

Abstract
Recent evidence suggests that transmembrane domain (TMD) interactions are essential for HIV-1 Vpu-mediated antagonism of the restriction factor BST-2/tetherin. We made Vpu TMD mutants to study the mechanism of BST-2 antagonism. Vpu-I17A, -A18F, - W22L, and -S23L co-localized with BST-2 within endosomal membranes while effectively enhancing virion release and down-regulating surface BST-2. However, Vpu-A18H was confined to an endoplasmic reticulum (ER)-like distribution, resulting in impaired down-regulation of BST-2 and reduced virion release. Brefeldin A confined wild type Vpu to the ER, resulting in a similarly impaired phenotype, as did the addition of a C-terminal ER-retention signal to Vpu. We determined the half-life of cell-surface BST-2 to be ~8 hours, whereas Vpu mediated an ~80% reduction of surface BST-2 within 6 hours, suggesting that TMD interactions between Vpu and BST-2 occur within post-ER membranes to directly and rapidly remove BST-2 from the cell surface and relieve restricted virion release.
Keywords: Vpu, BST-2, Tetherin, CD4, Endoplasmic reticulum, brefeldin A
Introduction
Several interferon (IFN)-inducible innate host restriction factors reduce the efficiency of Human Immunodeficiency Virus type-1 (HIV-1) replication in vitro, including APOBEC3G (A3G) (Sheehy et al., 2002), APOBEC3F (A3F) (Huthoff and Towers, 2008), TRIM5α (Stremlau et al., 2004), and BST-2/CD317/tetherin (Neil, Zang, and Bieniasz, 2008; Van Damme et al., 2008). BST-2 traps fully mature HIV-1 virus particles on the surface of infected cells, although the exact mechanism of restriction is unclear (Neil, Zang, and Bieniasz, 2008; Perez-Caballero et al., 2009). The sequestration of virus particles by BST-2 is not unique to HIV-1 and other retroviruses; BST-2 has been demonstrated to trap other enveloped viruses including KSHV, Lassa, as well as Ebola and Marburg virus-like particles (VLPs) (Jouvenet et al., 2009; Kaletsky et al., 2009; Mansouri et al., 2009; Sakuma et al., 2009). KSHV, Ebola, SIV, HIV-2, and HIV-1 have each evolved proteins to counteract the restrictive properties of BST-2 including K5 (KSHV) (Bartee, McCormack, and Fruh, 2006; Mansouri et al., 2009), Ebola glycoprotein (Kaletsky et al., 2009), Nef (SIV) (Jia et al., 2009), Env (HIV-2 and SIV) (Gupta et al., 2009b; Le Tortorec and Neil, 2009) and Vpu (HIV-1) (Neil, Zang, and Bieniasz, 2008; Van Damme et al., 2008).
BST-2 is a 30-36-kDa, 180-amino acid, heterogeneously glycosylated dimeric type II transmembrane protein (Kupzig et al., 2003; Ohtomo et al., 1999). The ectodomain of BST-2 consists of α-helices that form a coiled-coil (Hinz et al., 2010; Schubert et al., 2010). The extracellular domain also contains two N-linked glycosylation sites and is modified by a glycosyl phosphatidyl inositol (GPI) membrane-anchor at the C-terminus (Kupzig et al., 2003). The N-terminal cytoplasmic domain contains a conserved YxY sequence important for clathrin-mediated endocytosis (Masuyama et al., 2009; Rollason et al., 2007), non-human primate BST-2 orthologues contain a DDIWK sequence for responsiveness to SIV Nef (Jia et al., 2009; Zhang et al., 2009), and human BST-2 also contains a KxxK sequence whose lysine residues are ubiquitinated by K5 (Mansouri et al., 2009). Comparative analysis of BST-2 orthologs and site directed mutagenesis revealed that the transmembrane domain (TMD) of human BST-2 contains several residues that render the protein sensitive to Vpu (Gupta et al., 2009a; Jia et al., 2009; McNatt et al., 2009; Rong et al., 2009), suggesting that TMD-TMD interactions may play a significant role in the mechanism of the antagonism of BST-2 by Vpu.
Vpu is a 16-kDa, 81-amino acid, homo-oligomeric type I transmembrane phosphoprotein that causes the ER-associated degradation (ERAD) of newly synthesized CD4 (Cohen et al., 1988; Schubert et al., 1996a; Strebel, Klimkait, and Martin, 1988). Vpu also enhances virion release (Schubert et al., 1996a; Schubert et al., 1996b). The cytoplasmic domain (CD) of Vpu contains a DSGxxS phosphoserine motif that recruits β-TrCP and an SCF E3 ubiquitin ligase complex that induces the ERAD of CD4 (Margottin et al., 1998; Schubert et al., 1996a). The CD of Vpu plays a significant role in down-regulating BST-2 from the cell surface and relieving BST-2-associated restriction, also via a mechanism partially involving the recruitment of β-TrCP and the SCF E3 ubiquitin ligase complex (Douglas et al., 2009; Mitchell et al., 2009; Tokarev, Munguia, and Guatelli, 2011; Van Damme et al., 2008). However, the specific pathway used to down-regulate BST-2 is unclear, with evidence for an ERAD-like mechanism associated with degradation of BST-2 (Mangeat et al., 2009), endosomal trafficking mechanisms associated with specific removal of BST-2 from the cell surface (Mitchell et al., 2009), and entrapment of BST-2 within the trans-Golgi network (Dube et al., 2010; Hauser et al., 2010).
The TMD of Vpu forms homo-oligomeric cation-selective ion channels which were proposed to enhance virion release, a notion supported by the observation that the TMD sequence is required for both the enhancement of virion release and ion channel activity (Lopez et al., 2002; Schubert et al., 1996a; Schubert et al., 1996b). However, the discovery of BST-2 as the restriction factor antagonized by Vpu rendered the functional significance of Vpu’s ion channel activity unclear. The TMD of Vpu is required for the down-regulation of BST-2 from the cell surface as well as for counteraction of BST-2 mediated restriction (Van Damme et al., 2008). The recent evidence that specific BST-2 TMD residues are necessary for sensitivity to Vpu further support the notion that the TMDs of Vpu and BST-2 interact (Dube et al., 2010; Gupta et al., 2009a; Jia et al., 2009; McNatt et al., 2009; Rong et al., 2009).
Here, we show that mutating the TMD of Vpu affects both the protein’s subcellular localization and its ability to antagonize BST-2. Several of the Vpu mutants tested (Vpu-I17A, -A18F, -W22L, and -S23L) effectively co-localized with BST-2, enhanced virion release, and down-regulated BST-2 from the cell surface. Two of these mutants, Vpu-W22L and -S23L, were previously evaluated for their ion channel activity (Mehnert et al., 2008). Vpu-S23L was reportedly defective (Mehnert et al., 2008), indicating that ion channel activity may be unrelated to BST-2 antagonism. The Vpu mutant A18H inefficiently enhanced virion release, inefficiently down-regulated BST-2 from the cell surface, appeared to be trapped in the ER, and co-localized poorly with BST-2, although it was able to down-regulate CD4 from the cell surface. We could recapitulate the A18H phenotype in the case of wild type Vpu using the fungal metabolite brefeldin A (BFA) to block egress of Vpu from the ER, or by appending an ER-retention signal to the C-terminus of Vpu. We determined that BST-2 on the cell surface is relatively long-lived, with a half-life of approximately 8 hours, whereas Vpu mediated an 80% reduction of surface BST-2 as soon as six hours after the onset of expression. These data together suggest that the Vpu-mediated antagonism of BST-2 occurs independently of ion channel activity, within a post-ER compartment, and likely involves primarily the targeting of existing rather than newly synthesized BST-2 to directly decrease surface levels and relieve restricted virion release.
Results
Rationale of site-directed mutations and the frequency of Vpu polymorphisms in group M isolates of HIV-1
To evaluate the contribution of Vpu TMD domain residues, four Vpu TMD mutant-proteins were initially generated: Vpu-I17A, -A18H, -W22L, and -S23L. These four mutants were chosen based on previous literature suggesting potential effects on the ion-channel activity of Vpu (Mehnert et al., 2008; Park et al., 2003). Although Vpu-I17A and Vpu-A18H have not been characterized for ion-channel activity, the proximity of the mutations to a hinge region or kink in the middle of the α-helical Vpu TMD suggested that these mutations might alter channel activity (Park et al., 2003). Vpu-W22L was previously characterized as a functional ion-channel, although the durations between the open and closed conformations in artificial lipid bilayers were prolonged compared to the wild type Vpu (Mehnert et al., 2008). In contrast, Vpu-S23L was previously characterized as defective for ion-channel activity (Mehnert et al., 2008).
