

Abstract
Macrolide antibacterial agents inhibit parasite proliferation by targeting the apicoplast ribosome. Motivated by the long-term goal of identifying antiparasitic macrolides that lack antibacterial activity, we have systematically analyzed the structure-activity relationships among erythromycin analogues and have also investigated the mechanism of action of selected compounds. Two lead compounds, N-benzyl-azithromycin (11) and N-phenylpropyl-azithromycin (30), were identified with significantly higher antiparasitic activity and lower antibacterial activity than erythromycin or azithromycin. Molecular modeling based on the co-crystal structure of azithromycin bound to the bacterial ribosome suggested that a substituent at the N-9 position of desmethyl-azithromycin could improve selectivity due to species-specific interactions with the ribosomal L22 protein. Like other macrolides, these lead compounds display a strong “delayed death phenotype”; however, their early effects on T. gondii replication are more pronounced.
INTRODUCTION
Apicomplexan parasites such as Plasmodium falciparum and Toxoplasma gondii are ubiquitous human pathogens. Malaria, caused by Plasmodium spp., is responsible for more than one million deaths per year, primarily in children, whereas T. gondii, the etiologic agent of toxoplasmosis, causes serious morbidity and mortality in pregnant women and immunocompromised individuals undergoing chemotherapy or with AIDS 1–3. There is considerable interest in developing safer drugs against these protozoan infections, ones that are appropriate for therapy or prophylaxis in infants and pregnant women. At the same time, emerging resistance towards existing chemotherapeutic agents is also prompting the search for new antiparasitic drugs.
Apicomplexan parasites harbor a plastid (bacterial-like) organelle called the apicoplast that is recognized as a particularly attractive drug target 4. Small molecules that prevent protein biosynthesis on the plastid ribosome show antiparasitic activity. For example, tetracyclines such as doxycycline, which bind to the 30S subunit in bacterial ribosome, are commonly used as antimalarial prophylactic agents 5–7 (even though doxycycline is contraindicated during pregnancy and in children less than 8 years of age). Similarly, antibiotics that target the 50S subunit such as lincosamides, chloramphenicol and macrolides also display moderate antiparasitic activities 8. All of these compounds show a characteristic “delayed death phenotype”, presumably due to their ability to induce a time-dependent reduction in the copy number of the apicoplast genome 4.
Macrolide antibiotics such as erythromycin are widely used to treat respiratory tract infections caused by Gram-positive bacteria. Although laboratory and clinical studies have demonstrated that azithromycin, a semi-synthetic erythromycin analogue, has some efficacy against Toxoplasma 9 as well as Plasmodium 10–12, macrolides have not yet found extensive use as antiparasitic agents, presumably due to sub-optimal efficacy and concerns regarding the potential emergence of resistance against this frontline antibacterial agent. Identification of a parasite-specific macrolide with higher potency than erythromycin or azithromycin would therefore represent a significant advance in the field of parasitology. Due to the stability of azithromycin in the acidic gastric environment, its high serum half-life and tissue penetration, and its well-documented safety profile in children and pregnant women 13–15, this semi-synthetic macrolide was identified as a particularly attractive starting point for our investigations. Our studies were significantly aided by the recently solved high-resolution crystal structures of prototypical bacterial 50S ribosomes in complex with a number of macrolide antibiotics 16– 24.
The structures of azithromycin bound to the 50S ribosomal subunit from the eubacterium Deinococcus radiodurans (D50S) 22 and to that of the archaeon Haloarcula marismortui (H50S) 23 have been solved. In D50S, two azithromycin molecules bind cooperatively to the tunnel. The macrolactone ring (through hydrophobic interactions) and the desosamine sugar (through hydrogen bonding) of the first azithromycin (AZ-1) interact with the minor groove edges of nucleotides A2058Ec and A2059Ec of domain V of the 23S RNA subunit (Ec = E. coli numbering). The binding mode of AZ-1 is in accordance with that of the entire macrolide antibiotic family. The second molecule (AZ-2) reaches to domain II of the 23S RNA, and also engages ribosomal proteins L4 and L22 through hydrophobic interactions. Together, the two molecules effectively block the exit tunnel of the ribosome. In H50S, G2058Ec (instead of A) hinders the binding of one azithromycin molecule, and displaces the remaining macrolactone ring towards the tunnel wall. This residue is representative of eukaryotic ribosomal RNA sequences. As a result of this displacement, azithromycin partially blocks the exit tunnel of the archeon ribosome, consistent with its lower binding affinity to this target. Thus, in principle, azithromycin can achieve species selectivity as a result of subtle differences between the structures of prokaryotic and eukaryotic ribosomes 25. We sought to exploit these differences in our design of an antiparasitic macrolide. Specifically, multiple sequence alignment revealed high sequence identity in domain V residues of the T. gondii and B. subtilis 23S RNA that are likely to interact with azithromycin (Table S1). However, domain II and the ribosomal proteins L4 and L22 are less conserved in regions that are presumably relevant to macrolide binding. We therefore prepared and evaluated a set of erythromycin analogues that (a) have the potential of interacting with domain II, L4, and/or L22; and (b) are unlikely to be prohibitively expensive to synthesize on a large scale.
RESULTS
Synthesis of erythromycin and azithromycin analogues
Modifications at the C-9 ketone of erythromycin or the N-9 amine of azithromycin are relatively easy to introduce, and are likely to point toward L4 and L22. Alkylation at the C-6 position also has precedence (as in the case of clarithromycin), and is known to introduce beneficial conformational changes to the macrolide ring as well as to afford interactions with domain II 22, 26, 27.
To modify the C-9 position of erythromycin, we synthesized a series of erythromycin oxime-ether analogues, modeled after the antibacterial agent roxithromycin 28, 29. The N-9 position of azithromycin was modified by varying alkyl or arylalkyl substituents on the nitrogen atom. Both classes of compounds included analogues with unmodified or alkylated 6-OH groups.
The synthesis of erythromycin oxime ethers and azithromycin analogues followed earlier synthetic precedents (Figure 1B) 29–34. Briefly, 9(E)-erythromycin A-oxime (1a) was quantitatively obtained from erythromycin A by treatment with hydroxylamine in presence of acetic acid. The synthesis of erythromycin A 9-oxime ethers (4 – 7) accomplished in a single step by treating oxime 1a with corresponding alkyl halides and potassium tert-butoxide.
Figure 1.

(A) Structures of erythromycin, clarithromycin and azithromycin; (B) Synthesis of erythromycin A oxime ethers (4 – 7) and 15-membered azithromycin analogues (8 – 31). (a) 50% NH2OH (aq.), AcOH in iso-propanol; (b) i - toluene-ρ-sulphonyl chloride in MeOH, ii – NaBH4 in MeOH; (c) corresponding aldehyde, AcOH, NaBH3CN in DMF; (d) i - pyridine·HCl, cyclohexane diethyl ketal in CH3CN, ii – pyridine·HCl, hexamethyldisilazane in CH3CN, iii – allyl tert-butyl carbonate, Pd2(dba)3, dppb in THF, iv – AcOH in CH3CN and H2O; (e) corresponding alkyl halides, 1M potassium tert-butoxide in THF;
For the synthesis of azithromycin analogues, 9(E)-erythromycin A-oxime (1a) was activated with sulfonyl chlorides for the Beckmann rearrangement, thus converting a ring-expanded imino-ether compound. Subsequent reduction afforded a 15-membered aza-macrolide, often referred to as an azalide. Azithromycin (3) was obtained by methylating this secondary amine at the 9a position, whereas other azalides were obtained via reductive amination with aldehydes and cyano-borohydride 35.
Biological activities of erythromycin analogues
As an initial test of the biological activities of our erythromycin analogues, we measured their antiparasitic, antibacterial, and cytotoxic activities (Table 1). Half-maximal inhibitory concentrations (IC50) were measured in a parasite growth assay using an engineered strain of Toxoplasma gondii 2F-1 YFP2 36. Parasite growth was monitored over the course of 4 days. Separately, the minimum inhibitory concentrations (MIC) of each compound against a representative Gram-positive bacterium Bacillus subtilis were measured in standard two-fold dilution assays. Last, the half-maximal cytotoxic concentration (CC50) of each compound was measured using uninfected human fibroblast cultures as the test system. Based on these results, two of the most promising compounds (11 and 18) were azithromycin analogues in which the ring nitrogen was alkylated with a benzyl substituent. Compared to azithromycin, they had markedly improved antiparasitic activity as well as reduced antibacterial activity. Oxime ethers, including compounds 5 and 7 also had good antiparasitic activity, although they retained strong antibacterial activity. The antibacterial activity of azalides drops when either the ring nitrogen harbors a substituent other than a methyl group 37–39, or the 6-OH group is alkylated, or both.
Table 1.
Antiparasitic, antibacterial and cytotoxic activities of semisynthetic erythromycin analogues.
