

Abstract
There is growing evidence that chronic exposure of humans to inorganic arsenic, a potent environmental oxidative stressor, is associated with the incidence of type 2 diabetes (T2D). One critical feature of T2D is insulin resistance in peripheral tissues, especially in mature adipocytes, the hallmark of which is decreased insulin-stimulated glucose uptake (ISGU). Despite the deleterious effects of reactive oxygen species (ROS), they have been recognized as a second messenger serving an intracellular signaling role for insulin action. Nuclear factor erythroid 2-related factor 2 (NRF2) is a central transcription factor regulating cellular adaptive response to oxidative stress. This study proposes that in response to arsenic exposure, the NRF2-mediated adaptive induction of endogenous antioxidant enzymes blunts insulin-stimulated ROS signaling and thus impairs ISGU. Exposure of differentiated 3T3-L1 cells to low-level (up to 2 μM) inorganic arsenite (iAs3+) led to decreased ISGU in a dose- and time-dependent manner. Concomitant to the impairment of ISGU, iAs3+ exposure significantly attenuated insulin-stimulated intracellular ROS accumulation and AKT S473 phosphorylation, which could be attributed to the activation of NRF2 and induction of a battery of endogenous antioxidant enzymes. In addition, prolonged iAs3+ exposure of 3T3-L1 adipocytes resulted in significant induction of inflammatory response genes and decreased expression of adipogenic genes and glucose transporter type 4 (GLUT4), suggesting chronic inflammation and reduction in GLUT4 expression may also be involved in arsenic-induced insulin resistance in adipocytes. Taken together our studies suggest that prolonged low-level iAs3+ exposure activates the cellular adaptive oxidative stress response, which impairs insulin-stimulated ROS signaling that is involved in ISGU, and thus causes insulin resistance in adipocytes.
Keywords: arsenic, diabetes, insulin, ROS, NRF2, oxidative stress
Introduction
There is growing evidence that environmental inorganic arsenic (iAs) exposure is associated with the incidence of Type 2 diabetes (T2D), a metabolic disease attributed to a combination of genetic and environmental factors. In T2D patients, insulin resistance in target tissues, including the adipose tissue, muscle and liver, is the central pathophysiological event in the development of the disease. While a number of mechanisms, including increased levels of free fatty acids, inflammatory cytokines and adipokines, hyperinsulinemia and mitochondrial dysfunction, have been suggested to be responsible for insulin resistance [1], compelling evidence indicates that increased oxidative stress and inflammatory response play prominent roles [2; 3; 4; 5]. Accumulating data, including our own studies in vitro [6; 7], in vivo [8], and in humans [9], reveal that iAs exposure results in oxidative stress, suggesting that oxidative stress may be involved in the development of insulin resistance in humans exposed to iAs.
A key cellular component that defends cells against oxidative toxicity is the Nuclear factor erythroid 2 related factor 2 (NRF2), a transcription factor that is directly responsible for both basal and inducible expression of many antioxidant/detoxification enzymes [10]. The NRF2-mediated antioxidant induction has been recognized as one of the most important cellular adaptive responses to oxidative stress. However, this NRF2-driven induction of endogenous antioxidant enzymes - meant to maintain intracellular redox homeostasis and limit oxidative damage - may also have an adverse effect, i.e., to block the reactive oxygen species (ROS) that function as physiological signaling molecules [11; 12]. In adipocytes, ROS, in particular hydrogen peroxide (H2O2), are involved in insulin signal transduction leading to phosphorylation of protein kinase B (PKB/AKT) and glucose uptake [13]. Thus, persistent activation of NRF2 in response to chronic exposure to iAs and/or its trivalent metabolites may blunt insulin-triggered H2O2 signaling, resulting in reduced insulin sensitivity in adipocytes. Here we report that prolonged exposure to low-levels of inorganic arsenite (iAs3+) activates NRF2-mediated antioxidant response, impairs insulin signaling, such as phosphorylation of AKT S473, and suppresses insulin-stimulated glucose uptake (ISGU).
Materials and Methods
Chemicals
Sodium arsenite, dexamethasone, 3-isobutylmethylxanthine, insulin and Oil Red O (ORO) were obtained from Sigma (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) with high glucose, fetal bovine serum (FBS), 10 % bovine serum albumin (BSA), phosphate buffered saline (PBS), calf serum, TRIzol and blue-fluorescent DAPI nucleic acid stain were purchased from Invitrogen (Carlsbad, CA). [3H]-2-deoxy-D-glucose ([3H]-2-DG) was obtained from PerkinElmer, Inc. (Waltham, MA).