To assess the conservation of these residues, an alignment of Vpu TMDs was performed using all of the available 1563 HIV-1 group M sequences from the Los Alamos National Lab HIV Sequence Database (Table 1). The alignment indicated that residues A18 and W22 are highly conserved, with frequencies greater than 95%, while residues I17 and S23 occur at lower frequencies, 63% for I17 and 25% for S23. Three of the generated substitutions exhibited frequencies of less than 1% [A18H (0%), W22L (0%), and S23L (0.06%)], while the I17A substitution appeared as a naturally occurring polymorphism with a frequency of 8.2%. Notably, residue 23 occurred as an isoleucine in 11% of sequences.
Site-specific amino acid composition at Vpu positions 17,18, 22, and 23 based on 1563 sequences*
| p17 | Frequency (N) | p18 | Frequency (N) | p22 | Frequency (N) | p23 | Frequency (N) |
|---|---|---|---|---|---|---|---|
| I | 63 (991) | A | 95 (1479) | W | 99(1551) | T | 61 (956) |
| L | 27 (426) | V | 4 (55) | X | <1 (3) | S | 25 (387) |
| A | 8 (128) | T | 1 (19) | G | <1 (1) | I | 11 (117) |
| V | <1 (10) | G | <1 (6) | R | <1 (1) | A | 1 (22) |
| X | <1 (4) | X | <1 (3) | Y | <1 (1) | V | <1 (10) |
| S | <1 (2) | C | <1 (1) | X | <1 (4) | ||
| M | <1 (1) | N | <1 (1) | F | <1(3) | ||
| T | <1 (1) | D | <1 (1) | ||||
| G | <1 (1) | ||||||
| L | <1 (1) | ||||||
| P | <1 (1) | ||||||
| 117A 8% | A18H 0% | W22L 0% | S23L <1% |
Amino acid composition calculated using all available HIV-1 M group sequences in the Los Alamos National Lab HIV Sequence Database (one sequence included per host). Positions are numbered according to the NL4-3 reference sequence. Residues at each site are listed from most to least frequent. Numbers in parentheses indicate total number of sequences in the database with the listed amino acid. Gaps and stop codons were excluded from analysis.
Vpu-A18H inefficiently down-regulates the expression of BST-2 at the cell surface
The precise mechanism by which Vpu antagonizes the restriction factor BST-2 to enhance HIV-1 virion release has yet to be elucidated, although substantial evidence indicates a correlation between the Vpu-mediated down-regulation of BST-2 from the cell surface and the enhancement of virion release. To further test this correlation, we assayed the ability of each of the Vpu TMD mutant-proteins to down-regulate BST-2 from the cell surface. Cells were co-transfected with a plasmid expressing codon-optimized Vpu along with a plasmid expressing GFP to indicate transfection efficiency. Cells were stained for surface BST-2 and analyzed by two-color flow cytometry (Figure 1A, in which the relative cell number vs. BST-2 fluorescence intensity is shown for the GFP-positive cells). The levels of surface BST-2 were reduced approximately 10-fold by the expression of wild type Vpu after 24 hours. Similar levels of down-regulation of surface BST-2 were achieved with the expression of the Vpu TMD mutant-proteins Vpu-I17A, -W22L, and -S23L (Figure 1C). The expression of Vpu-2/6, which contains two substitutions (S52N and S56N) in the DSGXXS motif within the cytoplasmic domain, resulted in an intermediate down-regulation of surface BST-2 expression consistent with previous findings (Figure 1A and C). Most interestingly, Vpu-A18H was dramatically impaired in the down-regulation of surface BST-2. The majority of cells expressing Vpu-A18H showed surface BST-2 levels comparable to those observed in cells transfected with the empty vector, although modest down-regulation of surface BST-2 was detectable at high expression (GFP) levels (Figure 1A and data not shown).
Figure 1. Vpu-A18H inefficiently down-regulates surface BST-2 expression.

Cells (HeLa) were transfected with either an empty vector plasmid or a plasmid expressing codon-optimized Vpu (160ng), along with a plasmid expressing GFP (80ng) as a transfection marker. The next day, the cells were simultaneously stained for surface BST-2 and surface CD4 then analyzed by three-color flow cytometry. (A) Histograms represent the relative cell number vs. BST-2 fluorescence intensity for the GFP-positive cells. The transfection efficiency, as indicated by the percentage of GFP-positive cells, was approximately 30% for each of the transfections. The black line represents the level of surface BST-2 down-regulation achieved when Vpu is present. The shaded grey peak represents the levels of surface BST-2 present when transfected with empty vector. (B) Histograms represent the relative cell number vs. CD4 fluorescence intensity for the GFP-positive cells. The black line represents the level of surface CD4 down-regulation achieved when Vpu is present. The shaded grey peak represents the levels of surface CD4 present when transfected with empty vector. (C) The bar graph indicates the fractional BST-2 down-regulation achieved by each Vpu tested. The values were determined as 1-[(Vpu APC-MFI - isotype APC-MFI) / (empty vector APC-MFI – isotype APC-MFI)]; a value of 1.0 indicates complete down-regulation of surface BST-2 (MFI equal to that of the isotype control), whereas a value of 0.0 indicates no down-regulation. (D) The bar graph indicates the fractional CD4 down-regulation achieved by each Vpu tested. The values were determined as 1-[(Vpu PE-MFI - isotype PE-MFI) / (empty vector PE-MFI – isotype PE-MFI)]; a value of 1.0 indicates complete down-regulation of surface CD4 (MFI equal to that of the isotype control), whereas a value of 0.0 indicates no down-regulation.
Vpu down-regulates CD4 in an endoplasmic reticulum associated degradation (ERAD)-dependent manner (Binette et al., 2007; Magadan et al., 2010; Willey et al., 1992). We tested this phenotype to confirm that each Vpu mutant was not grossly misfolded. Cells were co-transfected with a plasmid expressing codon-optimized Vpu along with a plasmid expressing GFP to indicate transfection efficiency. The cells were then stained for CD4 and analyzed by two-color flow cytometry (Figure 1B and D). Each Vpu TMD mutant down-regulated CD4 from the cell surface, with the exception of Vpu 2/6, confirming the previously published characterization of the Vpu 2/6 mutant (Schubert et al., 1996a). Vpu-I17A, -A18H, -W22L, and –S23L all exhibited CD4 down-regulation similar to or better than wild type Vpu (Figure 1D).
Vpu-A18H enhances virion release inefficiently
As the Vpu TMD mutants induced varying degrees of down-regulation of cell surface BST-2, we next characterized their abilities to enhance virion release. Cells were transfected with either wild type pNL4-3 provirus or pvpuDEL-1 (ΔVpu) provirus complemented in trans with a plasmid expressing the codon-optimized version of each Vpu mutant-protein. The amount of Vpu expression plasmid was empirically determined as that which was just sufficient to rescue the release of ΔVpu to wild type levels. The wild type NL4-3 yielded a fractional p24 release of 35% compared to 4.2% in the case of ΔVpu, indicating an 8-fold enhancement of virion release by Vpu expressed in cis within the viral genome (Figure 2A). When the ΔVpu provirus was co-transfected with plasmids expressing either wild type Vpu or the Vpu mutant-proteins, the fractional release of p24 was 29.2% for wild type Vpu, 28.8% for Vpu-I17A, 24.9% for Vpu-W22L, and 33.3% for Vpu-S23L, indicating a 6- to 8-fold enhancement of virion release (Figure 2A). Complementation of ΔVpu with Vpu-2/6 resulted in a fractional p24 release of 12.7%, an intermediate, 3-fold enhancement consistent with previous data [Figure 2A, (Mitchell et al., 2009)].
Figure 2. Vpu-A18H inefficiently enhances virion release.

(A) Cells (HeLa) were transfected with either a proviral plasmid expressing wild type HIV-1NL4-3 or a vpu-negative proviral plasmid vpuDEL-1 (ΔVpu) (1.6μg). Each of the Vpu mutant-proteins was expressed in trans (160ng) with ΔVpu to assess their activity in virion release. The next day, the fraction of the total p24 capsid antigen produced by the cells that was secreted into the media was measured by ELISA. The average values from two independent experiments are graphed; the error bars indicate the actual values obtained from each experiment. (B) Cells were transfected with plasmid expressing the indicated codon-optimized Vpu (160ng). The next day cells were harvested and the cell lysates were analyzed by immunoblot for Vpu, BST-2, and β-actin.
Complementation of ΔVpu with Vpu-A18H resulted in a fractional p24 release of only 7.1%, a 1.7-fold enhancement relative to ΔVpu alone (Figure 2A). Protein expression was examined by immunoblot to ensure that each of the Vpu TMD mutants was expressed (Figure 2B). Notably, the extent to which each Vpu mutant facilitated down-regulation of surface BST-2 expression correlated with the extent to which it enhanced virion release. These data supported the hypothesis that the removal of BST-2 from the cell surface, the site of virion assembly and budding, is the mechanism by which Vpu enhances virion release. The data also indicated that the A18H substitution impairs the activity of Vpu as a BST-2 antagonist.