| Cmpd. # | IC50 (μM) T. gondii | MIC (μM) B. subtilis | CC50 (μM) | ||
|---|---|---|---|---|---|
| 1 | Erythromycin | 93 ± 10 | 0.04 | 475 | |
| 2 | Clarithromycin | 31 ± 5 | 0.04 | 260 | |
| 3 | Azithromycin | 20 ± 2 | 0.15 | 174 | |
|
| |||||
|
R1
| |||||
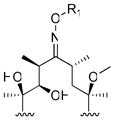
|
4 |
|
14 ± 2 | 0.04 | 22 |
| 5 |
|
1.6 ± 0.2 | 0.04 | 7 | |
| 6 |
|
2.7 ± 1 | 0.04 | 16 | |
| 7 |
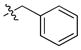
|
1.4 ± 0.1 | 0.04 | 3 | |
|
| |||||
|
R2
| |||||
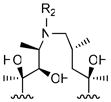
|
8 |
|
65 ± 7 | 1.2 | 386 |
| 9 |
|
16 ± 3 | 0.6 | 139 | |
| 10 |
|
13 ± 2 | 0.3 | 103 | |
| 11 |
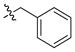
|
2 ± 0.2 | 0.3 | 32 | |
| 12 |
|
85 ± 3 | 10 | 480 | |
| 13 |
|
12 ± 0.3 | 1.2 | 222 | |
|
| |||||
|
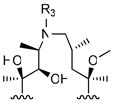
R3
| |||||
|
14 |
|
106 ± 11 | 2.5 | 460 |
| 15 |
|
89 ± 7 | 5 | 454 | |
| 16 |
|
20 ± 2 | 1.2 | 106 | |
| 17 |
|
13 ± 1 | 0.8 | 84 | |
| 18 |
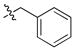
|
2 ± 0.1 | 0.3 | 13 | |
| 19 |
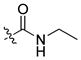
|
59 ± 4 | 10 | 409 | |
| 20 |
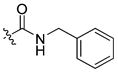
|
16 ± 0.4 | 1.5 | 62 | |
|
| |||||
|
R4
| |||||
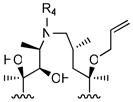
|
21 |
|
40 ± 1 | 2.5 | 74 |
| 22 |
|
25 ± 0.7 | 10 | 96 | |
| 23 |
|
15 ± 0.6 | 10 | 42 | |
| 24 |
|
12 ± 0.4 | 5 | 38 | |
| 25 |
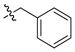
|
5 ± 0.3 | 6 | 14 | |
| 26 |
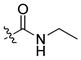
|
33 ± 2 | 5 | 90 | |
| 27 |
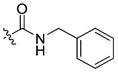
|
6 ± 0.4 | 1.5 | 28 | |
To quantify the antiparasitic activity of each compound, cultures of human foreskin fibroblast (HFF) cells grown in 96-well plates were infected with T. gondii cells 2F-1 YFP2 (T. gondii YFP) 36 expressing the yellow fluorescent protein under control of a constitutive promoter. Each compound was titrated into infected cultures at varying dilutions. Infected cultures were incubated for 4 days (2 infection cycles) in the continuous presence of compounds. Antibacterial activity was measured as the minimum inhibitory concentration (MIC) against the representative Gram-positive bacterium B. subtilis. Two-fold incremental dilutions of each compound were added to freshly inoculated cultures of B. subtilis. The lowest concentration at which no growth was observed was recorded as the MIC value. Cytotoxicity was determined in parallel with antibacterial and antiparasitic activity with MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxylmethoxylphenyl)-2H-tetrazolium) assay. Low passage HFF cells (2 × 103 cells/well) were seeded into the 96-well culture plates, and incubated for 24h at 37 °C. Then, the HFF cells were exposed to the various concentrations of macrolides for 2 days. After incubation, the macrolide treated medium was aspirated, and fresh medium and MTS were added in order. The plates were incubated for 4h at 37 °C, and the optimal density was measured at 490nm. CC50 was determined by cytotoxic dose of macrolides reducing cell viability by 50%.
Differential effects of alternative azalides on T. gondii
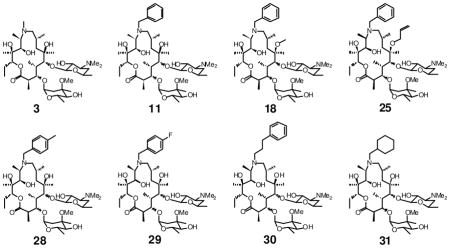
Macrolides are known to exhibit peculiar kinetics with respect to their antiparasitic activity, a phenomenon that is sometimes referred to as the “delayed death phenotype” 4, 40, 41. For example, azithromycin is a weak inhibitor of parasite replication during the first cycle of fibroblast infection (IC50 = 29 μM; Table 2). However, the resulting parasites are severely compromised in their ability to replicate in a second fibroblast infection cycle, even when the second infection cycle is performed in the absence of any antibiotic. Indeed, exposure of T. gondii to less than 1 μM azithromycin in the first cycle results in a >2-fold reduction in parasite titers by the end of the second infection cycle (Table 2). Using azithromycin as a control, we therefore quantified the effects of compounds 11, 18 and 25 on parasite proliferation in each of two successive infection cycles (Table 2). By using two slightly different protocols in these experiments, we were also able to investigate the effect of each compound on the biology of extracellular versus intracellular T. gondii. In one protocol, fibroblasts were infected with T. gondii YFP cells in the presence of individual macrolides. The infected cultures were then incubated for 48 h (1st cycle). Thereafter, the parasites released by lysed fibroblasts were harvested and inoculated into fresh fibroblast cultures (2nd cycle). No macrolide antibiotic was added to the 2nd cycle cultures. In an alternative protocol, T. gondii YFP cells were first allowed to infect the host fibroblast cells for 6 h. Thereafter the fibroblasts were washed to remove parasites that had not invaded the host cells, and replenished with fresh media containing macrolides. After 42 h (i.e., at the end of the 1st cycle), the released parasites were harvested as before, and used to inoculate fresh fibroblast cultures for a 2nd cycle, again without supplementary macrolide. In both protocols, the IC50 values of compound 11 were markedly higher after the 1st cycle than the 2nd cycle, whereas neither compound 18 nor 25 showed this delayed death phenotype. These observations suggested that, whereas compound 11 is genuinely a more potent analogue of azithromycin, the observed antiparasitic activities of compounds 18 and 25 may result from a different mode of action. We therefore focused our attention on the preparation and evaluation of additional analogues of 11.
Table 2.
Antiparasitic, antibacterial and cytotoxic activities of selected azalides.
 | ||||||
|---|---|---|---|---|---|---|
| IC50 (μM)
|
MICa (μM) | CC50b (μM) | ||||
|
0h |
6h |
|||||
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | |||
| 3 | 29 ± 2 | 0.6 ± 0.07 | 44 ± 5 | 0.6 ± 0.03 | 0.15 | 174 |
|
| ||||||
| 11 | 5 ± 1 | 0.5 ± 0.10 | 9 ± 1 | 0.5 ± 0.05 | 0.3 | 32 |
|
| ||||||
| 18 | 7 ± 0.3 | 5 ± 0.2 | 10 ± 1 | 4 ± 0.5 | 0.3 | 13 |
|
| ||||||
| 25 | 7 ± 1 | 10 ± 1 | > 10 | > 10 | 6.0 | 14 |
|
| ||||||
| 28 | 5 ± 1 | 0.6 ± 0.04 | 9 ± 1 | 0.7 ± 0.05 | 0.3 | 38 |
|
| ||||||
| 29 | 6 ± 1 | 0.5 ± 0.07 | 9 ± 1 | 0.7 ± 0.01 | 0.3 | 31 |
|
| ||||||
| 30 | 4 ± 1 | 0.6 ± 0.06 | 8 ± 1 | 0.6 ± 0.01 | 0.6 | 33 |
|
| ||||||
| 31 | 7 ± 1 | 0.6 ± 0.10 | 12 ± 2 | 0.6 ± 0.07 | 0.3 | 36 |
“0 h” data refers to results from experiments in which macrolide and parasite were simultaneously added to fibroblast cultures, whereas “6 h” data was obtained from experiments in which parasites were first allowed to infect before addition of the macrolide.
MIC: minimum inhibitory concentration against B. subtilis.
CC50: cytotoxic concentration at which human foreskin fibroblast viability drops by 50%
Structure-activity relationship (SAR) analysis of compound 11
Based on the above data, we designed a focused set of N-9 substituted aryl azalides. Our primary goal was to explore whether subtle steric or electronic changes in the N-9 substituent could affect antiparasitic activity. Representative analogues with electron donation into (28) or withdrawal from (29) the benzyl ring were targeted. We also sought to examine whether improved activity could result from increasing the flexibility of the linker to the aromatic ring (30). In addition, the need for an aromatic group at this position was tested by preparing and assaying cyclohexyl analogue 31.
To prepare these analogues, intermediate 8 was reacted with the corresponding aldehydes, and the resulting Schiff’s bases were reduced as before. The macrolides were assayed as above, using azithromycin and 11 as controls. In addition, their MIC values against B. subtilis and cytotoxicity towards uninfected human foreskin fibroblasts (calculated as CC50) were also measured. The overall in vitro pharmacological profiles of these compounds are summarized in Table 2. Compound 31 has slightly higher IC50 values compared to other benzylated compounds 28 – 30 at the 1st infectious cycles. Whereas the 2nd cycle antiparasitic activities of compounds 11 and 28 – 31 are similar to azithromycin, the new analogues have substantially better growth inhibitory activity than azithromycin in the 1st infection cycle. The bulkier substituent at the N9 position in the macrocyclic ring also conferred a modest increase in mammalian cell cytotoxicity, although the therapeutic index (CC50/IC50) remained unchanged owing to improved growth inhibition in the 1st cycle. One of the new analogues (30) showed a further reduction in antibacterial activity, which may mitigate potentially deleterious effects on commensal microflora in animal studies involving chronic dosing. The lowest dose of azithromycin that significantly protects mice against death due to acute toxoplasmosis is 200 mg/kg/day 9. Thus, compounds 11 and 28 – 31 could potentially be administered at lower doses due to increased antiparasitic activities. In all cases the therapeutic index appears to be adequately high for experimental animal use compared to azithromycin and clarithromycin.
Further investigations into the mode of action of selected azalides
Several additional experiments were performed to understand better the mechanism of delayed death of apicomplexan parasites that are treated with macrolide antibiotics. First, we monitored parasite replication non-invasively over the course of the 1st and 2nd infectious cycles using T. gondii RHΔhwgprt GFP. This strain expresses the green fluorescent protein, and can therefore be visualized and counted in individual parasitophorous vacuoles of infected fibroblasts. In the 1st infection cycle, fibroblasts infected with T. gondii were exposed to azithromycin (5 or 50 μM) or analogue 11 (5 μM). Both infection protocols described earlier (i.e., drug addition at 0 h and 6 h) were used. In these infection protocols, a small but measurable inhibitory effect was observed in the 1st cycle, whereas much stronger inhibition of T. gondii replication was observed in the 2nd cycle (Figure 2). Consistent with the population-averaged IC50 values reported in Table 2, compound 11 was more active than azithromycin in the 1st infection cycle, whereas both macrolides had comparable activities in the 2nd cycle. Intracellular T. gondii parasites divided approximately every 8 hours (or shorter, given that parasites that had already lysed out of the host cell were not counted), In contrast, when parasites were treated with azithromycin (50 μM) or 5 μM compound 11, the doubling time increased to ~ 9.5 h in the first infection cycle. At all concentrations tested in the 2nd infection cycle, parasite doubling times had increased to > 16 h. Thus, it appears that the antiparasitic activity of macrolides is, at least in part, due to their ability to inhibit the replication rates of protozoan parasites inside mammalian cells.
Figure 2.

