 3 months of baroreflex activation therapy in 45 patients with resistant hypertension.34 All subjects received optimal doses of antihypertensive medications with the median number of drugs being 5. During this time, medications were kept constant for 2 months before entry and during the 3 months of therapy. Baseline arterial pressure and heart rate were 179/105 mmHg and 80 beats/min, respectively. After 3 months of continuous, bilateral device-based therapy, there was a statistically significant reduction in arterial pressure by 21/12 mmHg and in heart rate by 8 beats/min. Furthermore, in a cohort of subjects followed for 2 years of therapy, these results were sustained and possibly even improved. After 1 year of baroreflex activation (n=26), reductions in arterial pressure and heart rate were 30/8 mmHg and 8 beats/min, respectively. The corresponding values after 2 years (n=17) were 33/22 mmHg and 11 beats/min. There were no significant changes in the number of antihypertensive drugs taken by the patients during the 2 year time period. Optimal stimulation parameters were identified on an individual basis as part of the acute dose-response testing that occurs at follow-up, and all subjects were stimulated bilaterally. In contrast, the majority of patients enrolled in the Pivotal Trial (discussed next), have been programmed for unilateral activation.
Thus, in patients with resistant hypertension, chronic baroreflex activation decreased arterial pressure beyond what could be achieved with drug treatment. Further, the procedure and device were well tolerated with favorable measures of safety during 2 years of therapy. With these encouraging results, an FDA approved randomized, double-blind placebo controlled US/European Pivotal Trial (identifier NCT00442286) has been ongoing since 2007. After all patients were fully enrolled and implanted (n=326), the Data Safety Monitoring Board informed investigators that the 6-month efficacy endpoint was not likely to be met: the predetermined responder analysis comparing therapy and control groups underestimated the magnitude of pressure reduction observed in the control group. Results of patients through 1 year of therapy are strikingly consistent with results from the forerunner DEBuT trial discussed above. Patients continue to be followed and full trial results will be presented in 2011, after completion of all endpoint-related follow-up visits.
Greater insight into the physiological responses to baroreflex activation in patients with resistant hypertension has been provided by two studies in subgroups of patients from the DEBut-HT trial. In one study, measurement of muscle sympathetic nerve activity (MSNA) by microneurography provided direct evidence for reduced MSNA during acute baroreflex activation; in this study, acute depressor responses were correlated with reductions in MSNA, suggesting a causal relationship.35 However, to date, chronic determinations of MSNA in patients treated with baroreflex activation therapy are lacking. As afferent impulses during electrical stimulation of the carotid sinus are continuous by design and not pulse synchronous, another important observation in this study was that baroreflex activation did not impair physiological baroreflex regulation. This observation is consistent with findings made during simultaneous 24-hour recordings of arterial pressure and heart rate in dogs.15 These recordings in dogs indicated that the sensitivity of spontaneous baroreflex control of heart rate actually increased during chronic baroreflex activation, despite the pattern of continuous, nonpulsatile electrical stimulation of the carotid sinus. Enhanced baroreflex sensitivity during continuous activation of the baroreflex may partially account for the absence of postural hypotension during device-based therapy in patients with resistant hypertension.34
Twenty-four hour recordings of hemodynamics have provided further insight into dynamic baroreflex control of circulatory function during chronic electrical stimulation of the carotid sinus. In a second subgroup of patients from the DEBut-HT study population, 24-hour recordings of ECG showed increased heart rate variability, along with sustained reductions in arterial pressure and heart rate, when determined after 3 months of baroreflex activation therapy.36 Continuous hemodynamic measurements in dogs indicate that chronic baroreflex activation not only leads to increased heart rate variability, but to decreased blood pressure variability as well.15 Taken together, these dynamic measures of circulatory function are consistent with inhibition of sympathetic activity and augmentation of parasympathetic activity during chronic electrical stimulation of the baroreflex. By decreasing blood pressure variability and associated target organ damage,37 and diminishing the likelihood of adverse cardiac events, these dynamic alterations in baroreflex function may contribute to the clinical benefit of baroreflex activation.
Conclusions
The experimental studies reviewed above have provided comprehensive insight into the mechanisms that account for the chronic lowering of blood pressure during suppression of central sympathetic outflow and, more specifically, during prolonged activation of the baroreflex. Although not directly tested, these studies suggest that baroreflex-mediated suppression of RSNA chronically increases renal excretory function and, in so doing, permits sodium balance during sustained reductions in arterial pressure. This increase in renal excretory function during baroreflex activation is critically dependent upon diminished adrenergic stimulation of renin secretion, a response that offsets pressure-dependent renin release. However, there are clearly additional mechanisms beyond renal sympathoinhibition that promote sodium excretion during sustained activation of the baroreflex.