Cell culture, differentiation and treatments
3T3-L1 preadipocytes were obtained from ATCC (Manassas, VA) and maintained in high-glucose DMEM with 50 unit/ml penicillin, 50 μg/ml streptomycin, and 10% calf serum. All the cells were maintained at 37 °C in a 5% CO2 environment. Adipogenic differentiation was induced by using 1 μM dexamethasone, 0.5 mM 3-isobutylmethylxanthine and 5 μg/ml insulin in DMEM with 10% FBS (DMI protocol) as described previously [14]. Differentiation of preadipocytes to mature adipocytes was confirmed by ORO staining of lipid vesicles [14]. Fully-differentiated 3T3-L1 cells were exposed to low levels of iAs3+ (up to 2 μM) for 7 days followed by immediate sample collection and measurements. To determine insulin-stimulated phosphorylation of AKT S473, following 7-day iAs3+ treatment, 3T3-L1 adipocytes were first starved for 4 hr in serum-free DMEM containing 1 % BSA while the same concentrations of iAs3+ were maintained in the media. The starved cells were then challenged with insulin (5 ng/ml in DMEM) for 5 min, and followed by immediate cell lysate isolation.
Glucose uptake assay
Glucose uptake in 3T3-L1 adipocytes was measured as described previously [15]. Briefly, after 4- or 7-day treatment with iAs3+, 3T3-L1 adipocytes were plated in a 12-well plate and starved for 4 hr as above. Glucose uptake was determined by the accumulation of [3H]-2-DG in the cells by a 20 min incubation with 0.5 μCi [3H]-2-DG in 1 ml glucose-free DMEM with 50 ng/ml insulin or vehicle (medium). The radioactivity of cell lysates was determined in 5 ml of Ecolume (MP Biomedicals, LLC, Solon, OH) using Tri-carb Liquid Scintillation Analyzer (Packard Instrument Co. Inc. Meriden, CT) and normalized with protein level. ISGU was then determined as follows after non-specific binding of [3H]-2-DG with cell surface was subtracted: (Radioactivity with insulin - Radioactivity with Vehicle)/Radioactivity with Vehicle.
Quantitative real-time RT–PCR analysis
Total RNA was isolated with TRIzol. Quantitative real-time RT-PCR was performed as described previously [16]. The primers (sequences are shown in Table S1, Supplemental Materials) were designed using Primer Express 4 (Applied Biosystems, Foster City, CA) and synthesized by MWG-BIOTECH Inc. (High Point, NC). Real-time fluorescence detection was carried out using an ABI PRISM 7900 HT Sequence Detector (Applied Biosystems).
Western blot analysis
Isolation of cell fractions and Western blotting were performed as described previously [6]. Antibodies for NRF2 (sc-13032; 1: 500), heme oxygenase 1 (HO-1, sc-136902; 1: 500) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibody for NAD(P)H: quinone oxidoreductase 1 (NQO1, 39-3700; 1: 1000) was from Invitrogen. Antibody for phosphorylated AKT S473 (p-AKT S473, #9271; 1: 500) and total AKT (t-AKT, #4685; 1: 500) was from Cell Signaling Technology, Inc. (Danvers, MA). Antibody for β-ACTIN (A1978; 1: 2000) was from Sigma.
Determination of total intracellular glutathione (GSH)
Cells were sonicated in cold PBS immediately after collection followed by centrifugation at 12,000 × g for 5 min. The resulting supernatants were used for measurement of total intracellular glutathione using the BIOXYTECH GSH/GSSG-412 kit (OxisResearch, Portland, OR) [17].
Intracellular peroxide determination
Intracellular peroxide levels in 3T3-L1 adipocytes were measured by fluorescence microscopy using the fluorescent probe 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Invitrogen) as described previously [18]. The final concentration of CM-H2DCFDA used was 2 μM and loading time was 30 min. The fluorescence images were obtained by using an Axio Observer Z1 fluorescence microscope (Carl Zeiss, Inc., Jena, Germany) and quantified by using ImageJ 1.44 (http://rsbweb.nih.gov/ij/).