Vpu-A18H exhibits a cellular distribution consistent with residence in the endoplasmic reticulum and a reduced interaction with BST-2
To investigate the inability of the A18H mutant to down-regulate surface BST-2 expression and enhance virion release, we examined the subcellular distribution of the protein, as well as the other Vpu mutant-proteins characterized above, using immunofluorescence microscopy. Cells were transfected to express each of the mutants, and then 24 hours later fixed, permeabilized, and stained for intracellular Vpu and BST-2. As described previously (Neil, Zang, and Bieniasz, 2008; Van Damme et al., 2008), BST-2 was distributed throughout the cytoplasm in punctate structures characteristic of endosomes. In some cells the protein was relatively concentrated in a juxtanuclear location. Wild type Vpu was distributed similarly to BST-2, and the two proteins co-localized extensively in both juxtanuclear and peripheral endosomal compartments (Figure 3A).
Figure 3. Vpu-A18H exhibits a cellular distribution consistent with residence in the endoplasmic reticulum.

(A) Cells (HeLa) were transfected with plasmid expressing the indicated codon-optimized Vpu (160ng). The next day the cells were fixed, permeabilized, and co-stained for Vpu (green) and BST-2 (red). (B) Pearson correlation coefficients were determined for the co-localization of BST-2 and Vpu for each of the Vpu mutants tested. The bar graph represents at least 10 cells with at least 6 z-planes each and indicates the level of co-localization between BST-2 and each Vpu mutant with standard deviation. (C) Cells (HEK293T) were transfected with a 1:2 plasmid ratio of BST-2 to the indicated Vpu and BST-2 was immunoprecipitated. The immunoprecipitated proteins were analyzed via immunoblot. The upper panel shows the loading control β-actin and input (expression) controls for BST-2 and Vpu. The lower panel shows the immunoprecipitation of BST-2 and the co-immunoprecipitation of each Vpu mutant.
All of the Vpu TMD mutant-proteins exhibited similar cellular distributions and appeared to co-localize to a high degree with BST-2, with the exception of Vpu-A18H (Figure 3A). Vpu-A18H exhibited a ring-like perinuclear as well as a reticular cytoplasmic distribution, consistent with residence in the ER, including the nuclear envelope. Accordingly, Vpu-A18H co-localized with BST-2 to a lesser degree than did wild type Vpu or the other Vpu mutants (Figure 3B, which shows quantitative image analysis and comparison of the Pearson coefficient of correlation between the signals for Vpu and BST-2).
To further explore the nature of the impaired antagonism of BST-2 exhibited by Vpu-A18H, co-immunoprecipitation was performed to measure the degree of interaction between the Vpu proteins and BST-2. Cells were co-transfected with a plasmid expressing each of the codon-optimized Vpu TMD mutants together with a plasmid expressing BST-2. 24 hours post-transfection the cells were lysed; BST-2 was immunoprecipitated; and the captured proteins were analyzed by immunoblot. Vpu-I17A, -W22L, -S23L, and Vpu 2/6 co-immunoprecipitated with BST-2 to the same extent as wild type Vpu, while Vpu-A18H exhibited a reduced interaction (Figure 3C). This reduced interaction could be solely the consequence of the mislocalization of Vpu-A18H, or the A18H substitution might also directly impair a putative direct interaction with the BST-2 TMD. In support of the latter possibility, the Vpu-A18F protein also interacted somewhat less efficiently with BST-2 (Figure 3C), although its subcellular localization appeared normal (Figure 6A).
Figure 6. Vpu-KKDQ is impaired as a BST-2 antagonist while Vpu-A18F appears similar to wt Vpu.

(A) Cells (HeLa) were transfected with plasmid expressing the indicated codon-optimized Vpu (160ng). The next day the cells were fixed, permeabilized, and co-stained for Vpu (green) and BST-2 (red). (B) Cells (HeLa) were transfected with either an empty vector plasmid or a plasmid expressing codon-optimized Vpu (160ng), along with a plasmid expressing GFP (80ng) as a transfection marker. The next day, the cells were stained for surface BST-2 and analyzed by two-color flow cytometry. Histograms represent the relative cell number vs. BST-2 fluorescence intensity for the GFP-positive cells. The transfection efficiency, as indicated by the percentage of GFP-positive cells, was approximately 30% for each of the transfections. The black line represents the level of surface BST-2 down-regulation achieved when Vpu is present. The shaded grey peak represents the levels of surface BST-2 present when transfected with empty vector. (C) Cells (HeLa) were transfected as described in 6B. The next day, the cells were stained for surface CD4 and analyzed by two-color flow cytometry. Histograms represent the relative cell number vs. CD4 fluorescence intensity for the GFP-positive cells. The black line represents the level of surface CD4 down-regulation achieved when Vpu is present. The shaded grey peak represents the levels of surface CD4 present when transfected with empty vector. (D) The bar graph indicates the fractional BST-2 and CD4 down-regulation achieved by each Vpu tested. The values were determined as 1-[(Vpu-MFI - isotype MFI) / (empty vector-MFI – isotype-MFI)]; value of 1.0 indicates complete down-regulation of surface BST-2 or CD4, whereas a value of 0 indicates no down-regulation. (E) Cells (HeLa) were transfected with either a proviral plasmid expressing wild type HIV-1NL4-3 or a vpu-negative proviral plasmid vpuDEL-1 (ΔVpu) (1.6μg). Each of the Vpu mutant-proteins was expressed in trans (160ng) with ΔVpu to assess their activity in virion release. The next day, the fraction of the total p24 capsid antigen produced by the cells that was secreted into the media was measured by ELISA. The average values from two independent experiments are graphed; the error bars indicate the actual values obtained from each experiment. (F) Cells (HEK293T) were transfected as in Figure 3C. The immunoprecipitated proteins were analyzed via immunoblot. The upper panel shows β-actin as the loading control and input (expression) controls for BST-2 and Vpu. The lower panel shows the immunoprecipitation of BST-2 and the co-immunoprecipitation of each Vpu mutant.
To confirm that the distribution of Vpu-A18H was consistent with residence in the ER, we co-stained cells for Vpu and calnexin, a calcium-binding protein located primarily in the ER. Vpu-A18H and calnexin both exhibited a ring-like perinuclear and reticular cytoplasmic distribution, and the two proteins co-localized substantially (Figure 4A). In contrast, wild type Vpu and calnexin co-localized less extensively, and wild type Vpu exhibited, as noted above, a more endosomal distribution (Figure 4A).
Figure 4. Brefeldin A induces wild type Vpu to adopt a subcellular distribution similar to that of Vpu-A18H.

(A) Cells (HeLa) were transfected as in Figure 3. In the lower panel BFA (10μg/ml) was added into the cells immediately following the 4-hour transfection. The next day the cells were fixed, permeabilized, and co-stained for Vpu (green) and calnexin or BST-2 as indicated (red). (B) Cells (HeLa) were transfected with either an empty vector plasmid or a plasmid expressing codon-optimized Vpu or Vpu-A18H (160 ng of plasmid expressing codon-optimized Vpu), along with a plasmid expressing GFP (80ng) as a transfection marker. Immediately following transfection, BFA (10 μg/ml) was added to the cells (+BFA) or the cells were left untreated (−BFA). The next day, the cells were stained for surface BST-2 and analyzed by two-color flow cytometry. The horizontal black line represents the isotype control. (C) Cells were transfected with either a proviral plasmid expressing wild type HIV-1NL4-3 or a vpu-negative proviral plasmid vpuDEL-1 (Δvpu; 1.6μg). Vpu or Vpu-A18H was expressed in trans (160ng of plasmid expressing codon-optimized Vpu) along with Δvpu. BFA was added immediately post-transfection (+BFA) or the cells were left untreated. The next day, the fraction of the total p24 capsid antigen produced by the cells that was secreted into the media was measured by ELISA. The average values from two independent experiments are graphed; the error bars indicate the actual values obtained from each experiment.
To confine Vpu to the ER by non-mutational means, we utilized the ER transport inhibitor brefeldin A (BFA). BFA inhibits guanine nucleotide exchange factors for ARF-1, blocking the formation of vesicles that transport proteins out of the ER. BFA caused wild type Vpu to adopt a similar sub-cellular distribution to Vpu-A18H and to co-localize more closely with calnexin (Figure 4A). These microscopic data supported the conclusion that Vpu-A18H mislocalizes to the ER, as does wild type Vpu in BFA-treated cells. This mislocalization might explain Vpu-A18H’s impaired down-regulation of cell-surface BST-2, its inability to enhance virion release, and its reduced interaction with BST-2.