Effect of azithromycin (3) and 11 on cell division of T. gondii. The number of GFP parasites per parasitophorous vacuoles was scored at 12-h intervals, assuming that each vacuole starts with a single T. gondii cell. Each data point is an average of three independent experiments. Average parasite counts were obtained by counting at least 50 randomly selected vacuoles in each well. Only the data for cultures exposed to antibiotics at 0 h is shown; the data for cultures in which drug was added at 6 h was similar, and is therefore not shown.
We also sought to test whether macrolides reduced the frequency of successful infections in the 1st infection cycle or not. The density of parasitophorous vacuoles was estimated at 24 h post-infection, using both the “0 h” and “6 h” infection protocols described above. Azithromycin as well as compounds 11 and 28–31 were tested in parallel experiments. As seen in Figure 3, at 10 μM concentrations, azalides 11 and 28–31 (but not azithromycin) show a marked decrease in the density of parasitophorous vacuoles, but only when added concurrently with the parasite inoculum (i.e., at 0 h). Vacuole density remains unaffected by all macrolides in pre-infected fibroblasts (i.e., when the antibiotic is added at 6 h). Together, these findings demonstrate that the new macrolides reported here are able to attenuate the infectivity of extracellular T. gondii even in the 1st infection cycle. Thus, their overall antiparasitic effects are presumably the cumulative result of two modes of actions.
Figure 3.

Effect of macrolides on T. gondii infectivity. The number of vacuoles per microscope field was scored 24 h into the 1st infection cycle. At least 20 randomly selected microscope fields from each of three experiments were scored to obtain each data point. For the treatment of compound 11 and 28 – 31 at 0 h procedure, differences between 1 and 10 μM were highly significant (* p<0.001).
Macrolides induce a comparable loss of apicoplast DNA in clindamycin-sensitive and clindamycin-resistant T. gondii
We sought to verify that the new macrolides described in this report were indeed inhibitors of apicoplast replication. In these experiments, we also chose to evaluate other clinically relevant ribosome inhibitors, such as the 16-membered macrolide spiramycin as well as chloramphenicol and the lincosamide clindamycin 8, 40, 41. All these antibiotics show the delayed death phenotype. Pyrimethamine, an antiparasitic antibiotic that inhibits nuclear DNA replication 42, was used as a negative control.
Intracellular parasites (“6 h” infection protocol) were treated with varying concentrations (0.1 μM, 1 μM and 10 μM) of azithromycin (3), 11, 25 or 30, as well as 1 μM clindamycin and 5 μM spiramycin throughout the 1st infection cycle. After 42h, extracellular parasites released from the first cycle were reinoculated into fresh HFF cell monolayers in the absence of additional antibiotics. Real-time polymerase chain reaction (RT-PCR) was used to quantify the copy number ratio of the apicoplast genome versus nuclear genome; a single-copy nuclear gene (UPRT) and a single-copy plastid genome (ycf24) were targeted for amplification 43.
As seen in Table 3, pyrimethamine did not affect the ratio of plastid to nuclear DNA in either infection cycle. Also, as predicted from the data in Table 2, the apicoplast copy number was only marginally affected upon addition of compound 25, confirming that its antiparasitic activity was likely due to a distinct mode of action. In contrast, apicoplast replication in T. gondii RHΔhxgprt was significantly impaired in even the 1st infection cycle in the presence of azithromycin, compounds 11 and 30, clindamycin, and spiramycin. In each case, further decrease in apicoplast copy number was observed in the 2nd infection cycle. Dose dependence was also observed in each case.
Table 3.
Copy number ratios of apicoplast/nuclear genome in response to antibiotic treatment.
| 1st cycle (48 h)
|
2nd cycle (48 h)
|
||||
|---|---|---|---|---|---|
| T. gondii RHΔ | T. gondii ClinR-4 | T. gondii RHΔ | T. gondii ClinR-4 | ||
| Control | - | 20 ± 3 | 26 ± 4 | 21 ± 3 | 21 ± 5 |
|
| |||||
| 3 | 10 μM | 10 ± 2 | 10 ± 3 | 3 ± 1 | 4 ± 1 |
|
| |||||
| 1 μM | 7 ± 1 | 8 ± 3 | 3 ± 1 | 3 ± 1 | |
|
| |||||
| 0.1 μM | 7 ± 4 | 18 ± 2 | 15 ± 5 | 13 ± 4 | |
|
| |||||
| 11 | 10 μM | 13 ± 3 | 11 ± 2 | 6 ± 2 | 5 ± 1 |
|
| |||||
| 1 μM | 7 ± 2 | 8 ± 1 | 5 ± 1 | 3 ± 0.3 | |
|
| |||||
| 0.1 μM | 13 ± 5 | 23 ± 2 | 17 ± 7 | 15 ± 4 | |
|
| |||||
| Clindamycin | 1 μM | 6 ± 1[a] | 12 ± 2[a] | 3 ± 1[b] | 9 ± 1[b] |
|
| |||||
| Spiramycin | 5 μM | 8 ± 3 | 8 ± 1 | 5 ± 0.4 | 3 ± 0.2 |
|
| |||||
| Pyrimethamine | 1 μM | 29 ± 8 | 27 ± 2 | 22 ± 2 | 23 ± 4 |
Control, azithromycin (3), compound 11, and reference compounds (clindamycin, spiramycin and pyrimethamine) were evaluated via real-time quantitative PCR assay (RT-PCR). The copy numbers were determined based on ratio of two genes synthesized from primer sets of a plastid gene (ycf24, Genbank accession U87145) and a nuclear gene (UPRT, U10246). T. gondii RHΔhxgprt and T. gondii ClinR-4 44 were allowed to infect a monolayer of HFF cells. After 6h, the medium was changed, and the parasites were incubated for 42 h in the presence of different concentrations of compounds. At 48 h post-infection, parasites (T. gondii RHΔhxgprt and T. gondii ClinR-4 44) were isolated and reinoculated into the fresh HFF monolayer, and incubated for 48 h in the absence of compounds.
Statistical analysis (by the t test) indicates that the genome ratios in clindamycin-treated T. gondii RHΔhwgprt are distinct from those of clindamycin-treated T. gondii ClinR-4 in both cycle (p < 0.001).
We also sought to determine whether the clindamycin-resistant T. gondii ClinR-4 strain 44 was resistant to azithromycin or compound 11. Both growth inhibition assays and quantitative RT-PCR assays were performed using the “0h” and “6h” infection protocols described above. Because T. gondii ClinR-4 lacked the YFP transgene, parasite cells were counted with a hemocytometer under the microscope. At both 1 μM and 10 μM concentrations, neither azithromycin nor compound 11 showed a noticeable difference between the wild-type and mutant T. gondii strains (data not shown). A similar conclusion was also obtained when the apicoplast/nuclear DNA ratio was quantified in the presence of macrolides 3 (azithromycin), 11 and spiramycin (Table 3). Thus, it appears that drug resistance of the T. gondii ClinR-4 strain is specific for the lincosamides. This mutant possesses a 1857(Ec) G→U point mutation in the apicoplast 50S rRNA, a site that is known to influence peptide translation 44. In contrast, macrolides block the exit tunnel of the 50S subunits of both wild-type and mutant ribosomes.
Homology modeling of the apicoplast and bacterial ribosomes with bound macrolides
Because the crystal structures of ribosomes from T. gondii and B. subtilis were unavailable, a molecular modeling approach was used to analyze potential binding interactions between these ribosomes and the N-9 amine substituent of azithromycin. Multiple sequence alignments of the 50S ribosomal RNA and the L4 and L22 proteins were performed, and homology models of the target ribosomal RNAs were generated based on the published structure of D. radiodurans ribosomal RNA 22, 50 using the MOE program (Molecular Operating Environment, Chemical Computing Group Inc., Montreal, Canada). These homology models included two molecules of 11, parts of the rRNA near the azithromycin binding site, and the complete L4 and L22 proteins. Models were minimized using the Amber99 force field to remove any steric clashes. In the T. gondii rRNA model (Figure 4B), the phenyl ring of Phe98 was positioned ca. 6 Å from the phenyl ring of 11-2. The model also suggested that the adenine ring of A1441Tg was close to the phenyl ring of 11-2. In contrast, in the B. subtilis model, the equivalent amino acid was Pro87, located ca. 9 Å from the N-phenyl moiety of 11-2 (Figure 4C).
Figure 4. Homology models of the macrolide binding region of D. radiodurans (A), T. gondii (B), and B. subtilis (C) ribosomes, each with two copies of compound 11 (designated 11-1 and 11-2).

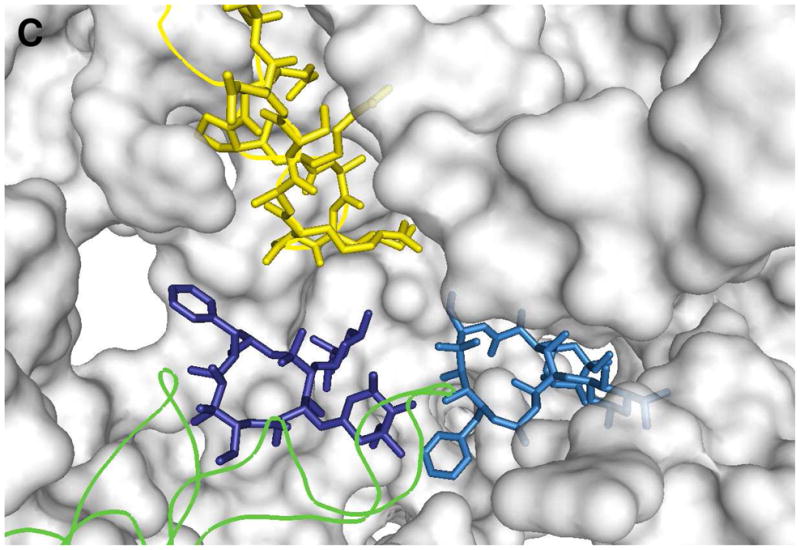
Because two azithromycin molecules are bound to the co-crystal structure of the D. radiodurans ribosome, two molecules of 11 (11-1 and 11-2) were used in the generation of our homology models. Structures 11-1 and 11-2 are light blue and dark blue, respectively. RNA fragments are shown as orange tubes or gray surfaces, and L4 and L22 proteins are in green and yellow, respectively. Pymol1.0 was used to generate the figures. For details, see text.