Older age, obesity, sleep apnea, and some classes of antihypertensive drugs are commonly associated with both increased sympathetic activity and resistant hypertension. Thus, increased sympathetic activity may have an unappreciated role in the pathogenesis of resistant hypertension. If this hypothesis is correct, it would account for the impressive antihypertensive responses to suppression of central sympathetic outflow by baroreflex activation in patients with resistant hypertension. Similarly, striking therapeutic blood pressure responses have also been reported in patients with resistant hypertension when subjected to a targeted approach for disrupting renal sympathetic drive to the kidneys--endovascular radiofrequency ablation of the renal sympathetic nerves.38, However, the therapeutic benefit of these devices in hypertension, and in other states of sympathetic activation such as heart failure,39 may also be dependent upon their ability to not only suppress RSNA, but sympathetic outflow to other regions as well.
It remains to be seen if and how rapidly baroreflex activation will be adopted in clinical practice. Various factors will influence this, including magnitude of pressure reduction, adverse outcome risk reduction, long-term safety profile, implant invasiveness, battery life, and lifetime treatment costs. As with device-based therapies in general, there will be an evolution of technology and evidence basis that will allow the health care system to determine the breath of appropriate indications.
Supplementary Material
Acknowledgments
The authors thank Eric Lovett for his helpful comments in reviewing this manuscript.
Sources of Funding The preclinical studies using electrical stimulation of the carotid sinus were supported by National Heart, Lung, and Blood Institute Grant HL-51971 and CVRx, Inc.
Footnotes
Conflicts of Interest/Disclosure Thomas E. Lohmeier Consultant fees, Scientific Advisory Board,-CVRx
References
- 1.Schwartz SI, Griffith LSC, Neistadt A, Hagfors N. Chronic carotid sinus nerve stimulation in the treatment of essential hypertension. Am J Surg. 1967;114:5–15. doi: 10.1016/0002-9610(67)90034-7. [DOI] [PubMed] [Google Scholar]
- 2.Tuckman J, Reich T, Lyon AF, Goodman B, Mendlowitz M, Jacobson JH. Hypertension 16: Neural Control of Arterial Pressure. New York: American Heart Association; 1968. Electrical stimulation of the sinus nerves in hypertensive patients. clinical evaluation and physiological studies; pp. 23–26. [Google Scholar]
- 3.Lohmeier TE, Barrett AM, Irwin ED. Prolonged activation of the baroreflex: a viable approach for the treatment of resistant hypertension? Curr Hypertens Rev. 2005;7:193–198. doi: 10.1007/s11906-005-0009-0. [DOI] [PubMed] [Google Scholar]
- 4.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jones DW, Materson BJ, Oparil S, Wright JT, Jr, Roccella EJ. Seventh report of the National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica A, Ferdinand K, Giles TD, Falkner B, Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;117:e510–526. doi: 10.1161/CIRCULATIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- 6.Lohmeier TE, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Prolonged activation of the baroreflex produces sustained hypotension. Hypertension. 2004;43:306–311. doi: 10.1161/01.HYP.0000111837.73693.9b. [DOI] [PubMed] [Google Scholar]
- 7.Cowley AW., Jr Long-term control of arterial pressure. Physiol Rev. 1992;72:231–300. doi: 10.1152/physrev.1992.72.1.231. [DOI] [PubMed] [Google Scholar]
- 8.Lohmeier TE, Hildebrandt DA, Warren S, May PJ, Cunningham JT. Recent insights into the interactions between the baroreflex and the kidneys in hypertension. Am J Physiol Regulatory Integrative Comp Physiol. 2005;288:R828–R836. doi: 10.1152/ajpregu.00591.2004. [DOI] [PubMed] [Google Scholar]
- 9.Lohmeier TE, Drummond HA. The baroreflex in the pathogenesis of hypertension. In: Lip GYH, Hall JE, editors. Comprehensive Hypertension. Philadelphia, PA: Elsevier; 2007. pp. 265–279. [Google Scholar]