Immunofluorescence staining
Cells were grown on glass coverslips in 6-well plate. After 7-day treatment with iAs3+, cells were fixed with 2 % formaldehyde overnight and permeabilized with 0.3 % Triton X-100 in PBS for 10 min at room temperature. After blocking in 10 % serum in PBS for 1 hr, cells were then incubated with anti-glucose transporter type 4 (GLUT4, sc-7938; 1: 50) antibody overnight at 4 °C. Coverslips were washed and incubated with Goat anti-rabbit IgG-FITC secondary antibody (sc-2012; 1:50). After DAPI staining, the coverslips were mounted on glass slides and examined using an Axio Observer Z1 fluorescence microscope.
Statistical analysis
All statistical analyses were performed using Graphpad Prism 4 (GraphPad Software, San Diego, CA), with p < 0.05 taken as significant. Data are expressed as mean ± SEM. For comparisons among groups, one-way or two-way ANOVA with Bonferroni post hoc testing was performed.
Results
Prolonged iAs3+ exposure suppressed insulin-stimulated phosphorylation of AKT S473 and glucose uptake in 3T3-L1 adipocytes
3T3-L1 cells, derived from mouse embryo fibroblasts, have been used as a model of adipogenic differentiation and insulin action. Upon hormonal stimulation, the cells undergo growth arrest and initiate differentiation manifested by the morphological and enzymatic characteristics of mature adipocytes, such as large lipid droplet (Supplemental Materials Fig. S1). During differentiation, 3T3-L1 cells increase the expression of insulin receptors and their capacity to bind insulin ligand [19]. Thus, 3T3-L1 adipocytes are a well accepted cell model to study insulin signaling.
The effect of chronic iAs3+ exposure on insulin sensitivity was investigated by measuring phosphorylation of AKT S473 and glucose uptake in 3T3-L1 adipocytes challenged with insulin following a 7-day iAs3+ exposure at non-cytotoxic concentrations (Fig. S1). As shown in Fig. 1A, acute insulin stimulation resulted in a dramatic increase in AKT S473 phosphorylation in Control cells. In contrast, insulin-stimulated AKT S473 phosphorylation was attenuated in iAs3+-exposed cells despite their moderately elevated basal levels in the absence of insulin. In keeping with the attenuation of AKT phosphorylation, iAs3+ exposure led to reduced ISGU in a time- and dose-dependent fashion (Fig. 1B).
Fig 1.
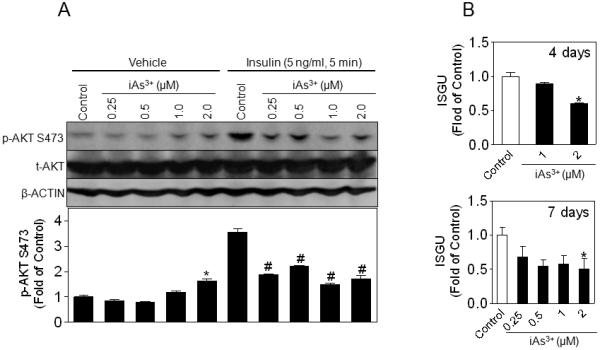
Prolonged low-level iAs3+ exposure inhibits insulin-stimulated phosphorylation of AKT S473 and ISGU in 3T3-L1 adipocytes. Confluent 3T3-L1 preadipocytes were differentiated using dexamethasone, 3-isobutylmethylxanthine and insulin for 7 days followed by a 7-day iAs3+ exposure. (A) Immunoboltting of p-AKT S473 and t-AKT in whole cell lysates, with β-ACTIN as a loading control. Lower panel is the quantitative result of p-AKT S473. n = 3; * p < 0.05 vs. Control with Vehicle (medium); # p < 0.05 vs. Control with Insulin. (B) ISGU measured in 3T3-L1 adipocytes that had been exposed to low-level iAs3+ for 4 days (upper panel) and 7 days (lower panel). n = 3; *p < 0.05 vs. Control.