DISCUSSION
Although its function is still unclear, the apicoplast has received much attention as an attractive target for new drugs against apicomplexan parasites such as Plasmodium spp. and Toxoplasma gondii 4. Macrolide antibacterial agents bind to the large (50S) ribosomal subunit of the protozoan apicoplast, and thereby also exhibit antiparasitic activity. Through the systematic design, synthesis and biological characterization of a series of erythromycin analogues, we have not only established prototypical structure-activity relationships but have also identified two promising azithromycin analogues with markedly higher antiparasitic activity and lower antibacterial activity than clinically available macrolides. In addition to providing potentially valuable new probes for investigating apicoplast biology in vivo, these compounds demonstrate the feasibility of engineering ribosome-binding antibiotics that are capable of discriminating between prokaryotic and selected eukaryotic pathogens.
Among the clinically available macrolides tested, azithromycin proved to be the best starting point for new antiparasitic agent discovery. The other macrolides (erythromycin, clarithromycin and roxithromycin) predominantly rely upon hydrogen bonds and hydrophobic contacts with the 23S rRNA in the peptidyl transferase cavity to achieve their antibiotic activities 21. Although contacts between these macrolides and ribosomal proteins have not been structurally observed 21, there is certainly scope for such interactions due to conformational changes in the flexible L22 hairpin at the exit tunnel 27, 45. Indeed, mutations in ribosomal proteins L4 and L22 that result in resistance towards erythromycin or other macrolides have been identified in several bacterial strains 46–49. In contrast, azithromycin appears to inhibit protein translation by a markedly different mechanism. In the crystal structure of Deinococcus radiodurans (D50S) 22, two molecules of azithromycin bind in a perpendicular orientation to block the protein exit tunnel. The molecule that binds in the non-conserved mode interacts with ribosomal proteins L4 and L22. We speculate that the maintenance of this 2:1 macrolide:ribosome stoichiometric ratio contributes significantly to the improved antiparasitic activity of azalides such as 11 and 30. Preservation of the hydrogen bond between the 6-OH of these azalides and C2565 in domain V of the 23S rRNA is critical for this atypical stoichiometry. At the same time, we hypothesize that the presence of an aromatic substituent at N-9 improves species selectivity because of potential interactions with ribosomal proteins L4 and/or L22 in T. gondii, which may differ in degree with those in B. subtilis (Table S1). The increased bulkiness of the N-9 substituent in compound 30 relative to 11 may enhance selectivity by reducing its affinity for the bacterial ribosome. Together, these results provide a starting point for a more intensive SAR investigation with the goal of maximizing antiparasitic activity in a macrolide that concomitantly lacks physiologically significant antibacterial activity.
Homology models of the T. gondii and B. subtilis ribosomal RNAs were generated based on the structure of the D. radiodurans ribosomal RNA (Figure 4A) 22, 50. These homology models included two molecules of 11, parts of the rRNA near the azithromycin binding site, and the complete L4 and L22 proteins. These models suggested that, whereas the N-phenyl moiety of 11-2 could not favorably interact with B. subtilis L22, favorable edge-to-face interactions with Phe98 in the T. gondii L22 were possible. It should be noted that the estimated distance between the two phenyl rings is larger than the optimal distance for an edge-to-face aromatic interaction (ca. 3.7 Å).51 Energy minimization alone is presumably insufficient to define the optimal arrangement of these aromatic functional groups.52 Favorable interactions may require the L22 loop region (Phe97 to Arg108) to be dynamic.53
Perhaps most importantly, three interesting conclusions can be drawn from our biological studies on azithromycin and its analogues. First, notwithstanding the well-documented delayed death phenotype of apicoplast inhibitors, compounds such as 11 and 30 induce rapid changes in T. gondii parasites. At low micromolar concentrations, they attenuate both apicoplast copy number (Table 3) and parasite growth rates (Table 2) within the 1st infection cycle. Thus, it appears that, even though defective apicoplasts exert their most overt effects in secondary cycles of infection, the biology of the protozoan cell starts to be altered on a much shorter timescale. Second, azalides affect both the infectivity of apicomplexan parasites in mammalian cells (Figure 3) as well as the kinetics of cell division in the resulting parasitophorous vacuoles (Figure 2). It is tempting to assume that both these features result from inhibition of the apicoplast ribosome, although this remains to be established. If not, then in vivo studies in animals will be required to establish which of these mechanisms is more critical to the observed antiparasitic activity of azalides. This in turn would guide the judicious selection of a primary assay for more intensive SAR efforts. Last but not least, the reason for the low but measurable cytotoxic activity of azalides against human fibroblasts (Table 2) needs to be better understood. Several investigators have demonstrated toxicity of azithromycin against cultured mammalian cells at concentrations in the 100 μM range 54–56. The precise mechanism for these cytotoxic effects is unknown. However, a closely related macrolide antibiotic megalomicin is known to inhibit intra-Golgi trafficking 57 and lysosomal function 58 at comparable concentrations. Deconvolution of the structural features of azalides that contribute to mammalian cell toxicity from those that enhance binding to the apicoplast ribosome could greatly enhance the chance of discovery of a new antiparasitic macrolide with a suitable safety profile for eventual clinical use.
EXPERIMENTAL PROCEDURES
Materials
Antibiotics used were purchased from TCI America (Boston, MA) or Sigma-Aldrich (Milwaukee, WI). Solvents and chemical reagents were from Fisher Scientific, Inc. (Springfield, NJ) or Sigma-Aldrich. Reactions were carried out under an N2 atmosphere in oven-dried glassware unless otherwise specified. Moisture- and oxygen-sensitive reagents were handled in an N2-filled dry box. The synthesis of erythromycin oxime ethers and azithromycin analogues followed earlier synthetic precedents.
General Methods
Analytical thin layer chromatography (TLC) was performed on precoated silica gel plates (60F254) and flash chromatography on silica gel (230–400 mesh). TLC spots were detected by UV and/or by staining with a water-based cerium molybdate stain. Inova400, Inova500, and Inova600 MHz NMR spectrometers were used to perform 1H and 13C NMR analysis, and spectra were recorded in CDCl3. 1H and 13C NMR spectra are reported as chemical shift in parts per million (multiplicity, coupling constant in Hz, integration). All final compounds were further introduced to liquid chromatography-mass spectrometry (LC-MS, Waters 2795 HPLC system) on a reverse phase C8 column (2.1 × 40 mm). Linear gradient elution was performed at 0.2 mL/min with CH3CN and H2O (both containing formic acid, 0.1%). The purity of each compound was calculated from the LC chromatogram at a wavelength of 210 nm. By this method, the purity of each compound was greater than or equal to 95.0%.
The procedures detailed below for compound 4, 9, 10 and 12 are representative of the procedure followed for synthesis of all of the erythromycin A 9-oxime ethers and azithromycin analogues. Erythromycin A 9-oxime (1a) and intermediate 8, 14 and 21 was prepared as described. Scale, yield and spectra are presented for each of the individual antibiotics assayed.
Erythromycin A 9-oxime, 1a
Erythromycin A (0.68 mmol, 0.5 g) was dissolved in iso-propanol (1.2 mL). 50% Hydroxylamine (aq.) (620 μL) and acetic acid (190 μL) were added to the reaction mixture, and the reaction was stirred for 15 h at 50 °C. After the reaction mixture was allowed to cool down to room temperature (rt), the reaction was quenched by saturated NaHCO3 (aq.). The iso-propanol was removed under rotovap, and chloroform was added to extract the product. The chloroform extract was washed with brine, and the product was vacuum-dried and used for the following reactions without further purification.
Erythromycin A 9-oxime methyl ether, 4
Erythromycin A 9-oxime, 1a (0.27 μmol, 200 mg) was dissolved in 1:1 mixture of THF and DMF (2 mL). 1-Iodomethane (0.32 μmol, 46 mg) and 1 M potassium tert-butoxide in THF (0.30 μmol, 300 μL) were added to the reaction mixture at 0 °C under N2. The reaction was stirred for 30 min at 0 °C, and further stirred for 2.5 h at rt under N2. The crude mixture was diluted into CH2Cl2 and was washed three times with saturated NaHCO3 (aq.), and was dried over Na2SO4. After evaporation of the solvent, the resulting product was purified by flash chromatography eluting with a step gradient ranging from 3% to 10% MeOH/CH2Cl2 containing 0.1% NH4OH (aq.) to yield 4 (23.4 mg, 12%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 5.10 (dd, J = 11.1, 2.2 Hz, 1H), 4.91 (d, J = 4.7 Hz, 1H), 4.47 – 4.37 (m, 2H), 4.04 (dd, J = 9.6, 1.3 Hz, 1H), 4.03 – 3.99 (m, 1H), 3.81 (s, 3H), 3.70 – 3.63 (m, 2H), 3.57 (d, J = 7.5 Hz, 1H), 3.48 (tdd, J = 11.4, 6.6, 4.7 Hz, 1H), 3.31 (s, 3H), 3.21 (dd, J = 10.3, 7.3 Hz, 1H), 3.11 (s, 1H), 3.02 (s, 1H), 2.90 (dq, J = 14.3, 7.0 Hz, 1H), 2.65 (q, J = 7.0 Hz, 1H), 2.42 (ddd, J = 12.3, 10.4, 3.9 Hz, 1H), 2.36 (d, J = 15.1 Hz, 1H), 2.28 (s, 6H), 2.25 – 2.19 (m, 1H), 2.18 – 2.15 (m, 1H), 2.02 – 1.95 (m, 1H), 1.91 (tdd, J = 15.2, 8.7, 6.4 Hz, 2H), 1.79 (s, 1H), 1.64 (ddd, J = 24.0, 8.2, 5.6 Hz, 2H), 1.59 – 1.52 (m, 2H), 1.50 – 1.43 (m, 1H), 1.46 (s, 3H), 1.30 (d, J = 6.2 Hz, 3H), 1.23 (s, 3H), 1.22 (d, J = 6.1 Hz, 3H), 1.18 (d, J = 5.4 Hz, 3H), 1.16 (d, J = 5.4 Hz, 3H), 1.12 (s, 3H), 1.10 (d, J = 7.5 Hz, 3H), 1.02 (d, J = 7.0 Hz, 3H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ 175.41, 171.71, 103.08, 96.40, 83.19, 79.99, 78.13, 77.75, 77.09, 75.48, 74.37, 72.82, 71.08, 70.64, 68.96, 65.65, 61.91, 49.62, 44.81, 40.41, 39.17, 37.90, 35.16, 32.97, 28.75, 27.17, 26.49, 21.63, 21.56, 21.21, 18.78, 18.74, 16.35, 16.27, 14.61, 10.83, 9.30. ESI mass spectrum; Calcd (MH+) 763.5; found 763.5.