- 10.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular diseases. Physiol Rev. 2010;90:513–557. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 11.Lohmeier TE, Dwyer TM, Hildebrandt DA, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Influence of prolonged baroreflex activation on arterial pressure in angiotensin hypertension. Hypertension. 2005;46:1194–1200. doi: 10.1161/01.HYP.0000187011.44201.2e. [DOI] [PubMed] [Google Scholar]
- 12.Lohmeier TE, Hildebrandt DA, Dwyer TM, Barrett AM, Irwin ED, Rossing MA, Kieval RS. Renal denervation does not abolish sustained baroreflex-mediated reductions in arterial pressure. Hypertension. 2007;49:373–379. doi: 10.1161/01.HYP.0000253507.56499.bb. [DOI] [PubMed] [Google Scholar]
- 13.Lohmeier TE, Dwyer TM, Irwin ED, Rossing MA, Kieval RS. Prolonged activation of the baroreflex abolishes obesity-induced hypertension. Hypertension. 2007;49:1307–1314. doi: 10.1161/HYPERTENSIONAHA.107.087874. [DOI] [PubMed] [Google Scholar]
- 14.Lohmeier TE, Hildebrandt DA, Dwyer TM, Iliescu R, Irwin ED, Cates AW, Rossing MA. Prolonged activation of the baroreflex decreases arterial pressure even during chronic adrenergic blockade. Hypertension. 2009;53:833–838. doi: 10.1161/HYPERTENSIONAHA.109.128884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lohmeier TE, Iliescu R, Dwyer TM, Irwin ED, Cates AW, Rossing MA. Sustained suppression of sympathetic activity and arterial pressure during chronic activation of the carotid baroreflex. Am J Physiol Heart Circ Physiol. 2010;299:H402–H409. doi: 10.1152/ajpheart.00372.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charkoudian N, Joyner MJ, Sokolnicki LA, Johnson CP, Eisenach JH, Dietx NM, Curry TB, Wallin BG. Vascular adrenergic responsiveness is inversely related to tonic activity of sympathetic vasoconstrictor nerves in humans. J Physiol. 2006;572:821–827. doi: 10.1113/jphysiol.2005.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guyton AC. Arterial Pressure and Hypertension. Philadelphia: Saunders; 1980. [Google Scholar]
- 18.Carroll RG, Lohmeier TE, Brown AJ. Chronic angiotensin II infusion decreases renal norepinephrine overflow in the conscious dog. Hypertension. 1984;6:675–681. doi: 10.1161/01.hyp.6.5.675. [DOI] [PubMed] [Google Scholar]
- 19.Lohmeier TE, Lohmeier JR, Haque A, Hildebrandt DA. Baroreflexes prevent neurally-induced sodium retention in angiotensin hypertension. Am J Physiol Regulatory Integrative Comp Physiol. 2000;279:R1437–R1448. doi: 10.1152/ajpregu.2000.279.4.R1437. [DOI] [PubMed] [Google Scholar]
- 20.Barrett CJ, Guild S-J, Ramchandra R, Malpas SC. Baroreceptor denervation prevents sympathoinhibition during angiotensin II-induced hypertension. Hypertension. 2005;46:1–5. doi: 10.1161/01.HYP.0000168047.09637.d4. [DOI] [PubMed] [Google Scholar]
- 21.Iliescu R, Lohmeier TE. Lowering of blood pressure during chronic suppression of central sympathetic outflow: insight from computer simulations. Clin Exp Pharmacol Physiol. 2009;37:e24–33. doi: 10.1111/j.1440-1681.2009.05291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirchheim HR, Finke R, Hackenthal E, Lowe W, Persson P. Baroreflex sympathetic activation increases threshold pressure for pressure-dependent renin release. Pfleugers Arch. 1985;405:127–135. doi: 10.1007/BF00584533. [DOI] [PubMed] [Google Scholar]
- 23.Yang HM, Lohmeier TE, Kivlighn SD, Smith MJ., Jr Sustained increases in plasma epinephrine concentration do not modulate renin release. Am J Physiol Endocrinol Metab. 1989;257:E57–E64. doi: 10.1152/ajpendo.1989.257.1.E57. [DOI] [PubMed] [Google Scholar]
- 24.Hall JE, Hildebrandt DA, Kuo J. Obesity hypertension: role of leptin and sympathetic nervous system. Am J Hypertens. 2001;14:103s–115s. doi: 10.1016/s0895-7061(01)02077-5. [DOI] [PubMed] [Google Scholar]
- 25.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension. 2006;48:1–10. doi: 10.1161/01.HYP.0000242642.42177.49. [DOI] [PubMed] [Google Scholar]