Prolonged iAs3+ exposure activated NRF2 and the antioxidant response and dampened insulin-stimulated ROS production in 3T3-L1 adipocytes
The effect of chronic iAs3+ exposure on NRF2-mediated antioxidant response was investigated by measuring the expression of NRF2 and its downstream target antioxidant genes in fully differentiated 3T3-L1 cells. As shown in Fig. 2A and B, prolonged exposure (7 days) of 3T3-L1 adipocytes to iAs3+ resulted in a dose-dependent increase in the expression of NRF2 at mRNA and protein levels. Consistent with these findings, significant induction of NRF2 target genes, including γ-glutamate cysteine ligase catalytic subunit (GCLC) and regulatory subunit (GCLM), glutathione S-transferase (GST), NQO1 and HO-1, was also observed in iAs3+-treated cells (Fig. 2C and D, and Supplemental Materials Fig. S2). In addition, intracellular GSH levels showed a similar pattern to NRF2 and its target gene expression (Fig. 2E). Furthermore, the expression of many inflammatory factors, including tumor necrosis factor α (Tnfα), interleukin 6 (Il6), interleukin 1β (Il1β), cyclooxygenase 2 (Cox2) and inducible nitric oxide synthase (iNos), was also significantly induced in response to prolonged iAs3+ exposure (Supplemental Materials Fig. S3). These results clearly demonstrate prolonged, low-level iAs3+ exposure activates NRF2-mediated antioxidant response, as well as inflammatory response in 3T3-L1 adipocytes.
Fig. 2.
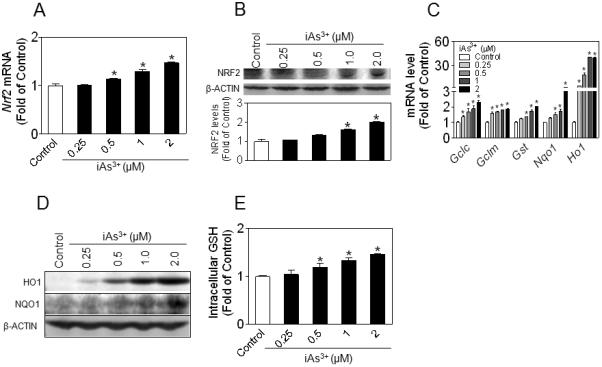
iAs3+ activates NRF2-mediated antioxidant response in 3T3-L1 adipocytes. 3T3-L1 adipocytes were exposed to iAs3+ for 7 days. (A) Gene expression of Nrf2. n = 3–6; *, p < 0.05 vs. Control. (B) Protein level of NRF2 in whole cell lysates. β-ACTIN is a loading control. Lower panel is the quantified result of immunoblots. n = 3; * p < 0.05 vs. Control. (C) Expression of NRF2-target genes measured by real-time RT-PCR. n = 3; * p < 0.05 vs. Control. (D) Protein expression of NRF2 downstream target HO1 and NQO1. (E) Intracellular GSH level. GSH levels in whole cell lysates were normalized by protein content and expressed as fold of Control. The intracellular GSH concentration in Control cells is 20.7 ± 2.2 μmol/g protein. n = 3; * p < 0.05 vs. Control.
Intracellular peroxide levels were determined to investigate whether iAs3+ exposure affects insulin-stimulated ROS signaling in 3T3-L1 adipocytes. Consistent with the premise that ROS is a second messengers mediating insulin signaling [13], as shown in Fig. 3, insulin significantly increased the intracellular peroxide level in Control cells. As expected, cells exposed to iAs3+ alone, a potent oxidative stressor, exhibited high basal peroxide levels. However, when the iAs3+-exposed cells were stimulated with insulin, no additional increase in the peroxide level was observed, suggesting that the elevated antioxidants after prolonged iAs3+ exposure (Fig. 2 and Supplemental Materials Fig. S2) may blunt the production of any signaling ROS that are stimulated by insulin. The abolished peroxide response is likely responsible, at least partially, for the mitigated phosphorylation of AKT S473 and ISGU observed in iAs3+-exposed cells (Fig. 1).
Fig. 3.
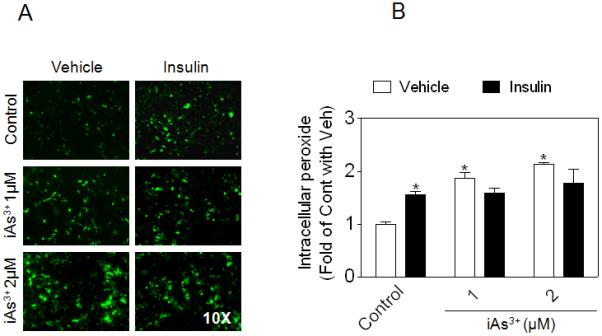
Effect of iAs3+ exposure on insulin-stimulated peroxide production in 3T3-L1 adipocytes. (A) Following a 7-day exposure to iAs3+, intracellular peroxide level was determined using CM-H2DCFDA under basal (no insulin) and insulin-challenged condition (50 ng/ml insulin; 30 min). (B) Quantification of (A). n = 2-5. * p < 0.05 vs. Control with vehicle.