9-Deoxo-9a-aza-9a-homoerythromycin A, 8
Erythromycin A 9-oxime (0.67 mmol, 0.5 g) dissolved in methanol (2.5 mL) was cooled to 5 °C, and toluene-ρ-sulphonyl chloride (0.80 mmol, 153 mg) was added to the reaction mixture followed by water. While the reaction was stirred for 5h at 5 °C, pH of the reaction was maintained at 6 ~ 7 by adding 20% NaOH (aq.). The crude mixture was diluted into water, adjusted pH at 6 with 1N HCl, and extracted with methylene chloride twice. After adjust pH at 8 with 20% NaOH (aq.), extraction with methylene chloride was performed to yield erythromycin A imino ether. Erythromycin A imino ether (0.27 mmol, 0.2 g) dissolved in methanol (5 mL) was cooled to 4 °C, and sodium borohydride (5.4 mmol, 204 mg) was added in portions. The reaction was stirred for 36 h at rt. Dry ice was added to the reaction mixture until precipitation was completed, and the precipitate was filtered off. The filtrate was concentrated, and redissolved in chloroform. To the mixture, water and 1N-HCl were added to acidify the reaction at pH 2.5, and allowed to stir for 15 min. After adjusting pH 6 with 20% NaOH (aq.), chloroform was used for extraction. The extraction was repeated at pH 6.5 and 8.3. The extract at pH 8.3 was concentrated, and dry ether was added. After stirring for 2h in the ice bath, the precipitant was vacuum-dried to yield 8 (300 mg, 61%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 5.09 (d, J = 4.6 Hz, 1H), 4.71 (dd, J = 10.3, 2.2 Hz, 1H), 4.42 (d, J = 7.3 Hz, 1H), 4.32 (dd, J = 4.3, 1.9 Hz, 1H), 4.07 (dq, J = 12.3, 6.2 Hz, 1H), 3.64 (d, J = 7.4 Hz, 1H), 3.49 (tdd, J = 11.2, 6.4, 4.6 Hz, 1H), 3.44 (d, J = 1.7 Hz, 1H), 3.38 (s, 1H), 3.33 (s, 3H), 3.22 (dd, J = 10.2, 7.4 Hz, 1H), 3.08 – 2.99 (m, 3H), 2.92 (s, 1H), 2.76 (qd, J = 7.4, 4.8 Hz, 1H), 2.59 – 2.53 (m, 1H), 2.43 (ddd, J = 12.3, 10.4, 3.8 Hz, 1H), 2.34 (d, J = 15.2 Hz, 1H), 2.27 (s, 6H), 2.26 – 2.23 (m, 1H), 2.17 (d, J = 10.5 Hz, 1H), 1.96 – 1.89 (m, 1H), 1.88 (ddd, J = 14.2, 7.6, 2.3 Hz, 1H), 1.82 (t, J = 11.3 Hz, 1H), 1.73 (d, J = 14.3 Hz, 2H), 1.68 – 1.62 (m, 1H), 1.57 (dd, J = 15.2, 5.0 Hz, 1H), 1.52 – 1.42 (m, 1H), 1.36 (dd, J = 14.9, 7.7 Hz, 1H), 1.31 (d, J = 6.2 Hz, 3H), 1.28 (s, 3H), 1.23 (s, 3H), 1.21 (d, J = 6.1 Hz, 4H), 1.19 (d, J = 7.5 Hz, 4H), 1.13 (d, J = 6.5 Hz, 3H), 1.07 (s, 3H), 1.04 (d, J = 7.5 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H), 0.88 (t, J = 7.4 Hz, 4H). 13C NMR (500 MHz, CDCl3) δ 178.95, 103.10, 94.93, 83.43, 78.28, 78.04, 78.01, 73.93, 73.88, 73.16, 73.08, 70.97, 68.87, 65.92, 65.63, 57.47, 56.93, 49.58, 45.50, 42.33, 40.49, 34.87, 30.00, 28.83, 27.58, 22.04, 21.73, 21.51, 21.12, 18.40, 16.24, 15.04, 14.09, 11.28, 9.24. ESI mass spectrum; Calcd (MH+) 735.5; found 735.6.
N9-propyl azithromycin, 9
9-Deoxo-9a-aza-homoerythromycin A, 8 (204 μmol, 150 mg) and propionaldehyde (1021 μmol, 59 mg) was dissolved in DMF (1.5 mL). Acetic acid (2041 μmol, 123 mg) and sodium cyanoborohydride (306 μmol, 19 mg) were added to the reaction mixture. The reaction was stirred for 7 h at 70 °C. The crude mixture was diluted into CH2Cl2 and was washed three times with saturated NaHCO3 (aq.), and was dried over Na2SO4. After evaporation of the solvent, the resulting product was purified by flash chromatography eluting with a step gradient ranging from 3% to 10% MeOH/CH2Cl2 containing 0.1% NH4OH (aq.) to yield 9 (38 mg, 24%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 5.05 (d, J = 4.5 Hz, 1H), 4.70 (dd, J = 9.7, 2.1 Hz, 1H), 4.43 (d, J = 7.3 Hz, 1H), 4.20 (dd, J = 5.3, 2.3 Hz, 1H), 4.12 (s, 1H), 4.07 (dt, J = 15.3, 6.1 Hz, 1H), 3.76 (s, 1H), 3.64 (d, J = 6.9 Hz, 1H), 3.54 – 3.45 (m, 1H), 3.38 (s, 1H), 3.32 (s, 3H), 3.23 (dd, J = 10.1, 7.3 Hz, 1H), 3.02 (t, J = 8.7 Hz, 1H), 2.86 (td, J = 12.9, 4.5 Hz, 1H), 2.81 – 2.71 (m, 3H), 2.63 (s, 1H), 2.53 (td, J = 13.0, 4.3 Hz, 1H), 2.45 (td, J = 12.2, 3.8 Hz, 1H), 2.34 (d, J = 15.2 Hz, 1H), 2.28 (s, 6H), 2.20 (d, J = 9.8 Hz, 1H), 2.09 – 1.96 (m, 3H), 1.87 (dtd, J = 22.0, 7.6, 2.6 Hz, 1H), 1.73 (d, J = 14.2 Hz, 1H), 1.65 (dd, J = 11.0, 1.8 Hz, 1H), 1.57 (dd, J = 15.2, 4.9 Hz, 2H), 1.51 – 1.38 (m, 3H), 1.37 – 1.33 (m, 1H), 1.32 (s, 1H), 1.31 (s, 4H), 1.23 (t, J = 5.1 Hz, 5H), 1.21 (t, J = 6.7 Hz, 7H), 1.13 (d, J = 6.8 Hz, 3H), 1.06 (d, J = 9.1 Hz, 6H), 0.91 (d, J = 7.3 Hz, 3H), 0.88 (d, J = 7.4 Hz, 2H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ 178.23, 133.57, 103.24, 95.67, 95.27, 83.85, 78.61, 78.19, 77.86, 75.04, 74.26, 73.90, 72.98, 71.01, 68.97, 65.77, 65.73, 65.14, 61.83, 52.16, 49.57, 45.04, 42.07, 41.32, 40.50, 35.03, 29.82, 28.91, 28.48, 27.32, 22.54, 21.72, 21.67, 21.49, 20.46, 18.38, 16.57, 15.06, 12.25, 11.45, 9.61. ESI mass spectrum; Calcd (MH+) 777.5; found 777.5.
N9-allyl azithromycin, 10
9-Deoxo-9a-aza-homoerythromycin A, 8 (204 μmol, 150 mg) was mixed with allyl acetate (10204 μmol, 1021 mg). TEA (539 μmol, 55 mg) and tetrakis(triphenylphosphine)palladium(0) (20 μmol, 24 mg) was added to the reaction mixture at rt under N2. The reaction was stirred for 7 h at 80 °C, and further stirred for 15 h at rt under N2. The crude mixture was diluted into CH2Cl2 and was washed three times with saturated NaHCO3 (aq.), and was dried over Na2SO4. After evaporation of the solvent, the resulting product was purified by flash chromatography eluting with a step gradient ranging from 3% to 10% MeOH/CH2Cl2 containing 0.1% NH4OH (aq.) to yield 4 (41 mg, 26%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 6.00 (dq, J = 10.0, 6.8 Hz, 1H), 5.15 – 5.02 (m, 3H), 4.70 (dd, J = 10.0, 2.0 Hz, 1H), 4.43 (d, J = 7.3 Hz, 1H), 4.38 (s, 1H), 4.24 – 4.20 (m, 1H), 4.07 (dq, J = 12.4, 6.1 Hz, 1H), 3.75 (s, 1H), 3.64 (d, J = 6.9 Hz, 1H), 3.64 – 3.57 (m, 1H), 3.50 (dd, J = 9.8, 5.4 Hz, 1H), 3.41 – 3.34 (m, 1H), 3.32 (s, 3H), 3.23 (dd, J = 10.1, 7.4 Hz, 1H), 3.02 (t, J = 9.2 Hz, 2H), 2.79 (dt, J = 10.8, 7.1 Hz, 3H), 2.70 (s, 1H), 2.48 – 2.41 (m, 1H), 2.35 (d, J = 15.1 Hz, 1H), 2.27 (s, 6H), 2.18 (d, J = 10.2 Hz, 1H), 2.04 – 1.97 (m, 3H), 1.87 (dqd, J = 15.2, 7.5, 2.5 Hz, 1H), 1.71 (d, J = 14.3 Hz, 1H), 1.67 – 1.62 (m, 1H), 1.57 (dd, J = 15.2, 5.0 Hz, 1H), 1.52 – 1.42 (m, 1H), 1.31 – 1.32 (m, 6H), 1.23 (s, 3H), 1.19 – 1.22 (m, 6H), 1.14 (d, J = 6.8 Hz, 3H), 1.07 (s, 3H), 1.06 (d, J = 7.5 Hz, 3H), 0.88 (t, J = 7.5 Hz, 3H), 0.86 (d, J = 5.9 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ 178.38, 136.45, 117.66, 103.18, 95.21, 83.77, 78.46, 78.20, 77.80, 74.88, 74.36, 74.10, 73.00, 70.99, 68.95, 65.80, 65.75, 61.83, 53.57, 49.58, 45.12, 41.96, 41.52, 40.50, 34.98, 28.87, 27.80, 27.15, 22.19, 21.72, 21.60, 21.50, 18.39, 16.49, 15.01, 11.42, 9.85, 9.54. ESI mass spectrum; Calcd (MH+) 775.5; found 775.4.