Vpu fails to down-regulate the expression of BST-2 at the cell surface and inefficiently enhances virion release in the presence of brefeldin A
The preceding data led to the hypothesis that the failure of Vpu to reach post-ER compartments is associated with a loss of BST-2 antagonism. To test this hypothesis, we assayed Vpu for the ability to down-regulate surface BST-2 expression and enhance fractional p24 release in the presence of BFA. In the absence of BFA, wild type Vpu effectively down-regulated the levels of surface BST-2 (compare surface BST-2 levels in empty vector control to wild type Vpu, in Figure 4B). In contrast, Vpu-A18H essentially failed to down-regulate surface BST-2 (Figure 4B). BFA alone caused a modest reduction in cell surface BST-2 expression levels (Figure 4B; compare empty vector control surface BST-2 levels in upper and lower panels), but this was not as great as the reduction caused by Vpu. Importantly, Vpu was unable to further reduce the levels of BST-2 in the presence of BFA (Figure 4B, lower panel). We attempted to evaluate the down-regulation of CD4 from the cell surface by Vpu in the presence of BFA; however, BFA treatment alone reduced surface CD4 to undetectable levels (data not shown). Notably, previously reported data suggests that Vpu-mediated degradation of CD4 is actually enhanced in the presence of BFA (Willey et al., 1992). Together, these data suggested that Vpu is ineffective at down-regulating BST-2 from the surface of cells treated with BFA, potentially because it is confined to the ER, even though this treatment enhances the degradation of CD4 by Vpu.
We next determined the influence of BFA on virion release. Consistent with its inhibitory effect on the down-regulation of BST-2 by Vpu, BFA also inhibited the enhancement of virion release by Vpu. In the absence of BFA, Vpu enhanced the fractional p24 release by 8-fold, consistent with previous data (Figure 4C). However, in the presence of BFA, Vpu enhanced fractional p24 release by only 1.5-fold (Figure 4C; compare “wt Vpu+ BFA” to “ΔVpu+BFA”). Interestingly, although BFA inhibited the release of virus when Vpu was expressed, it enhanced (by 2-fold) the fractional release of Δvpu virus (Figure 4C). This increase was consistent with the reduced levels of surface BST-2 induced by BFA, resulting in less effective restriction. Overall, these data on the effect of BFA on virion release in the presence and absence of Vpu are similar to those previous reported, which were also obtained using HeLa (BST-2 expressing) cells (Schubert and Strebel, 1994). Vpu-A18H enhanced fractional p24 release by 2.6-fold in the absence of BFA and had almost no activity in the presence of BFA (Figure 4C; compare “ΔVpu+BFA” to “A18H Vpu+BFA”). The decreased ability of wild type Vpu to enhance virion release in the presence of BFA, along with phenotype of the Vpu-A18H mutant, suggests that the confinement of Vpu to the ER inhibits its ability to antagonize BST-2.
Endogenously expressed BST-2 exhibits a relatively long steady-state half-life at the cell surface
The preceding data suggested that Vpu antagonizes BST-2-mediated restriction within a post-ER compartment. This hypothesis is in contradistinction to counteraction of restriction by means of ER-associated degradation of BST-2 (Mangeat et al., 2009). Implicit in the latter model is the notion that the net removal of BST-2 from the cell surface is a consequence of a blockade to the deposition of newly synthesized BST-2, together with the removal of surface BST-2 at an endogenous turnover rate. To test this, we compared the endogenous rate of turnover of BST-2 at the cell surface with the rate of removal by Vpu. To determine the endogenous half-life of surface BST-2, cells were incubated with BFA or cycloheximide (CHX) for various times, and then stained for surface BST-2 and analyzed by flow cytometry (Figure 5A). BFA was used to block the egress of BST-2 from the ER; this treatment decreased surface levels to 50% of their initial levels after 9 hours. CHX was used to block de novo synthesis of BST-2 and yielded similar results; the half-life of surface BST-2 was 8-9 hours (Figure 5A).
Figure 5. Vpu-mediated down-regulation occurs more rapidly than can be accounted for by the endogenous rate of turnover of BST-2 at the cell surface.
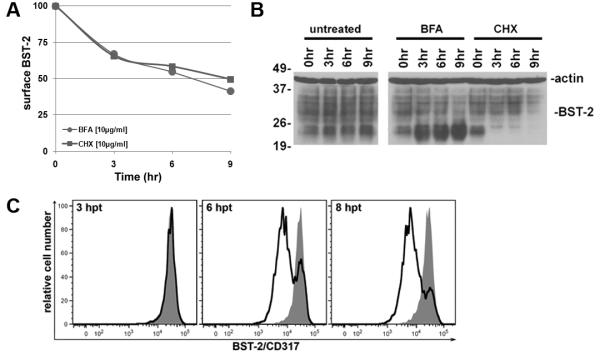
(A) Cells (HeLa) were exposed for increasing times to BFA (10 μg/ml) or CHX (10 μg/ml) to block deposition of newly synthesized BST-2 on the plasma membrane or de novo synthesis of BST-2. The cells were stained for BST-2 at the indicated time points and analyzed by flow cytometry. The data graphed are the mean fluorescence intensities of BFA- or CHX-treated cells normalized to cells not treated with BFA or CHX harvested at the same time. (B) The total cellular levels of BST-2 were examined by immunoblot at the indicated time points post treatment with the indicated drug (BFA of CHX). BST-2 levels are shown along with β-actin loading controls. (C) The down-regulation of surface BST-2 mediated by Vpu was measured as previously described via flow cytometry but at early time points. The two-color plots from cells harvested 3, 6, 8 hours after the initiation of transfection are shown; GFP positive events were 0.4, 1.8, and 3.4% at each respective time point.
To examine further the steady-state half-life of endogenously expressed BST-2, we measured the total cellular levels by immunoblot at various times in the presence of BFA or CHX. In untreated cells, BST-2 levels were constant throughout the duration of the experiment, and the ratio of fully glycosylated BST-2 (26-37 kDa) to the unglycosylated and/or high mannose forms (19-26 kDa) was stable (Figure 5B). In the presence of BFA, unglycosylated and/or high mannose forms of BST-2 accumulated as early as 3 hour after BFA treatment (Figure 5B), suggested that BFA effectively blocks egress from the ER. Furthermore, fully glycosylated BST-2 levels decreased only after 9 hours of BFA-treatment, suggesting that endogenous, fully glycosylated BST-2 is relatively stable (Figure 5B). Treatment with CHX resulted in the disappearance of the unglycosylated and/or high mannose forms of BST-2 after 3 hours, indicating an effective block to de novo synthesis (Figure 5B). However, the levels of mature fully glycosylated BST-2 did not diminish until 9 hours of CHX-treatment, suggesting again that endogenous mature BST-2 is relatively long-lived (Figure 5B).
We next examined the kinetics of Vpu-mediated removal of BST-2 from the cell surface. Cells were co-transfected with a plasmid expressing codon-optimized Vpu along with a plasmid expressing GFP as a marker of transfection efficiency, as described above. Three hours after the initiation of the transfection, GFP expression was minimal and no appreciable down-regulation of BST-2 was observed (Figure 5C). However, by 6 hours after initiation of the transfection, we observed a 5-fold reduction in the levels of BST-2 at the cell surface (Figure 5C). These data indicate that the rate and extent of Vpu-mediated down-regulation is much faster than can be accounted for by the natural half-life of BST-2 on the cell surface. The data suggest that Vpu acts to directly remove BST-2 from the cell surface.
ER confinement of Vpu results in impaired antagonism of BST2
The previous experiments suggested that confinement of Vpu to the ER, either through chemical means (BFA) or mutagenesis (A18H), results in impaired antagonism of BST-2. However, the phenotype of Vpu-A18H could be due not only to mislocalization but also to a reduced ability to interact with BST-2. To resolve this, we tested two more Vpu mutants, Vpu-KKDQ and Vpu-A18F. Vpu-KKDQ is identical to wild type Vpu except that it has a transmembrane protein specific ER-retention signal (KKXX) added at the C-terminus (Shikano and Li, 2003). Vpu-A18F is a single residue mutant that we thought might localize normally but interact poorly with BST-2. Immunofluorescence microscopy confirmed that Vpu-KKDQ exhibited an ER-like distribution and co-localized to an extent with calnexin; this distribution was similar to Vpu-A18H (Figure 6A). This suggested that the C-terminal ER-retention signal KKDQ effectively confines Vpu-KKDQ to the ER. In contrast, Vpu-A18F exhibited a sub-cellular distribution more typical of the endosomal system and was similar to wild type Vpu (Figure 6A).