Effect of prolonged iAs3+ exposure on the expression of adipogenic genes and GLUT4 in 3T3-L1 adipocytes
Peroxisome proliferator activated receptor γ (PPARγ) and CCAAT-enhancer-binding protein α (C/EBPα) are expressed abundantly in adipose tissue and is considered to be the dominant transcriptional regulator of adipogenic differentiation and adipocyte function [14]. As shown in Fig. 4A, prolonged exposure of 3T3-L1 adipocytes to low levels of iAs3+ resulted in markedly reduced gene expression of Pparγ1, Pparγ2, Cebpα and Glut4. In keeping with the reduced gene expression of Glut4, immunofluorescence microscopy also showed a substantial decrease in the GLUT4 protein level in iAs3+-exposed 3T3-L1 adipocytes (Fig. 4B).
Fig. 4.
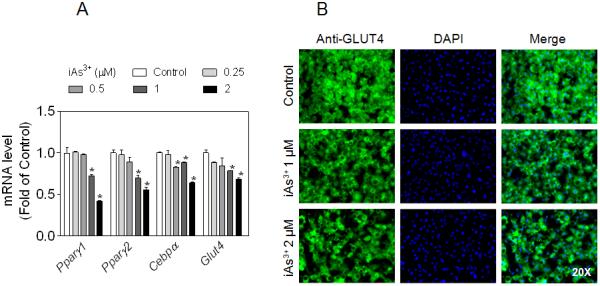
Effect of prolonged iAs3+ exposure on the expression of adipogenic genes and glucose transporter GLUT4 in 3T3-L1 adipocytes. Cells were exposed to low-level iAs3+ for 7 days. (A) Gene expression of Pparγ, Cebpα and Glut4. n = 3. *p < 0.05 vs. Control. (B) Representative images of immunostaining of GLUT4. Left panels, immunofluorescent images of GLUT4; Middle panels, nuclear DAPI staining; Right panels, merged images of GLUT4 and DAPI.
Discussion
While the precise mechanisms for the diabetogenic effect of iAs are still largely undefined, recent experimental studies indicated that acute exposure to iAs or its metabolites impairs ISGU and results in insulin resistance [15; 20; 21]. The present study reveals that prolonged low-level iAs3+ exposure of 3T3-L1 adipocytes causes oxidative stress and inflammatory response and impairs insulin-stimulated phosphorylation of AKT S473 and ISGU. The concurrence of iAs3+-induced activation of NRF2-mediated antioxidant response, attenuation of insulin-stimulated peroxide production, AKT S473 phosphorylation and ISGU suggests that chronic iAs exposure-induced insulin resistance in adipocytes may result, at least partially, from activation of NRF2 and subsequent induction of antioxidant enzymes, which might dampen ROS-mediated signaling transduction by insulin.
Traditionally, ROS have been thought of as useless by-products of respiratory metabolism in mitochondria, and are generally believed to be deleterious to biological systems. In the field of T2D research, oxidative stress from excessive ROS production is recognized as a chief culprit in the development of insulin resistance in insulin-responsive cells [5]. Despite the deleterious effects of ROS, growing evidence indicates that they can function as a second messenger serving an intracellular signaling role [22]. With respect to T2D, it has been shown that ROS, in particular H2O2, is involved in insulin signal transduction in insulin-responsive cells [23]. In adipocytes, NADPH oxidase has been identified as an important source of ROS production in response to insulin stimulation [23]. Based on these studies it can be argued that augmentation of the cellular ROS-scavenging capacity, as a result of the adaptive response to oxidative stress, may actually impair the ‘beneficial’ aspects of ROS signaling that contribute to insulin action. Thus, we propose that NRF2-mediated adaptive antioxidant response plays paradoxical roles in insulin-responsive cells: (1) it protects these cells from oxidative damage; but (2) it also blunts insulin-triggered ‘ROS signaling’ and thus results in reduced insulin sensitivity in insulin-responsive cells. Clearly, the second premise may be relevant to low-dose iAs-induced insulin resistance in adipocytes as oxidative damage may not be significant at these low doses.