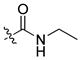
N9-ethyl amide azithromycin, 12
9-Deoxo-9a-aza-homoerythromycin A, 8 (88 μmol, 65 mg) was dissolved in toluene (1.8 mL). Ethyl isocyanate (91 μmol, 6 mg) was added to the reaction mixture, and the reaction was stirred for 1 h at rt. After evaporation of the solvent, the crude mixture was diluted into CH2Cl2 and was washed three times with saturated NaHCO3 (aq.), and was dried over Na2SO4. The resulting product was purified by flash chromatography eluting with a step gradient ranging from 3% to 10% MeOH/CH2Cl2 containing 0.1% NH4OH (aq.) to yield 12 (33 mg, 47%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 4.99 (dd, J = 9.3, 2.7 Hz, 1H), 4.83 (d, J = 4.2 Hz, 1H), 4.66 (s, 1H), 4.45 (d, J = 7.2 Hz, 1H), 4.05 (td, J = 12.7, 6.3 Hz, 1H), 4.01 – 3.98 (m, 1H), 3.75 (s, 1H), 3.59 (dd, J = 9.9, 4.8 Hz, 1H), 3.49 (d, J = 4.4 Hz, 1H), 3.44 (d, J = 14.3 Hz, 1H), 3.32 (dd, J = 10.5, 6.8 Hz, 1H), 3.27 (s, 3H), 3.24 – 3.16 (m, 3H), 3.02 (d, J = 7.3 Hz, 1H), 2.66 (dt, J = 12.4, 6.2 Hz, 1H), 2.59 – 2.49 (m, 2H), 2.29 (s, 6H), 2.34 – 2.25 (m, 9H), 2.14 (s, 2H), 1.91 (ddd, J = 14.1, 7. 5, 3.0 Hz, 2H), 1.70 – 1.61 (m, 2H), 1.55 (dd, J = 15.1, 4.9 Hz, 1H), 1.46 (ddd, J = 14.4, 9.2, 7.3 Hz, 1H), 1.30 (d, J = 6.9 Hz, 3H), 1.26 (d, J = 6.1 Hz, 6H), 1.24 – 1.20 (m, 9H), 1.13 (s, 2H), 1.11 (t, J = 7.2 Hz, 3H), 1.04 (t, J = 7.0 Hz, 6H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ 159.86, 104.64, 96.84, 79.23, 78.23, 77.60, 74.68, 74.59, 74.17, 72.95, 70.84, 69.52, 66.36, 64.77, 49.46, 46.28, 41.36, 40.56, 35.91, 35.15, 29.41, 28.25, 22.46, 21.63, 21.33, 21.23, 17.91, 17.45, 15.46, 15.08, 12.57, 11.57, 10.40. ESI mass spectrum; Calcd (MH+) 806.5; found 806.6.
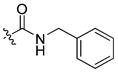
C6-methoxl 9-Deoxo-9a-aza-9a-homoerythromycin A, 14
Clarithromycin (5.4 mmol, 4 g), 50% hydroxylamine (7 mL), and acetic acid (5 mL) in iso-propanol was stirred for 5 days at 40 °C to yield 6-methoxy erythromycin A 9-oxime as a white solid (2.6 g, 64 %). C6-methoxyl erythromycin A 9-oxime (3.4 mmol, 2.6 g), toluene-ρ-sulphonyl chloride (6.81 mmol, 1.3 g), and pyridine (6.81 mmol, 0.5 g) in THF was stirred for 20 h at 40 °C to yield C6-methoxy erythromycin A 9,11-imino ether (2 g, 80%). C6-methoxyl erythromycin A 9,11-imino ether (2.7 mmol, 2 g) in MeOH was reduced by adding acetic acid and sodium borohydride (18.8 mmol, 0.7 g) to yield 14 as a white powder (460 mg, 23 %). 1H NMR (500 MHz, CDCl3) δ 4.95 (d, J = 4.0 Hz, 1H), 4.86 (dd, J = 10.8, 2.0 Hz, 1H), 4.47 (d, J = 7.3 Hz, 1H), 4.08 – 3.98 (m, 2H), 4.00 (dd, J = 7.2, 1.6 Hz, 1H), 3.82 (d, J = 6.4 Hz, 1H), 3.69 – 3.58 (m, 1H), 3.51 (dd, J = 9.8, 5.4 Hz, 2H), 3.35 (s, 3H), 3.32 (s, 3H), 3.18 (dd, J = 10.3, 7.3 Hz, 2H), 3.07 – 2.98 (m, 2H), 2.86 (dt, J = 14.8, 7.5 Hz, 1H), 2.76 (dd, J = 13.1, 6.5 Hz, 1H), 2.45 – 2.38 (m, 1H), 2.34 (d, J = 15.1 Hz, 1H), 2.27 (s, 6H), 2.22 (d, J = 10.7 Hz, 1H), 2.11 (t, J = 10.5 Hz, 1H), 1.92 – 1.82 (m, 3H), 1.70 – 1.61 (m, 3H), 1.58 (dd, J = 9.0, 5.6 Hz, 1H), 1.50 (ddd, J = 14.2, 10.9, 7.2 Hz, 1H), 1.38 – 1.34 (m, 3H), 1.33 – 1.28 (m, 1H), 1.30 (d, J = 6.2 Hz, 3H), 1.25 (s, 3H), 1.24 – 1.19 (m, 9H), 1.12 (t, J = 8.5 Hz, 1H), 1.07 (s, 3H), 1.06 (d, J = 7.6 Hz, 2H), 1.04 – 1.01 (m, 1H), 0.99 (d, J = 7.0 Hz, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ 177.62, 102.61, 96.05, 79.91, 79.16, 77.99, 73.77, 72.96, 72.74, 71.04, 68.85, 66.14, 65.71, 58.21, 57.23, 51.82, 49.55, 45.30, 40.46, 35.08, 29.83, 28.70, 28.05, 22.01, 21.65, 21.60, 20.90, 20.40, 18.75, 16.27, 16.03, 12.85, 11.06, 9.35. ESI mass spectrum; Calcd (MH+) 749.5; found 749.5.
C6-alloxyl 9-deoxo-9a-aza-9a-homoerythromycin A, 21
Erythromycin A 9-oxime, 1a (6.7 mmol, 5 g), pyridine hydrochloride (10 mmol, 1.2 g), and cyclohexane, diethyl ketal (16.7 mmol, 2.9 g) in acetonitrile were stirred for 16 h under N2 to yield erythromycin A 9-(O-ethoxy-cyclohexyl) oxime. The oxime (6.2 mmol, 5.4 g), pyridine hydrochloride (9.3 mmol, 1.1 g), and hexamethyl disilazane (25 mmol, 5.2 g) in acetonitrile were stirred for 2 h to yield 2′, 4″-O-bis(TMS) erythromycin A-9-(1-(1-ethoxy)cyclohexyl) oxime (5.8 g, 92%). The protected oxime (2 mmol, 2 g), allyl tert-butyl carbonate (2.4 mmol, 0.4 g), Pd2(dba)3 (29.6 μmol, 37 mg) and dppb (39.2 μmol, 17 mg) in THF was refluxed for 3 h under N2. After reflux, the protection groups were removed by acetic acid (5.6 mL) in acetonitrile (20 mL) and H2O (4 mL) to yield C6-alloxyl erythromycin A 9-oxime (1.2 g, 80%). C6-alloxyl erythromycin A 9-oxime (1.14 mmol, 900 mg), toluene ρ-sulphonyl chloride (1.71 mmol, 326 mg), and pyridine (2.28 mmol, 180 mg) in THF were stirred for 21 h at 45 °C to yield imino ether product (590 mg, 67 %). C6-alloxyl erythromycin A 9, 11-imino ether (713 μmol, 550 mg) in MeOH was reduced by adding acetic acid and sodium cyanoborohydride (3.6 mmol, 223 mg) to yield 21 as a white powder (240 mg, 44 %). 1H NMR (500 MHz, CDCl3) δ 6.03 (ddd, J = 22.6, 10.6, 5.4 Hz, 1H), 5.25 (dd, J = 17.3, 1.8 Hz, 1H), 5.10 (dd, J = 10.5, 1.7 Hz, 1H), 5.04 (dd, J = 11.0, 2.1 Hz, 1H), 4.86 (d, J = 4.5 Hz, 1H), 4.32 (d, J = 7.2 Hz, 1H), 4.15 (dd, J = 9.4, 1.7 Hz, 1H), 4.05 – 3.95 (m, 2H), 3.90 (dd, J = 12.2, 5.4 Hz, 2H), 3.72 (d, J = 8.3 Hz, 1H), 3.47 (ddd, J = 10.9, 6.2, 1.9 Hz, 1H), 3.40 (d, J = 2.6 Hz, 1H), 3.35 – 3.27 (m, 3H), 3.16 (dt, J = 11.8, 5.8 Hz, 2H), 3.00 (t, J = 9.7 Hz, 1H), 2.90 (s, 1H), 2.86 – 2.81 (m, 2H), 2.44 (ddd, J = 12.4, 10.3, 3.8 Hz, 1H), 2.35 (d, J = 15.0 Hz, 1H), 2.32 – 2.23 (m, 2H), 2.27 (s, 6H), 2.21 – 2.17 (m, 1H), 2.09 – 1.98 (m, 3H), 1.92 (dtd, J = 12.7, 7.4, 5.2 Hz, 2H), 1.67 – 1.63 (m, 1H), 1.59 – 1.52 (m, 2H), 1.52 – 1.45 (m, 2H), 1.39 (s, 3H), 1.29 (d, J = 6.2 Hz, 3H), 1.26 – 1.21 (m, 12H), 1.15 (d, J = 7.4 Hz, 3H), 1.09 (s, 3H), 1.03 (d, J = 6.4 Hz, 3H), 0.87 – 0.83 (m, 6H). 13C NMR (500 MHz, CDCl3) δ 174.74, 136.79, 115.08, 103.96, 103.34, 97.91, 92.09, 83.17, 82.65, 81.03, 78.27, 77.64, 73.60, 72.68, 71.25, 69.03, 67.93, 65.77, 65.70, 63.32, 51.23, 49.79, 49.54, 45.85, 40.83, 40.48, 39.58, 35.88, 29.84, 28.75, 26.78, 25.32, 21.65, 21.51, 21.25, 19.79, 18.61, 17.27, 16.27, 15.25, 10.86, 9.74. ESI mass spectrum; Calcd (MH+) 775.5; found 775.7.