We evaluated the capacity of Vpu-KKDQ and Vpu-A18F to down-regulate BST-2 from the cell surface. Vpu-KKDQ was defective for the down-regulation of BST-2, appearing even more impaired than Vpu-A18H (Figure 6B and D). In contrast, Vpu-A18F down-regulated BST-2 with an efficiency similar to the wild type (Figure 6B and D). We also evaluated the capacity of Vpu-KKDQ and Vpu-A18F to down-regulate CD4 from the cell surface. Vpu-KKDQ was minimally impaired for the down-regulation of CD4, appearing less impaired than Vpu-A18H (Figure 6C and D). In contrast, Vpu-A18F down-regulated CD4 with efficiency similar to wild type Vpu (Figure 6C and D).
We next examined each Vpu’s ability to enhance virion release (Figure 6D). Vpu-KKDQ was virtually inactive for enhancement of virion release, while VpuA18H enhanced virion release less than 2-fold (Figure 6E). In contrast, Vpu-A18F enhanced virion release by 6-fold, an activity almost equal to that of wild type Vpu (Figure 6E). Western blot confirmed that each of these Vpu mutants was expressed, although Vpu-KKDQ was expressed at levels slightly lower than that of the wild type (Figure 6F and Figure 3C regarding Vpu-A18F). Immunoprecipitation data indicated that the interaction between BST-2 and Vpu-KKDQ or Vpu-A18H was reduced relative to wild type Vpu (Figure 6F). These data suggest that Vpu cannot effectively antagonize BST-2 when confined to the ER, even though it can still down-regulate CD4. The data also suggest that even when the Vpu TMD sequence is intact, confinement to the ER reduces the potential for Vpu to bind to BST-2.
To assess whether the Vpu-A18H TMD was intrinsically impaired in its potential to interact with the TMD of BST-2, we performed molecular dynamic (MD) simulations on the individual TMDs within hydrated lipid bilayers followed by a packing protocol to generate dimeric assemblies (Figure 7) (Kruger and Fischer, 2008; Kruger and Fischer, 2009). The TMDs maintained a stable helical conformation throughout a short 10 ns MD simulation after generating the ideal helices; the RMSD values leveled off after an initial rise within the first 1000 ps and remained in a range of 0.1 to 0.2 nm for BST-2 and 0.15 and 3.0 nm for the Vpu and Vpu-A18H helices (Supplemental Figure 1).
Figure 7. Interaction potential of BST-2 with Vpu and Vpu-A18H.
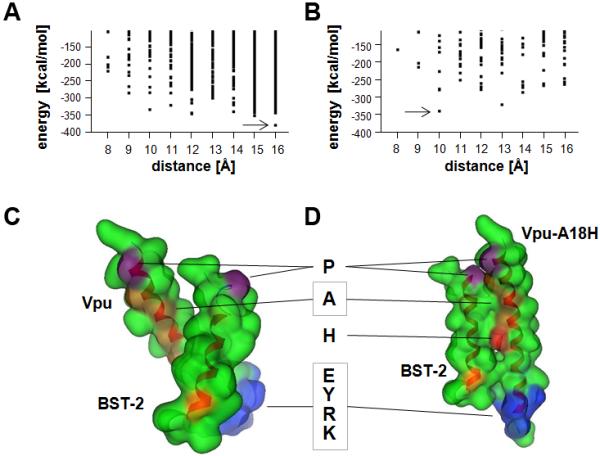
Interaction energies of the BST-2/Vpu (A) and BST-2/Vpu-A18H (B) TMD assemblies in relation to their interhelical distance as determined by a packing protocol using MOE script language. Dots represent individual structures. Arrows indicate the lowest energy structures. Representation of the lowest energy structures of BST-2/Vpu (C) and BST-2/Vpu-A18H (D) in the ‘Gaussian contact’ mode of the MOE suite. The backbone is indicated in a ribbon representation; hydrophobic residues are shown in green, prolines in violet, alanines in orange, BST-2-G25 in yellow-orange, His-18 in red, and a hydrophilic motif of Vpu, EYRK, in blue. The boxed “A” indicates four alanines in the Vpu (A7, A10, A14, and A18) that are on one face of the TMD helix.
Packing of BST-2 with wild type Vpu led to a lowest energy structure with an interhelical distance of about 16 Å, rotational angle of 90 °, a positive tilt of 60 °, and a potential energy of −381.8 kcal/mol (Figure 7A, Suppl. 1C). The five most stable BST-2/wild type Vpu structures had potential energies of less than or equal to −347.1 kcal/mol. Replacing alanine-18 with an uncharged histidine yielded a lowest energy structure of −341.4 cal/mol with a minimum distance of 10 Å, rotational angle of 90 ° and a tilt of 18 ° (Figure 7B, Suppl. 1D). The next most stable BST-2/Vpu-A18H assembly had a minimum distance of 13 Å and a potential energy of −323.1 kcal/mol. All other structures of BST-2/Vpu-A18H were above −300 kcal/mol. The packing of BST-2 with wild type Vpu in these simulations was driven by contact of hydrophobic residues (Figure 7C), whereas in the lowest energy structure of BST-2/Vpu-A18H the histidine faces the backbone of BST-2 (Figure 7D). Overall, these simulations indicated that Vpu-A18H has the potential to interact with BST-2 via its TMD, although the interaction is likely to be somewhat less stable than in the case of wild type Vpu.
Discussion
We show here that a single amino acid substitution in the transmembrane domain of Vpu (A18H) is sufficient to cause abnormal retention of the protein in the ER. This mislocalization is associated with defects in the ability of Vpu to remove the interferon-induced tethering factor BST-2 from the cell surface and in the ability of Vpu to enhance the release of virions. Mimicry of these phenomena by treatment of cells with the fungal metabolite BFA, which blocks the egress of proteins from the ER, or by the addition of an ER retention signal to the C-terminus of Vpu, supports the notion that Vpu counteracts BST-2 from within post-ER membrane systems. In support of a model in which Vpu directly removes BST-2 from the plasma membrane, we observed that the half-life of BST-2 on the cell surface is too long to allow ER-degradation or sequestration along the biosynthetic/exocytic pathway to fully account for the speed and extent of Vpu-mediated down-regulation. The conservation of residue A18 among group M HIV-1 Vpu’s, and its frequent absence from group O and SIV Vpu’s that are unable to counteract BST-2, supports a general importance of this TMD residue for Vpu function [Figure 8 and (Sauter et al., 2009)].
Figure 8. Amino acid frequencies at p18 in HIV-1 and SIVcpz Vpu proteins.
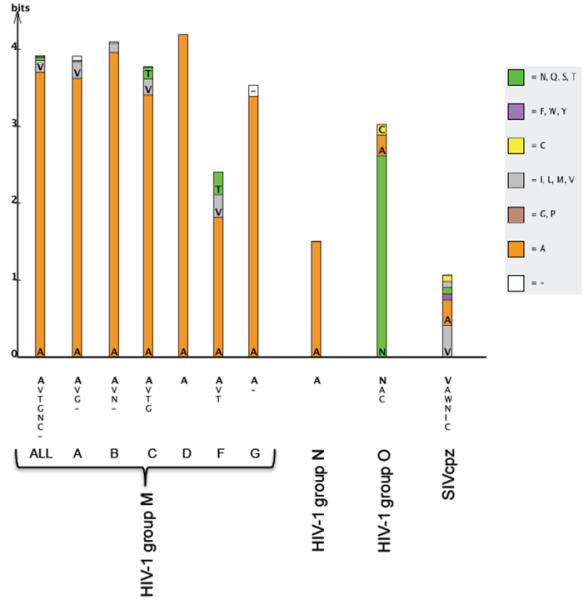
The overall height of each bar indicates level of sequence conservation (difference between maximum possible entropy and observed entropy) at Vpu position 18 within each lentiviral lineage (correcting for small sample size). The relative heights within the stack indicate the relative frequency of each amino acid, based on all available sequences in the Los Alamos HIV Sequence Database (only a single sequence per host individual was included in the analysis). Alanine is represented by the color orange, and the remaining residues are colored according to the “Cinema” amino acid labeling scheme as shown. A complete list of residues appearing in each lineage is included under each stack, ordered by frequency (hyphens represent deletions). Frequencies within the entire HIV-1 M-group including recombinants are represented in the first column (“ALL”), and subsequent columns represent group M subtypes A-G, group N, group O, and SIVcpz. Group O HIV-1 and SIVcpz Vpu proteins infrequently encode A18 are generally ineffective antagonists of BST-2 (Sauter et al., 2009).
Prior to the identification of BST-2 as the restriction factor counteracted by Vpu, the cation selective ion-channel activity of Vpu homo-oligomers was presumed to be the determinant of enhanced virion release (Lopez et al., 2002; Schubert et al., 1996a; Schubert et al., 1996b). Even after the role of the Vpu TMD in the down-regulation and antagonism of BST-2 was reported (Van Damme et al., 2008), the notion that ion channel activity could underlie an effect of Vpu on the endosomal trafficking of BST-2 remained plausible.