Consistent with the idea that activation of NRF2 may be involved in the development of insulin resistance, elevated levels of antioxidants have been observed in ob/ob mice [24], subjects at risk of T2D [25] and diabetic patients [26; 27]. More importantly, emerging in vivo data suggest that overexpression of H2O2-scavenging enzymes, such as catalase and glutathione peroxidase 1, actually increased insulin resistance and the likelihood of developing diabetes despite potentially improved handling of oxidative stress [28; 29]. Together with our previous study [17] showing that low levels of iAs3+ impair glucose-stimulated insulin secretion in pancreatic β-cells, which is also associated with the adaptive cellular oxidative stress response, iAs-induced adaptive induction of antioxidants stress may be an important general mechanism for the development of T2D.
GLUT4 is an insulin-regulated glucose transporter highly expressed in the adipose tissue and skeletal muscle [30]. Transgenic mice expressing high levels of GLUT4 in the adipose tissue are insulin sensitive and glucose tolerant. In contrast, conditional depletion of GLUT4 in the adipose tissue causes insulin resistance [30]. In the absence of insulin, GLUT4 is sequestered in the lipid bilayers of intracellular vesicles of fat and muscle cells. Insulin induces the translocation of GLUT4 from the intracellular store to the plasma membrane and thus increases glucose uptake. Although insulin-stimulated GLUT4 translocation, a major downstream event of insulin-stimulated AKT activation, is one of the most important mechanisms in regulating ISGU, the observed downregulation of GLUT4 expression by iAs (Fig. 4) suggests that another mechanism may also be at work to contribute to iAs-induced insulin resistance in adipocytes. Obviously, the exact mode of GLUT4 regulation by iAs at the transcriptional or translational level needs further investigation.
Taken together, this study indicates that low concentrations of iAs cause oxidative stress and cellular adaptive response involving NRF2 activation, in adipocytes, and such a cellular adaptive response to iAs is associated with impairment of insulin sensitivity. While the etiology of T2D is still unclear, to date, therapeutic approaches have focused on medication and lifestyle modification. The role of environmental exposures should also be considered in the future.
Supplementary Material
Acknowledgements
This work was supported in part by the National Institutes of Health Grants DK76788 and ES016005 (to J.P.) and the American Chemistry Council-Long Range Research Initiative and the DOW Chemical Company (to M.E.A.). The content is solely the responsibility of the authors. P.X., Y.H., Q.Z., C.G.W, M.E.A. and J.P. are employees of The Hamner Institutes for Health Sciences. The Hamner is a 501(c)3 not-for-profit organization that has a diverse research portfolio that includes funding from the American Chemistry Council, a trade association that represents chemical manufacturers.
Abbreviations
- BSA
bovine serum albumin
- C/EBPα
CCAAT-enhancer-binding protein α
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate, acetyl ester
- COX2
cyclooxygenase 2
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- GCLC
γ-glutamate cysteine ligase catalytic subunit
- GCLM
γ-glutamate cysteine ligase regulatory subunit
- GSH
Glutathione
- GST
glutathione S-transferase
- [3H]-2-DG
[3H]-2-deoxy-D-lucose
- HO-1
heme oxygenase 1
- H2O2
hydrogen peroxide
- iAs
inorganic arsenic
- IL1β
interleukin 1β
- IL6
interleukin 6
- GLUT4
glucose transporter type 4
- iAs3+
inorganic arsenite
- iNos
inducible nitric oxide synthase
- ISGU
insulin-stimulated glucose uptake
- NQO1
NAD(P)H: quinone oxidoreductase 1
- NRF2
Nuclear factor erythroid 2-related factor 2
- ORO
Oil Red O
- PKB/AKT
protein kinase B
- PBS
phosphate buffered saline
- PPARγ
peroxisome proliferator activated receptor γ
- ROS
reactive oxygen species
- T2D
type 2 diabetes
- TNFα
tumor necrosis factor α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have agreed to its content and there are no financial or other conflicts of interest.