Minimum Inhibitory Concentration (MIC) analysis of Bacillus subtilis
LB medium was used to grow B. subtilis cultures. A standard two-fold serial dilution method in a 96-well plate was used to determine the MIC of each antibiotic compound against B. subtilis. B. subtilis cultures were incubated in presence of antibiotics at 30 °C for 16h ~ 18h, and the optical density was monitored at 490 nm.
Toxoplasma gondii replication assay
Tachyzoites of T. gondii strains RHΔhxgprt YFP, GFP or clinR-4 were harvested from cultured human foreskin fibroblast (HFF) monolayers grown in 25 cm2 T-flasks, and inoculated onto confluent HFF cells in multi-well culture plates (6-well, 2×105 tachyzoites/9.6 cm2 well; 12-well, 2×104 parasite tachyzoites/3.8 cm2 well; and 96-well, 8×103 parasite tachyzoites/0.32 cm2 well). For the “0h” protocol, the test compound was added to the appropriate wells simultaneously with parasites, and the cultures were incubated for 48h before analysis or use in the 2nd infection cycle. In the “6h” protocol, parasites were pre-incubated with HFF cells for 6h. Thereafter, the supernatant containing uninfected parasites was replaced with fresh medium containing the appropriate antibiotic dose, and cultures were incubated for 42h before analysis or use in the 2nd infection cycle. In experiments involving a 2nd infection cycle, purified extracellular tachyzoites harvested at the end of the 1st cycle were inoculated into fresh cultures without antibiotic compounds. Infected cultures were incubated at 37 °C, 5% CO2 in DMEM (Dulbecco’s modified eagle medium) buffer supplemented with 1% PS (penicillin-streptomycin) and 10 % fetal bovine serum. In all experiments, the reported data are from experiments performed at least in triplicate.
Monitoring parasite growth by fluorescence
Tachyzoites from the RHΔhxgprt of T. gondii with the YFP gene were inoculated and cultured in 96-well plates in the same manner as above. However, parasite growth was monitored in a SpectraMax Gemini EM fluorescent plate reader (Sunnyvale, CA). Both excitation (510 nm) and emission (540 nm) were read from the bottom.
Cytotoxicity assay
HFF cell cytotoxicity was determined by a standard MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxylmethoxylphenyl)-2H-tetrazolium) assay (CellTiter 96 Aqueous One Solution Assay; Promega). Low passage HFF cells (2 × 103 cells/well) were seeded into the 96-well culture plates, and incubated for 24h at 37 °C. Then, the HFF cells were exposed to the varying concentrations of macrolides for 2 days. After incubation, the macrolide treated medium was aspirated, and 100 μL DMEM was added followed by 20 μL MTS. The plates were incubated for 4h at 37 °C, and the optical density was measured at 490nm. CC50 was calculated as dose of each macrolide that reduced cell viability by 50%.
Parasite cell division assay
HFF cells were placed on glass cover-slips in 12-well plates, and tachyzoites of the RHΔhxgprt of T. gondii harboring a GFP gene were inoculated and cultured as above. The cover-slips were mounted onto glass microscope slides, and the number of parasites in individual parasitophorous vacuole was counted by fluorescence microscopy (Olympus BX60 microscope, 100×, N.A. 1.4/0.9 objective).
RT-PCR assay
Tachyzoites of the T. gondii strain RHΔhxgprt GFP and clinR-4 were cultured in 6-well plates as above, and then collected by centrifugation at 2,000 rpm for 10 min. The total DNA was extracted using the DNeasy kit (Qiagen, Hilden, Germany), and stored at -20 °C until use. PCR primers 5′-CCAATATGTAACATTTTAGTTCCAGTATCA-3′ (forward) and 5′-GGTCAGTAATAACTTGGAAATATCCTTCTAC-3′ (reverse) yielded a 130-bp nucleotide product from the apicoplast genome (ycf24), and primers 5′-AACAACTGCGGAGCCTAAGG-3′ (forward) and 5′-TGCACTCGAAGACACCTGATG-3′ (reverse) yielded an 89-bp fragment of the nuclear uracil phosphoribosyl transferase gene (UPRT). RT-PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems) with the following amplification conditions: 1 cycle at 94 °C for 20 s, 40 cycles of 94 °C for 3 s, and 60 °C for 30 s.
Supplementary Material
Acknowledgments
This research was supported by grants from the NIH (R01 GM 087934 to C.K., R01 AL 41014 to J.B. and 5U54MH074404, Hugh Rosen, Principal Investigator, which supported the work of J.Y.C. and WRR).
Abbreviations used
- AZ
azithromycin
- MIC
minimum inhibitory concentration
- IC50
half-maximal inhibitory concentrations
- CC50
half-maximal cytotoxic concentration
- YFP
yellow fluorescent protein
- GFP
green fluorescent protein
- RT-PCR
real time polymerase chain reaction
Footnotes
Supporting Information Available: Mutiple sequence alignment(Table S1), synthesis details of compounds 5–7, 11, 13, 15–20, and 22–31 and molecular modeling. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES and NOTES
- 1.Jones JL, Lopez A, Wilson M, Schulkin J, Gibbs R. Congenital toxoplasmosis: A review. Obstet Gynecol Surv. 2001;56:296–305. doi: 10.1097/00006254-200105000-00025. [DOI] [PubMed] [Google Scholar]
- 2.Luft BJ, Remington JS. Toxoplasmic Encephalitis in Aids. Clin Infect Dis. 1992;15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 3.Kasper LH, Buzoni-Gatel D. Some opportunistic parasitic infections in AIDS: Candidiasis, pneumocystosis, cryptosporidiosis, toxoplasmosis. Parasitol Today. 1998;14:150–156. doi: 10.1016/s0169-4758(97)01212-x. [DOI] [PubMed] [Google Scholar]
- 4.Fichera ME, Roos DS. A plastid organelle as a drug target in apicomplexan parasites. Nature. 1997;390:407–409. doi: 10.1038/37132. [DOI] [PubMed] [Google Scholar]
- 5.Baird JK. Drug therapy: Effectiveness of antimalarial drugs. New Engl J Med. 2005;352:1565–1577. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 6.White NJ. The treatment of malaria. New Engl J Med. 1996;335:800–806. doi: 10.1056/NEJM199609123351107. [DOI] [PubMed] [Google Scholar]
- 7.Lell B, Kremsner PG. Clindamycin as an antimalarial drug: Review of clinical trials. Antimicrob Agents Ch. 2002;46:2315–2320. doi: 10.1128/AAC.46.8.2315-2320.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfefferkorn ER, Borotz SE. Comparison of Mutants of Toxoplasma-Gondii Selected for Resistance to Azithromycin, Spiramycin, or Clindamycin. Antimicrob Agents Chemother. 1994;38:31–37. doi: 10.1128/aac.38.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araujo FG, Shepard RM, Remington JS. Invivo Activity of the Macrolide Antibiotics Azithromycin, Roxithromycin and Spiramycin against Toxoplasma-Gondii. Eur J Clin Microbiol. 1991;10:519–524. doi: 10.1007/BF01963942. [DOI] [PubMed] [Google Scholar]
- 10.Dunne MW, Singh N, Shukla M, Valecha N, Bhattacharyya PC, Dev V, Patel K, Mohapatra MK, Lakhani J, Benner R, Lele C, Patki K. A multicenter study of azithromycin, alone and in combination with chloroquine, for the treatment of acute uncomplicated Plasmodium falciparum malaria in India. J Infect Dis. 2005;191:1582–1588. doi: 10.1086/429343. [DOI] [PubMed] [Google Scholar]
- 11.Gingras BA, Jensen JB. Antimalarial Activity of Azithromycin and Erythromycin against Plasmodium-Berghei. Am J Trop Med Hyg. 1993;49:101–105. doi: 10.4269/ajtmh.1993.49.101. [DOI] [PubMed] [Google Scholar]
- 12.Heppner DG, Walsh DS, Uthaimongkol N, Tang DB, Tulyayon S, Permpanich B, Wimonwattrawatee T, Chuanak N, Laoboonchai A, Sookto P, Brewer TG, McDaniel P, Eamsila C, Yongvanitchit K, Uhl K, Kyle DE, Keep LW, Miller RE, Wongsrichanalai C. Randomized, controlled, double-blind trial of daily oral azithromycin in adults for the prophylaxis of Plasmodium vivax malaria in western Thailand. Am J Trop Med Hyg. 2005;73:842–849. [PubMed] [Google Scholar]
- 13.Girard AE, Girard D, English AR, Gootz TD, Cimochowski CR, Faiella JA, Haskell SL, Retsema JA. Pharmacokinetic and Invivo Studies with Azithromycin (Cp-62,993), a New Macrolide with an Extended Half-Life and Excellent Tissue Distribution. Antimicrob Agents Chemother. 1987;31:1948–1954. doi: 10.1128/aac.31.12.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray RH, Wabwire-Mangen F, Kigozi G, Sewankambo NK, Serwadda D, Moulton LH, Quinn TC, O’Brien KL, Meehan M, Abramowsky C, Robb M, Wawer MJ. Randomized trial of presumptive sexually transmitted disease therapy during pregnancy in Rakai, Uganda. Am J Obstet Gynecol. 2001;185:1209–1217. doi: 10.1067/mob.2001.118158. [DOI] [PubMed] [Google Scholar]
- 15.Wawer MJ, Sewankambo NK, Serwadda D, Quinn TC, Paxton LA, Kiwanuka N, Wabwire-Mangen F, Li CJ, Lutalo T, Nalugoda F, Gaydos CA, Moulton LH, Meehan MO, Ahmed S, Gray RH, Grp RPS. Control of sexually transmitted diseases for AIDS prevention in Uganda: a randomised community trial. Lancet. 1999;353:525–535. doi: 10.1016/s0140-6736(98)06439-3. [DOI] [PubMed] [Google Scholar]