However, the data presented here suggests that the enhanced virion release mediated by Vpu is likely independent of ion-channel activity. The I17A substitution studied here was intended to affect a hinge region or kink in the Vpu TMD α-helix (Park et al., 2003). Although the ion-channel activity of the Vpu-I17A has not been determined, Vpu-I17A effectively enhanced virion release and down-regulated BST-2 from the cell surface. Furthermore, Vpu-I17A exhibited a similar subcellular distribution to wild type Vpu and co-localized with BST-2 within endosomes. The frequency of the polymorphism I17A was the highest of the four mutants tested (8%), supporting our observations that the I17A substitution does not adversely affect Vpu function. Vpu-W22L was previously characterized as a functional ion-channel, although rather than oscillating rapidly between open and closed conformations, the transitions between the two conformations are prolonged (Mehnert et al., 2008). Nevertheless, Vpu-W22L efficiently down-regulated BST-2 from the cell surface; it efficiently enhanced virion release; and it co-localized extensively with BST-2. We observed that W22 was very highly conserved among group M Vpu’s (frequency greater than 99%). This conservation suggests that the very subtle defects of Vpu-W22L in the down-regulation of BST-2 from the cell surface and in the enhancement of virion release may be important in vivo.
Strikingly, Vpu-S23L was previously characterized as defective for ion-channel activity (Mehnert et al., 2008), yet it robustly down-regulated BST-2 from the cell surface; it enhanced virion release to the same extent as wild type Vpu; and it co-localized and interacted with BST-2. These results suggest that Vpu’s ion channel activity is not necessary for the down-regulation of BST-2 and the enhancement of virion release. The existence of naturally occurring polymorphisms at this position (in particular isoleucine in 11% of group M Vpu’s) further supports the notion that Vpu’s ion-channel activity may not be necessary for optimal virion release.
Vpu-A18H was first conceived to mimic the Influenza virus M2 protein ion-channel and allow Vpu’s channel activity to be modulated by adamantane drugs (Hout et al., 2006a; Hout et al., 2006b). The M2 protein forms a proton channel, and it facilitates an essential step of the Influenza virus life-cycle by promoting acidification and uncoating of the virion (Henkel et al., 1999; Henkel and Weisz, 1998; Sakaguchi, Leser, and Lamb, 1996). Although the ion-channel activity of Vpu-A18H has not been determined, its presence in a SHIV background rendered the virus susceptible to rimantadine when used at high concentrations during spreading infections (Hout et al., 2006a). This result seems at some odds with the data herein, in which the A18H substitution itself impaired Vpu’s ability to enhance virion release, even in the absence of an adamantane. To reconcile these findings, we note that Vpu-A18H retained a modest ability to down-regulate BST-2 from the cell surface at the very highest levels of transfection (as measured by GFP intensity). Furthermore, Vpu-A18H was not completely devoid of virologic activity; rather it enhanced virion release by only 2-fold compared to the 7- to 8-fold increase observed for the wild type protein.
Unexpectedly, microscopic data indicated that Vpu-A18H remains largely trapped in the ER and fails to efficiently access the trans-Golgi network and the endosomal system. How does a single amino acid substitution in the TMD affect the steady-state subcellular localization of Vpu? Structural analysis of the Vpu TMD in artificial lipid bilayers suggests that the A18H substitution increases the length of the α-helical TMD (Park and Opella, 2007). This in turn increases the tilt angle of the helix relative to the lipid bilayer, at least in artificial bilayers of fixed composition (Park and Opella, 2007). Since the ER, Golgi, and plasma membranes are of unequal thickness due to different contents of cholesterol, a change in TMD length could conceivably alter the accessibility of Vpu-A18H to different membrane compartments, but shorter rather than longer TMD lengths are associated with retention in the ER (Bretscher and Munro, 1993; Pelham and Munro, 1993; Yang et al., 1997).
Remarkably, a precedent exists for the ER-retention induced by the A18H substitution: the introduction of single charged residues or histidines within the TMD of the IL-2 receptor α chain (Tac antigen), a protein normally resident on the plasma membrane, induces ER-retention, although the mechanism of this effect is obscure (Bonifacino et al., 1991). Here, this single substitution in the Vpu TMD was similarly sufficient to induce ER-retention, and to adversely affect BST-2 antagonism.
Apparently, Vpu is rendered largely inactive for down-regulating BST-2 from the cell surface and enhancing virion release when it is trapped in the ER. However, since Vpu-A18H co-localized poorly with BST-2, an alternative explanation is that the A18H substitution directly disrupts the reported interaction between the two proteins (Douglas et al., 2009; Mangeat et al., 2009; Rong et al., 2009). Residue A18 is part of an AxxxAxxxA sequence that is relatively well conserved among group M and N Vpu’s (Figure 8), proteins that are generally active as BST-2 antagonists (Sauter et al., 2009). GxxxG and AxxxA motifs are thought to facilitate backbone interactions between α-helixes within lipid bilayers (Senes, Engel, and DeGrado, 2004), and so the A18H substitution could potentially affect directly the interaction with BST-2. Similarly, the increased tilt angle of the Vpu-A18H TMD could potentially disrupt the interaction with BST-2 (Park and Opella, 2007). In this regard, computational modeling indicated that Vpu-A18H has the potential to interact with BST-2 via its TMD, although the interaction is predicted to be somewhat less stable than wild type Vpu. In contrast to histidine, the substitution of A18 with phenylalanine (Vpu-A18F) minimally disrupted Vpu activity. This mutant localized normally, although its ability to interact with BST-2 was modestly impaired. Altogether, these data suggest that the primary defect in Vpu-A18H is subcellular mislocalization. The data also suggest that an alanine at position 18 is not required for function, although a histidine is clearly non-permissive. Recent data suggests that transmembrane domain residues A14 and W22 influence BST-2 antagonism more significantly than residue A18 (Vigan and Neil, 2010).
Blocking the egress of proteins from the ER by treating cells with BFA largely mimicked the effect of the A18H substitution. Morphologic and biochemical evidence suggested that BFA blocked the egress of both Vpu and BST-2 from the ER, and this rendered Vpu unable to down-regulate BST-2 from the cell surface or to enhance virion release. Interestingly, BFA itself modestly mimicked the effect of Vpu; such mimicry was expected since the block to egress from the ER modestly decreased the levels of BST-2 at the cell surface, though this effect was less than the effect of Vpu.
Consistent with the notion that Vpu antagonizes BST-2 from a post-ER compartment, we observed that the half-life of cell surface BST-2 is too long to enable degradation or trapping along the biosynthetic pathway to account for Vpu activity. The half-life of endogenously-expressed surface BST-2 in BFA- and CHX-treated cells was between 8 and 9 hours. Given the rapid rate of endocytosis of BST-2, in which 50% of the protein is internalized from the cell surface within 20-40 minutes (Mitchell et al., 2009), the long surface half life measured here suggests that internalized BST-2 is efficiently recycled to the plasma membrane, with only a fraction entering an endosomal degradative pathway. Put another way, 8-9 hours is the half-life of BST-2 on the cell surface when the transport of newly synthesized BST-2 to the plasma membrane is blocked or de novo synthesis is inhibited. Despite this, Vpu mediated a 5-fold decrease in the levels of BST-2 within as little as 6 hours. Given the 8-9 hour half-life of surface BST-2, Vpu cannot mediate such a rapid and extensive effect solely by degrading newly synthesized BST-2 via the ERAD pathway or by trapping it in the TGN or other exocytic membranes (Dube et al., 2010; Mangeat et al., 2009).
Instead, our data suggest that Vpu removes BST-2 directly from the plasma membrane to enhance virion release. This conclusion is consistent with the morphologic data indicating that Vpu functions from within post-ER, endosomal membranes. It is also consistent with the previous observation of a role for the endocytic clathrin adaptor AP-2 in the Vpu-mediated down-regulation of BST-2 and the hypothesis that Vpu inhibits the recycling of BST-2 (Mitchell et al., 2009). The data herein indicate that the down-regulation of BST-2 occurs very rapidly, a condition likely necessary so that Vpu, which is expressed relatively late as a Rev-dependent protein, can effectively remove BST-2 before the onset of viral budding.
Taken together, the data presented here show that Vpu-mediated down-regulation of surface BST-2 correlates with enhanced virion release and likely occurs independently of Vpu ion-channel activity. The Vpu-mediated antagonism of BST-2 likely occurs in post-ER endosomal compartments and at a rate consistent with the direct removal of BST-2 from the cell surface. This removal is extremely rapid, consistent with the hypothesis that it is the primary mechanism of the counteraction of restriction.