- 26.Shibao C, Gamboa A, Diedrich A, Ertl AC, Chen KY, Byrne DW, Farley G, Paranjape SY, Davis SN, Biaggioni I. Hypertension. 2007;49:17–33. doi: 10.1161/01.HYP.0000251679.87348.05. [DOI] [PubMed] [Google Scholar]
- 27.Fu Q, Zhang R, Witkowski S, Arbab-Zadeh A, Prasad A, Okazaki K, Levine BD. Persistent sympathetic activation during chronic antihypertensive therapy. A potential mechanism for long-term morbidity? Hypertension. 2005;45:513–521. doi: 10.1161/01.HYP.0000158312.63381.c1. [DOI] [PubMed] [Google Scholar]
- 28.De Champlain J, Karas M, Nguyen P, Cartier P, Wistaff R, Toal CB, Nadeau R, Larochelle P. Different effects of nifedipine and amlodipine on circulating catecholamine levels in essential hypertensive patients. J Hypertens. 1998;16:1357–1369. [PubMed] [Google Scholar]
- 29.Ligtenberg G, Blankestijn PJ, Oey PL, Klein IHH, Dijkhorst-Oei L-T, Boomsma F, Wieneke GH, van Huffelen AC, Koomans HA. Reduction of sympathetic hyperactivity by enalapril in patients with chronic renal failure. N Engl J Med. 1999;340:1321–1328. doi: 10.1056/NEJM199904293401704. [DOI] [PubMed] [Google Scholar]
- 30.Lohmeier TE, Hildebrandt DA, Hood WA. Renal nerves promote sodium excretion during long-term increases in salt intake. Hypertension. 1999;33:487–492. doi: 10.1161/01.hyp.33.1.487. [DOI] [PubMed] [Google Scholar]
- 31.Buckwalter JB, Clifford PS. α- Adrenergic vasoconstriction in active skeletal musclesduring dynamic exercise. Am J Physiol Heart Circ Physiol. 1999;277:H33–H39. doi: 10.1152/ajpheart.1999.277.1.H33. [DOI] [PubMed] [Google Scholar]
- 32.Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Post-junctional α-adrenoceptors and basal limb vascular tone in healthy men. J Physiol. 2002;540:1103–1110. 2002. doi: 10.1113/jphysiol.2001.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iliescu R, Dwyer TM, Irwin ED, Georgakopoulos D, Rossing MA, Lohmeier TE. Electrical activation of carotid baroreflex enhances the chronic blood pressure lowering effects of calcium channel blockade by abolishing sympathoexcitation. Hypertension. 2010;56:e71–72. abstract. [Google Scholar]
- 34.Scheffers IJM, Kroon AA, Schmidli J, Jordan J, Tordoir JJM, Mohaupt MG, Luft FC, Haller H, Menne J, Engeli S, Ceral J, Eckert S, Erglis A, Narkiewicz K, Philipp T, de Leeuw PW. Novel baroreflex activation therapy in resistant hypertension. J Am Coll Cardiol. 2010;56:1254–1258. doi: 10.1016/j.jacc.2010.03.089. [DOI] [PubMed] [Google Scholar]
- 35.Heusser K, Tank J, Engeli S, Diedrich A, Menne J, Eckert S, Peters T, Sweep FCGJ, Haller H, Pichlmaier, Luft F, Jordan J. Carotid baroreceptor stimulation, sympathetic activity, baroreflex function, and blood pressure in hypertensive patients. Hypertension. 2010;55:619–626. doi: 10.1161/HYPERTENSIONAHA.109.140665. [DOI] [PubMed] [Google Scholar]
- 36.Wustmann K, Kucera JP, Scheffers I, Mohaupt M, Kroon AA, de Leeuw PW, Schmidli J, Allemann Y, Delacretaz E. Effects of chronic baroreceptor stimulation on the autonomic cardiovascular regulation in patients with drug-resistant arterial hypertension. Hypertension. 2009;54:530–536. doi: 10.1161/HYPERTENSIONAHA.109.134023. [DOI] [PubMed] [Google Scholar]
- 37.Parati G. Blood pressure variability: its measurement and significance. J Hypertens. 2005;23:s19–s25. doi: 10.1097/01.hjh.0000165624.79933.d3. [DOI] [PubMed] [Google Scholar]
- 38.Symplicity HTN-2 investigators. Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Bohm M. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomized controlled trial. Lancet. 2010;376:1903–1909. doi: 10.1016/S0140-6736(10)62039-9. [DOI] [PubMed] [Google Scholar]
- 39.Zucker IH, Hackley JF, Cornish KG, Hiser BA, Anderson NR, Kieval R, Irwin ED, Serdar DJ, Peuler JD, Rossing RA. Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension. 2007;50:904–910. doi: 10.1161/HYPERTENSIONAHA.107.095216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.