- 16.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, Yonath A. Structure of functionally activated small ribosomal subunit at 3.3 angstrom resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 17.Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 18.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A. High resolution structure of the large ribosomal subunit from a mesophilic Eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 19.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 20.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 21.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 22.Schlunzen F, Harms JM, Franceschi F, Hansen HAS, Bartels H, Zarivach R, Yonath A. Structural basis for the antibiotic activity of ketolides and azalides. Structure. 2003;11:329–338. doi: 10.1016/s0969-2126(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 23.Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol Cell. 2002;10:117–128. doi: 10.1016/s1097-2765(02)00570-1. [DOI] [PubMed] [Google Scholar]
- 24.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330:1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 25.Auerbach T, Bashan A, Yonath A. Ribosomal antibiotics: structural basis for resistance, synergism and selectivity. Trends Biotechnol. 2004;22:570–576. doi: 10.1016/j.tibtech.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Ma ZK, Clark RF, Brazzale A, Wang SY, Rupp MJ, Li LP, Griesgraber G, Zhang SM, Yong H, Phan LT, Nemoto PA, Chu DTW, Plattner JJ, Zhang XL, Zhong P, Cao ZS, Nilius AM, Shortridge VD, Flamm R, Mitten M, Meulbroek J, Ewing P, Alder J, Or YS. Novel erythromycin derivatives with aryl groups tethered to the C-6 position are potent protein synthesis inhibitors and active against multidrug-resistant respiratory pathogens. J Med Chem. 2001;44:4137–4156. doi: 10.1021/jm0102349. [DOI] [PubMed] [Google Scholar]
- 27.Berisio R, Schluenzen F, Harms J, Bashan A, Auerbach T, Baram D, Yonath A. Structural insight into the role of the ribosomal tunnel in cellular regulation. Nat Struct Biol. 2003;10:366–370. doi: 10.1038/nsb915. [DOI] [PubMed] [Google Scholar]
- 28.Chantot JF, Bryskier A, Gasc JC. Antibacterial Activity of Roxithromycin - a Laboratory Evaluation. J Antibiot. 1986;39:660–668. doi: 10.7164/antibiotics.39.660. [DOI] [PubMed] [Google Scholar]
- 29.Gasc JC, Dambrieres SG, Lutz A, Chantot JF. New Ether Oxime Derivatives of Erythromycin-a - a Structure-Activity Relationship Study. J Antibiot. 1991;44:313–330. doi: 10.7164/antibiotics.44.313. [DOI] [PubMed] [Google Scholar]
- 30.Kobrehel GZ, Djokic S. (PLIVA) 11-Methyl-11-aza-4-0-cladinosyl-6-0-desosaminyl-15-ethyl-7,1 3,14-trihydroxy-3,5,7,9,12,14-hexamethyl-oxacyclopentadecane-2-one and derivatives thereof. 4 517 359. US. 1985
- 31.Bright GM. (Pfizer Inc.) N-Methyl 11-aza-10-deoxo-10-dihydro-erytromycin A, intermediates therefor. 4 474 768. US. 1984
- 32.Djokic S, Kobrehel G, Lazarevski G, Lopotar N, Tamburasev Z, Kamenar B, Nagl A, Vickovic I. Erythromycin Series. Part 11. Ring Expansion of Erythromycin A Oxime by the Beckmann Rearrangement. J Chem Soc Perk Trans I. 1986:1881–1890. [Google Scholar]
- 33.Djokic S, Kobrehel G, Lopotar N, Kamenar B, Nagl A, Mrvos D. Erythromycin Series. Part 13. Synthesis and Structure Elucidation of 10-Dihydro-10-Deoxo-11-Methyl-11-Azaerythromycin-A. J Chem Res. 1988;5:152–153. [Google Scholar]
- 34.Yang BWV, Goldsmith M, Rizzi JP. A Novel Product from Beckmann Rearrangement of Erythromycin-a 9(E)Oxime. Tetrahedron Lett. 1994;35:3025–3028. [Google Scholar]
- 35.Kirst HA, Wind JA, Leeds JP, Willard KE, Debono M, Bonjouklian R, Greene JM, Sullivan KA, Paschal JW, Deeter JB, Jones ND, Ott JL, Feltyduckworth AM, Counter FT. Synthesis and Structure-Activity-Relationships of New 9-N-Alkyl Derivatives of 9(S)-Erythromycylamine. J Med Chem. 1990;33:3086–3094. doi: 10.1021/jm00173a028. [DOI] [PubMed] [Google Scholar]
- 36.Gubbels MJ, Li C, Striepen B. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob Agents Chemother. 2003;47:309–316. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Djokic S, Kobrehel G, Lazarevski G. Erythromycin Series.12. Antibacterial Invitro Evaluation of 10-Dihydro-10-Deoxo-11-Azaerythromycin-a - Synthesis and Structure-Activity Relationship of Its Acyl Derivatives. J Antibiot. 1987;40:1006–1015. doi: 10.7164/antibiotics.40.1006. [DOI] [PubMed] [Google Scholar]
- 38.Bright GM, Nagel AA, Bordner J, Desai KA, Dibrino JN, Nowakowska J, Vincent L, Watrous RM, Sciavolino FC, English AR, Retsema JA, Anderson MR, Brennan LA, Borovoy RJ, Cimochowski CR, Faiella JA, Girard AE, Girard D, Herbert C, Manousos M, Mason R. Synthesis, In vitro and In vivo Activity of Novel 9-Deoxo-9a-Aza-9a-Homoerythromycin a Derivatives - a New Class of Macrolide Antibiotics, the Azalides. J Antibiot. 1988;41:1029–1047. doi: 10.7164/antibiotics.41.1029. [DOI] [PubMed] [Google Scholar]
- 39.Kujundzic NS, Kobrehel G, Kelneric Z. 9a-N-(N′-Carbamoyl) and 9a-N-(N′-Thiocarbamoyl) derivatives of 9-deoxo-9a-aza-9a-homoerythromycin A. 0 657 464. EP. 1996
- 40.Pfefferkorn ER, Nothnagel RF, Borotz SE. Parasiticidal Effect of Clindamycin on Toxoplasma-Gondii Grown in Cultured-Cells and Selection of a Drug-Resistant Mutant. Antimicrob Agents Chemother. 1992;36:1091–1096. doi: 10.1128/aac.36.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fichera ME, Bhopale MK, Roos DS. In-Vitro Assays Elucidate Peculiar Kinetics of Clindamycin Action against Toxoplasma-Gondii. Antimicrob Agents Chemother. 1995;39:1530–1537. doi: 10.1128/aac.39.7.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mccabe RE, Oster S. Current Recommendations and Future-Prospects in the Treatment of Toxoplasmosis. Drugs. 1989;38:973–987. doi: 10.2165/00003495-198938060-00008. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Q, Zhang M, Hong LX, Zhou KF, Lin YG. Evaluation of drug effects on Toxoplasma gondii nuclear and plastid DNA replication using real-time PCR. Parasitol Res. 2010;106:1257–1262. doi: 10.1007/s00436-010-1792-3. [DOI] [PubMed] [Google Scholar]
- 44.Camps M, Arrizabalaga G, Boothroyd J. An rRNA mutation identifies the apicoplast as the target for clindamycin in Toxoplasma gondii. Mol Microbiol. 2002;43:1309–1318. doi: 10.1046/j.1365-2958.2002.02825.x. [DOI] [PubMed] [Google Scholar]
- 45.Gregory ST, Dahlberg AE. Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23 S ribosomal RNA. J Mol Biol. 1999;289:827–834. doi: 10.1006/jmbi.1999.2839. [DOI] [PubMed] [Google Scholar]
- 46.Peric M, Bozdogan B, Jacobs MR, Appelbaum PC. Effects of an efflux mechanism and ribosomal mutations on macrolide susceptibility of Haemophilus influenzae clinical isolates. Antimicrob Agents Chemother. 2003;47:1017–1022. doi: 10.1128/AAC.47.3.1017-1022.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prunier AL, Trong HN, Tande D, Segond C, Leclercq R. Mutation of L4 ribosomal protein conferring unusual macrolide resistance in two independent clinical isolates of Staphylococcus aureus. Microb Drug Resist. 2005;11:18–20. doi: 10.1089/mdr.2005.11.18. [DOI] [PubMed] [Google Scholar]
- 48.Bogdanovich T, Bozdogan B, Appelbaum PC. Effect of efflux on telithromycin and macrolide susceptibility in Haemophilus influenzae. Antimicrob Agents Chemother. 2006;50:893–898. doi: 10.1128/AAC.50.3.893-898.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark C, Bozdogan B, Peric M, Dewasse B, Jacobs MR, Appelbaum PC. In vitro selection of resistance in Haemophilus influenzae by amoxicillin-clavulanate, cefpodoxime, cefprozil, azithromycin, and clarithromycin. Antimicrob Agents Chemother. 2002;46:2956–2962. doi: 10.1128/AAC.46.9.2956-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CMT, Fucini P. Translational regulation via L11: Molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 51.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Compound 30, which possesses two more extended carbons than 11, may have favorable interaction with Phe98.
- 53.Dynamic calculations were performed with fixed potential except the loop region of L22 (Lys94 – Arg108), 11, and adjacent amino acids and RNA fragments within 4 Å from the loop region. However, significant improvement of the interactions between the two phenyl groups was not observed because of packed environment near the loop region of L22 with fixed rRNA fragments.
- 54.Viluksela M, Vainio PJ, Tuominen RK. Cytotoxicity of macrolide antibiotics in a cultured human liver cell line. J Antimicrob Chemother. 1996;38:465–473. doi: 10.1093/jac/38.3.465. [DOI] [PubMed] [Google Scholar]
- 55.Vorbach H, Armbruster C, Robibaro B, Griesmacher A, El Menyawi I, Daxecker H, Raab M, Muller MM. Endothelial cell compatibility of azithromycin and erythromycin. J Antimicrob Chemother. 2002;49:407–409. doi: 10.1093/jac/49.2.407. [DOI] [PubMed] [Google Scholar]
- 56.Millrose M, Kruse M, Flick B, Stahlmann R. Effects of macrolides on proinflammatory epitops on endothelial cells in vitro. Arch Toxicol. 2009;83:469–476. doi: 10.1007/s00204-008-0388-5. [DOI] [PubMed] [Google Scholar]
- 57.Bonay P, Munro S, Fresno M, Alarcon B. Intra-Golgi transport inhibition by megalomicin. J Biol Chem. 1996;271:3719–3726. doi: 10.1074/jbc.271.7.3719. [DOI] [PubMed] [Google Scholar]
- 58.Bonay P, Fresno M, Alarcon B. Megalomicin disrupts lysosomal functions. J Cell Sci. 1997;110:1839–1849. doi: 10.1242/jcs.110.16.1839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.