Materials and Methods
Plasmids, antibodies, and reagents
pcDNA3.1 (Invitrogen, Carlsbad, CA) was used as an empty vector control. Proviral plasmid pNL4-3 was obtained from the National Institute of Health (NIH) AIDS Research & Reference Reagent Program. The ΔVpu mutant of pNL4-3 (vpuDEL-1) and the pcDNA3.1-based plasmid expressing codon optimized Vpu (pVphu) were provided by Dr. Klaus Strebel. Mutations in pVphu were introduced using the Stratagene (La Jolla, CA) QuikChange kit and confirmed by nucleotide sequencing. The plasmid expressing GFP (pCG-GFP) was provided by Dr. Jacek Skowronski. The murine monoclonal antibody to BST-2/HM1.24/CD317 was a gift from Chugai Pharmaceutical Co., Kanagawa, Japan. An isotype control antibody, IgG2a, a secondary goat anti-mouse IgG antibody conjugated to allophycocyanin (APC), murine monoclonal –antibody to BST-2 (hCD317 RS38E) directly conjugated to Alexa 647, and an isotype control Alexa 647-direct conjugate were obtained from BioLegend (San Diego, CA). The murine monoclonal antibody to human CD4 directly conjugated to PE and a PE-conjugated isotype control were obtained from BD Biosciences (San Jose, CA). Rabbit antiserum specific to HIV-1 Vpu and human BST-2 was obtained from the NIH AIDS Research & Reference Reagent Program and contributed by Dr. Klaus Strebel. The murine monoclonal antibody to Calnexin was obtained from BD Biosciences (San Jose, CA). The secondary antibodies used for immunofluorescence were obtained from Jackson ImmunoResearch (West Grove, PA). Brefeldin A was obtained from Sigma-Aldrich (St. Louis, MO).
Cells and transfections
The HeLa cells used in most of this study, clone P4.R5, express both CD4 and CCR5 were obtained from Dr. Ned Landau; these cells are a derivative of clone P4 and were maintained in DMEM supplemented with 10% FBS, penicillin/streptomycin, and puromycin. The HeLa cells used to evaluate the effectiveness of BFA in blocking newly synthesized CD4 from reaching the cell surface (clone Z24) do not express CD4; were provided by Dr. Chris Aiken; and were maintained in DMEM supplemented with 10% FBS, and penicillin/streptomycin. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the supplied directions. For the experiments evaluating BST-2 surface down-regulation and protein expression, 160ng of pVphu or pcDNA3.1 along with 40ng of pCG-GFP were used to transfect 1.2 million HeLa P4.R5 cells in wells of a 6-well plate. For experiments evaluating fractional virion release, 1.6μg of proviral plasmid pNL4-3 or vpuDEL-1 along with 160ng of the indicated pVphu plasmid were used to transfect 1.2 million HeLa P4.R5 cells in wells of a 6-well plate. For experiments simultaneously evaluating the surface down-regulation of BST-2 and CD4, 200ng of pVphu along with 100ng of pCG-GFP were used to transfect 0.5 million HeLa P4.R5 cells in wells of a 12-well plate.
Flow cytometry
For analysis of surface levels of BST-2, cells were stained before fixation in phosphate buffered saline (PBS) including sodium azide, and 2% FBS at 4C using an indirect method to detect BST-2. The primary murine HM1.24 monoclonal antibody (0.1μg/ml) was followed by a secondary goat anti-mouse IgG conjugated to APC. All samples were analyzed using two-color flow cytometry. The BST-2 gate was set using an isotype (IgG2a) control as the primary antibody, and the gate for GFP was set using non-GFP expressing cells. For simultaneous analysis of surface levels of BST-2 and CD4, cells were stained for 1hr at 4C before fixation in phosphate buffered saline (PBS) including sodium azide and 2% FBS using a a murine hCD317 monoclonal antibody directly conjugated to Alexa 647- (0.1μg/ml) and a murine hCD4 monoclonal antibody directly conjugated to PE (0.1μg/ml). All samples were analyzed using three-color flow cytometry. The BST-2 gate was set using an isotype Alexa 647 direct conjugate control;the CD4 gate was set using the isotype PE direct conjugate control; and the gate for GFP was set using non-GFP expressing cells. Data profiles and mean fluorescence intensity (MFI) values were generated using FlowJo software (Tree Star, Inc, Ashland, OR).
Virion-release assays
A p24 antigen capture ELISA (Perkin-Elmer) was used to determine the concentration of viral capsid protein in culture supernatants that were first clarified by centrifugation at 400 X g as well as the concentration of capsid protein in detergent lysates (0.5% Triton-X-100 in PBS) of the adherent cells. The fractional release of p24 capsid was determined as the concentration of p24 antigen in the supernatants divided by the total concentration of p24 antigen in both the supernatants and cell lysates.
Immunofluorescence microscopy
Cells were stained for BST-2, Calnexin, and HIV-1 Vpu using the antibodies above after fixation in 3% formaldehyde and permeabilization using 0.1% NP-40, both in PBS, as previously described. Images were obtained using a fluorescence microscope with the 100x objective (Olympus, Melville, NY). For each field, a Z-series of images was collected, and the data were processed using a nearest neighbors deconvolution algorithm (Slidebook software, Intelligent Imaging Innovations, Inc). Composite multi-color images of single optical sections were assembled using Adobe Photoshop software.
Western blot and immunoprecipitations
Cells were suspended in Laemmli buffer and boiled for 10 minutes. The samples were then resolved on 12% SDS-denaturing polyacylamide gels (Bio-Rad, Hercules, CA) and subsequently transferred to polyvinylidene difluoride membranes. Immunoblotting was performed with the antibodies to BST-2 and Vpu described above or with antibody to β-actin (Sigma). Detection was performed using a goat anti-mouse antibody conjugated to horseradish peroxidase (Bio-Rad) or a donkey anti-rabbit antibody conjugated to horseradish peroxidase (Pierce Protein Research Products, Rockford, IL), followed by development with enhanced chemiluminescence reagent (GE Healthcare, Piscataway, NJ).
For IP, 90-95% confluent 293T cells were transfected with 7.5 μg BST-2 and 15 μg of Vpu-expressing plasmids, in 10 cm2 dishes. 24 hours later, the cells were collected and lysed in buffer (50 mM TrisHCl, pH7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) for 30 minutes on ice and 30 minutes at room temperature (RT). Cell lysates were cleared by centrifugation at 16,000g at 4°C for 10 minutes, then incubated with anti-BST-2 antibody pre-bound to anti-mouse IgG coated magnetic Dynabeads (Invitrogen Dynal, Oslo, Norway), for 2 hours at 4°C. The ratio of cells/anti-BST-2 antibody/magnetic beads was 1×10 cm2 dish/2 μg anti-BST-2/200×105 beads. Beads were washed three times with buffer (lysis buffer except with 250 mM NaCl), bound proteins were eluted with 30 μl Laemmli buffer, and subjected to Western blot analysis.
Modeling interaction potential of BST-2 and Vpu-A18H
Ideal helices of BST-2 (LLGIGILVLLIIVILGVPLIIF) and Vpu-WT (MVPIIVAIVA10 LVVAIIIAIV20 VWSIVIIEYR30 KI, GenBank: AAB59750.1, www.ncbi.nlm.nih.gov) and the mutant Vpu-A18H containing the TMDs of the respective proteins were created using integrated protein builder of MOE (www.chemcomp.com) (Kruger and Fischer, 2009). After a short equilibration in a fully hydrated lipid bilayer using molecular dynamics (MD) simulations (GROMACS-3.3), an averaged structure was used to generate the dimers (see supplemental materials for details).
Determination of HIV-1 and SIV Vpu polymorphism frequencies
All available Vpu sequences were downloaded from the Los Alamos National Laboratory HIV Sequence Database (http://www.hiv.lanl.gov/). Single representative sequences for each host individual were chosen arbitrarily for analysis when multiple sequences were available. Multalin (Corpet, 1988) was used to generate all multiple sequence alignments. LogoBar v1.0 (Perez-Bercoff, Koch, and Burglin, 2006) was implemented to examine site-specific Shannon entropy and relative amino acid frequencies at Vpu position 18 in HIV-1 and SIV taxa. The height of each individual bar represents the difference between the maximum possible entropy and the entropy of the observed amino acid distribution (correcting for small sample size).