References
- 1.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–46. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 2.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. 2003;52:1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Scott JA, King GL. Oxidative stress and antioxidant treatment in diabetes. Ann N Y Acad Sci. 2004;1031:204–13. doi: 10.1196/annals.1331.020. [DOI] [PubMed] [Google Scholar]
- 4.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–8. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 6.Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–45. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- 7.Pi J, He Y, Bortner C, Huang J, Liu J, Zhou T, Qu W, North SL, Kasprzak KS, Diwan BA, Chignell CF, Waalkes MP. Low level, long-term inorganic arsenite exposure causes generalized resistance to apoptosis in cultured human keratinocytes: Potential role in skin co-carcinogenesis. Int J Cancer. 2005;116:20–6. doi: 10.1002/ijc.20990. [DOI] [PubMed] [Google Scholar]
- 8.Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T, Yamauchi H, Itoh K, Yamamoto M, Sun G, Waalkes MP, Kumagai Y. A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Radic Biol Med. 2003;35:102–13. doi: 10.1016/s0891-5849(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 9.Pi J, Yamauchi H, Kumagai Y, Sun G, Yoshida T, Aikawa H, Hopenhayn-Rich C, Shimojo N. Evidence for induction of oxidative stress caused by chronic exposure of Chinese residents to arsenic contained in drinking water. Environ Health Perspect. 2002;110:331–6. doi: 10.1289/ehp.02110331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–40. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, Collins S, Andersen ME. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244:77–83. doi: 10.1016/j.taap.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Pi J, Woods CG, Andersen ME. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol Appl Pharmacol. 2010;244:84–97. doi: 10.1016/j.taap.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–21. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, Yehuda-Shnaidman E, Lee C, Lau J, Kurtz TW, Chan JY. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 2010;285:9292–300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walton FS, Harmon AW, Paul DS, Drobna Z, Patel YM, Styblo M. Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: possible mechanism of arsenic-induced diabetes. Toxicol Appl Pharmacol. 2004;198:424–33. doi: 10.1016/j.taap.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Woods CG, Fu J, Xue P, Hou Y, Pluta LJ, Yang L, Zhang Q, Thomas RS, Andersen ME, Pi J. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicol Appl Pharmacol. 2009;238:27–36. doi: 10.1016/j.taap.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, Sun G, Andersen ME, Pi J. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect. 118:864–70. doi: 10.1289/ehp.0901608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–91. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 19.Reed BC, Lane MD. Expression of insulin receptors during preadipocyte differentiation. Adv Enzyme Regul. 1980;18:97–117. doi: 10.1016/0065-2571(80)90011-4. [DOI] [PubMed] [Google Scholar]
- 20.Paul DS, Hernandez-Zavala A, Walton FS, Adair BM, Dedina J, Matousek T, Styblo M. Examination of the effects of arsenic on glucose homeostasis in cell culture and animal studies: development of a mouse model for arsenic-induced diabetes. Toxicol Appl Pharmacol. 2007;222:305–14. doi: 10.1016/j.taap.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paul DS, Harmon AW, Devesa V, Thomas DJ, Styblo M. Molecular mechanisms of the diabetogenic effects of arsenic: inhibition of insulin signaling by arsenite and methylarsonous Acid. Environ Health Perspect. 2007;115:734–42. doi: 10.1289/ehp.9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–31. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakao C, Ookawara T, Sato Y, Kizaki T, Imazeki N, Matsubara O, Haga S, Suzuki K, Taniguchi N, Ohno H. Extracellular superoxide dismutase in tissues from obese (ob/ob) mice. Free Radic Res. 2000;33:229–41. doi: 10.1080/10715760000301401. [DOI] [PubMed] [Google Scholar]
- 25.Costa A, Iguala I, Bedini J, Quinto L, Conget I. Uric acid concentration in subjects at risk of type 2 diabetes mellitus: relationship to components of the metabolic syndrome. Metabolism. 2002;51:372–5. doi: 10.1053/meta.2002.30523. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Scholl TO, Leskiw MJ, Donaldson MR, Stein TP. Association of glutathione peroxidase activity with insulin resistance and dietary fat intake during normal pregnancy. J Clin Endocrinol Metab. 2003;88:5963–8. doi: 10.1210/jc.2003-030544. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Wildman RP, Hamm LL, Muntner P, Reynolds K, Whelton PK, He J. Association between inflammation and insulin resistance in U.S. nondiabetic adults: results from the Third National Health and Nutrition Examination Survey. Diabetes Care. 2004;27:2960–5. doi: 10.2337/diacare.27.12.2960. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Chen H, Epstein PN. Metallothionein and Catalase Sensitize to Diabetes in Nonobese Diabetic Mice: Reactive Oxygen Species May Have a Protective Role in Pancreatic {beta}-Cells. Diabetes. 2006;55:1592–604. doi: 10.2337/db05-1357. [DOI] [PubMed] [Google Scholar]
- 29.McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, Liu F, Lei XG. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci U S A. 2004;101:8852–7. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–52. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.