Supplementary Material
Acknowledgments
We thank Jason Munguia and Marissa Suarez for technical assistance; Klaus Strebel for providing the proviral plasmid mutant of HIV-1NL4-3 vpuDEL-1 and the pcDNA3.1-based plasmid expressing codon optimized Vpu (pVphu); Jacek Skowronski for pCG-GFP; and Chugai Pharmaceuticals for the antibody to HM1.24/BST-2. Antibody to Vpu was originated by Klaus Strebel was obtained from the National Institutes of Health AIDS Research & Reference Reagent Program. We thank Sherri Rostami, Deya Collier, and Nancy Keating for the p24 ELISAs; Neal Sekiya, Peggy O’Keefe, and Judy Nordberg for the flow cytometry; and Carol Ignacio and Parris Jordan for nucleotide sequencing. This work was supported by NIH Grant AI081668 to John Guatelli. Mark Skasko was supported by AIDS Training Grant T32AI007384 and an ARRA Supplement to NIH Grant AI081668. WBF and CCC acknowledge National Yang-Ming University, the government of Taiwan and the National Science Council of Taiwan (NSC) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartee E, McCormack A, Fruh K. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006;2(10):e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binette J, Dube M, Mercier J, Halawani D, Latterich M, Cohen EA. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology. 2007;4:75. doi: 10.1186/1742-4690-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Shah N, Klausner RD. Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J. 1991;10(10):2783–93. doi: 10.1002/j.1460-2075.1991.tb07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261(5126):1280–1. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- Cohen EA, Terwilliger EF, Sodroski JG, Haseltine WA. Identification of a protein encoded by the vpu gene of HIV-1. Nature. 1988;334(6182):532–4. doi: 10.1038/334532a0. [DOI] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16(22):10881–90. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J Virol. 2009;83(16):7931–47. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010;6(4):e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Hue S, Schaller T, Verschoor E, Pillay D, Towers GJ. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009a;5(5):e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Mlcochova P, Pelchen-Matthews A, Petit SJ, Mattiuzzo G, Pillay D, Takeuchi Y, Marsh M, Towers GJ. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc Natl Acad Sci U S A. 2009b doi: 10.1073/pnas.0907075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology. 2010;7:51. doi: 10.1186/1742-4690-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JR, Popovich JL, Gibson GA, Watkins SC, Weisz OA. Selective perturbation of early endosome and/or trans-Golgi network pH but not lysosome pH by dose-dependent expression of influenza M2 protein. J Biol Chem. 1999;274(14):9854–60. doi: 10.1074/jbc.274.14.9854. [DOI] [PubMed] [Google Scholar]
- Henkel JR, Weisz OA. Influenza virus M2 protein slows traffic along the secretory pathway. pH perturbation of acidified compartments affects early Golgi transport steps. J Biol Chem. 1998;273(11):6518–24. doi: 10.1074/jbc.273.11.6518. [DOI] [PubMed] [Google Scholar]
- Hinz A, Miguet N, Natrajan G, Usami Y, Yamanaka H, Renesto P, Hartlieb B, McCarthy AA, Simorre JP, Gottlinger H, Weissenhorn W. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe. 2010;7(4):314–23. doi: 10.1016/j.chom.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hout DR, Gomez LM, Pacyniak E, Miller JM, Hill MS, Stephens EB. A single amino acid substitution within the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein renders simian-human immunodeficiency virus (SHIV(KU-1bMC33)) susceptible to rimantadine. Virology. 2006a;348(2):449–61. doi: 10.1016/j.virol.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Hout DR, Gomez ML, Pacyniak E, Gomez LM, Fegley B, Mulcahy ER, Hill MS, Culley N, Pinson DM, Nothnick W, Powers MF, Wong SW, Stephens EB. Substitution of the transmembrane domain of Vpu in simian-human immunodeficiency virus (SHIVKU1bMC33) with that of M2 of influenza A results in a virus that is sensitive to inhibitors of the M2 ion channel and is pathogenic for pig-tailed macaques. Virology. 2006b;344(2):541–59. doi: 10.1016/j.virol.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Huthoff H, Towers GJ. Restriction of retroviral replication by APOBEC3G/F and TRIM5alpha. Trends Microbiol. 2008;16(12):612–9. doi: 10.1016/j.tim.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5(5):e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83(4):1837–44. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106(8):2886–91. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger J, Fischer WB. Exploring the conformational space of Vpu from HIV-1: a versatile adaptable protein. J Comput Chem. 2008;29(14):2416–24. doi: 10.1002/jcc.20986. [DOI] [PubMed] [Google Scholar]
- Kruger J, Fischer WB. Assembly of Viral Membrane Proteins. J. Chem. Theory Comput. 2009;5(9):2503–2513. doi: 10.1021/ct900185n. [DOI] [PubMed] [Google Scholar]
- Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4(10):694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. 2009;83(22):11966–78. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CF, Montal M, Blasie JK, Klein ML, Moore PB. Molecular dynamics investigation of membrane-bound bundles of the channel-forming transmembrane domain of viral protein U from the human immunodeficiency virus HIV-1. Biophys J. 2002;83(3):1259–67. doi: 10.1016/S0006-3495(02)73898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magadan JG, Perez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010;6(4):e1000869. doi: 10.1371/journal.ppat.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009;5(9):e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2009;83(19):9672–81. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1(4):565–74. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- Masuyama N, Kuronita T, Tanaka R, Muto T, Hirota Y, Takigawa A, Fujita H, Aso Y, Amano J, Tanaka Y. HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with alpha-adaptin. J Biol Chem. 2009;284(23):15927–41. doi: 10.1074/jbc.M109.005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5(2):e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert T, Routh A, Judge PJ, Lam YH, Fischer D, Watts A, Fischer WB. Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins. 2008;70(4):1488–97. doi: 10.1002/prot.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009;5(5):e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–30. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Hirano T, Tsuchiya M. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem Biophys Res Commun. 1999;258(3):583–91. doi: 10.1006/bbrc.1999.0683. [DOI] [PubMed] [Google Scholar]
- Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333(2):409–24. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Park SH, Opella SJ. Conformational changes induced by a single amino acid substitution in the trans-membrane domain of Vpu: implications for HIV-1 susceptibility to channel blocking drugs. Protein Sci. 2007;16(10):2205–15. doi: 10.1110/ps.073041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HR, Munro S. Sorting of membrane proteins in the secretory pathway. Cell. 1993;75(4):603–5. doi: 10.1016/0092-8674(93)90479-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Bercoff A, Koch J, Burglin TR. LogoBar: bar graph visualization of protein logos with gaps. Bioinformatics. 2006;22(1):112–4. doi: 10.1093/bioinformatics/bti761. [DOI] [PubMed] [Google Scholar]
- Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139(3):499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollason R, Korolchuk V, Hamilton C, Schu P, Banting G. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J Cell Sci. 2007;120(Pt 21):3850–8. doi: 10.1242/jcs.003343. [DOI] [PubMed] [Google Scholar]
- Rong L, Zhang J, Lu J, Pan Q, Lorgeoux RP, Aloysius C, Guo F, Liu SL, Wainberg MA, Liang C. The transmembrane domain of BST-2 determines its sensitivity to down-modulation by human immunodeficiency virus type 1 Vpu. J Virol. 2009;83(15):7536–46. doi: 10.1128/JVI.00620-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Leser GP, Lamb RA. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J Cell Biol. 1996;133(4):733–47. doi: 10.1083/jcb.133.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. Inhibition of Lassa and Marburg virus production by tetherin. J Virol. 2009;83(5):2382–5. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 2009;6(5):409–21. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert HL, Zhai Q, Sandrin V, Eckert DM, Garcia-Maya M, Saul L, Sundquist WI, Steiner RA, Hill CP. Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc Natl Acad Sci U S A. 2010;107(42):17951–6. doi: 10.1073/pnas.1008206107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Bour S, Ferrer-Montiel AV, Montal M, Maldarell F, Strebel K. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J Virol. 1996a;70(2):809–19. doi: 10.1128/jvi.70.2.809-819.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996b;398(1):12–8. doi: 10.1016/s0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- Schubert U, Strebel K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J Virol. 1994;68(4):2260–71. doi: 10.1128/jvi.68.4.2260-2271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14(4):465–79. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Shikano S, Li M. Membrane receptor trafficking: evidence of proximal and distal zones conferred by two independent endoplasmic reticulum localization signals. Proc Natl Acad Sci U S A. 2003;100(10):5783–8. doi: 10.1073/pnas.1031748100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebel K, Klimkait T, Martin MA. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science. 1988;241(4870):1221–3. doi: 10.1126/science.3261888. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427(6977):848–53. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Tokarev AA, Munguia J, Guatelli JC. Serine-Threonine Ubiquitination Mediates Downregulation of BST-2/Tetherin and Relief of Restricted Virion Release by HIV-1 Vpu. J Virol. 2011;85(1):51–63. doi: 10.1128/JVI.01795-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3(4):245–52. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigan R, Neil SJ. Determinants of Tetherin Antagonism in the Transmembrane Domain of the Human Immunodeficiency Virus Type-1 (Hiv-1) Vpu Protein. J Virol. 2010 doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey RL, Maldarelli F, Martin MA, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol. 1992;66(12):7193–200. doi: 10.1128/jvi.66.12.7193-7200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Ellenberg J, Bonifacino JS, Weissman AM. The transmembrane domain of a carboxyl-terminal anchored protein determines localization to the endoplasmic reticulum. J Biol Chem. 1997;272(3):1970–5. doi: 10.1074/jbc.272.3.1970. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6(1):54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.