

Abstract
Pasteurella multocida is a pathogen of veterinary and medical importance. Here, we report the 1.85 Å resolution crystal structure of the class C acid phosphatase from this organism (denoted rPmCCAP). The structure shows that rPmCCAP exhibits the same haloacid dehalogenase fold and dimeric assembly as the class C enzyme from Haemophilus influenzae. Formation of the dimer in solution is demonstrated using analytical ultracentrifugation. The active site is devoid of a magnesium ion due to the presence of citrate in the crystallization buffer. Absence of the metal ion minimally perturbs the active site structure, which suggests that the main role of the ion is to balance the negative charge of the substrate rather than stabilize the active site structure. The crystal lattice displays unusual crystal packing involving the C-terminal polyhistidine tag mimicking the substrate. Steady-state kinetic constants are determined for the substrates NMN, 5´-AMP, 3´-AMP, 2´-AMP, and p-nitrophenyl phosphate. The highest catalytic efficiency is observed with NMN. The production of polyclonal anti-rPmCCAP antibodies is demonstrated, and these antibodies are shown to cross-react with the H. influenzae class C phosphatase. The antibodies are used to detect PmCCAP in clinical P. multocida and Mannheimia haemolytica strains cultured from infected animals.
Keywords: X-ray crystallography, class C acid phosphatase, analytical ultracentrifugation, steady-state kinetics, polyhistidine affinity tag, haloacid dehalogenase fold
Introduction
Class C acid phosphatases (CCAPs) are outer membrane acid phosphatases that contain four essential Asp residues imbedded in the bipartite sequence motif of [IV]-[VAL]-D-[IL]-D-E-T-[VM]-L-X-[NT]-X(2)-Y in the N-terminal half of the polypeptide chain and [IV]-[LM]-X(2)-G-D-[NT]-L-X-D-F in the C-terminal half [1]. Because of their localization to the bacterial outer membrane, CCAPs are potential candidates for vaccine development, and some progress has been made toward creating a vaccine against nontypeable Haemophilus influenzae using catalytically inactive mutants of the H. influenzae CCAP [2–4]. Biochemical studies have shown that CCAPs typically hydrolyze a variety of aryl phosphatases as well as nucleoside 5´- and 3´-monophosphates [5–15]. Crystallographic studies of the recombinant CCAP from H. influenzae, a.k.a. rP4, have shown that CCAPs belong to the haloacid dehalogenase (HAD) structural superfamily and revealed the basis of selectivity for nucleoside monophosphate substrates [9, 15]. The structural and biochemical data also showed that the first of the four conserved Asp residues is the nucleophile that attacks the P atom of the substrate, the second one functions as the acid that protonates the leaving group, and all four Asp residues contribute to the binding of a Mg2+ ion.
Previously, it was shown that the Pasteurella multocida genome encodes a protein that shares 58 % global amino acid sequence identity with rP4 suggesting that the organism produces a CCAP [16]. This finding is potentially significant, because P. multocida is a pathogen of medical and veterinary importance [17]. The organism is a ubiquitous Gram-negative, facultative anaerobic bacterium and is often a constituent of the normal respiratory microbiota, but is also capable of causing pneumonia, osteomyelitis, and endocarditis and meningitis infections. In humans, P. multocida infections are common from dog and cat bites. P. multocida and other Pasteurellaceae, including Mannheimia haemolytica (formerly Pasteurella haemolytica), are the main causative agents of bovine respiratory disease, which causes annual losses in the U.S. animal agriculture industry of $750 million to $1 billion [18]. Although there has been some progress in understanding the factors involved in the pathogenesis of P. multocida, including the polysaccharide capsule, lipopolysaccharide, outer membrane proteins, fimbriae and adhesions, there remains a lack of knowledge about the host-pathogen relationship [17].
Given the value of outer membrane proteins as potential targets for vaccine development, we previously cloned and expressed a recombinant form of the predicted CCAP from P. multocida [16]. Here report the crystal structure of this protein (denoted rPmCCAP), show that the enzyme forms a dimer in solution and exhibits nucleotidase activity, and demonstrate the production of anti-rPmCCAP polyclonal antibodies.
Material and methods
Crystal structure determination
Methods for the expression, purification, and crystallization of rPmCCAP were reported previously [16]. Briefly, the crystals were grown in sitting drops at room temperature over a reservoir containing 20% (w/v) PEG 3350, 0.2 M ammonium citrate, and 10% (v/v) n-propanol. The space group is C2 with unit cell parameters of a = 80.0, b = 106.1, c = 89.7, and β = 93.1°. The 1.85 Å resolution X-ray diffraction data set used for structure determination was collected from an epitaxially-twinned crystal at beamline 4.2.2 of the Advanced Light Source as described previously [16].
The structure was solved using molecular replacement phasing as implemented in MOLREP [19]. The search model was a monomer extracted from the coordinates of rP4 (PDB code 3ET4), which shares 58 % amino acid sequence identity with rPmCCAP. A clear solution having three molecules in the asymmetric unit was obtained. Based on the method of Matthews [20, 21], the solvent content is estimated to be 44 % with a Vm value of 2.2 Å3/Da. The model from molecular replacement was refined with simulated annealing in PHENIX [22]. The refined model was used to calculate phases, which were input to ARP/wARP [23] for automated building and sequence assignment. The model from ARP/wARP was used as the starting point for several rounds of manual model building in COOT [24] followed by refinement in PHENIX. Refinement statistics for the final model are listed in Table 1. The pairwise root mean square deviations for Cα atoms of the three chains are 0.21 – 0.33 Å, indicating that the overall conformations are identical within experimental error.
Table 1.
Data collection and refinement statisticsa
| Space group | C2 |
|---|---|
| Wavelength (Å) | 1.00000 |
| Data collection resolution (Å) | 27 - 1.85 (1.95 - 1.85) |
| Unit-cell parameters (Å, °) | a = 80.0, b = 106.1, c = 89.7, β = 93.1° |
| No. of observations | 215684 |
| No. of unique reflections | 62294 |
| Rmerge(I) | 0.094 (0.272) |
| Average I/σ | 10.4 (3.1) |
| Completeness (%) | 97.8 (86.7) |
| Redundancy | 3.5 (2.5) |
| Refinement resolution (Å) | 27 - 1.85 (1.88 - 1.85) |
| Rcryst | 0.211 (0.302) |
| Rfreeb | 0.267 (0.385) |
| No. of protein residues | 754 |
| No. of protein atoms | 5814 |
| No. of water molecules | 583 |
| Average B-factor (Å2) | |
| Protein | 23.2 |
| Water | 27.1 |
| rmsdc | |
| Bonds (Å) | 0.006 |
| Angles (°) | 0.93 |
| Ramachandran plotd | |
| Favored (%) | 97.7 |
| Allowed (%) | 2.3 |
| Coordinate error (Å)e | 0.29 |
| PDB code | 3PCT |
Enzyme activity assays
Steady-state kinetic constants were determined using a discontinuous assay that measures the production of inorganic phosphate [25]. The assays were performed at 37 °C in 200 mM sodium acetate and 1 mM MgCl2 at pH 5.5 (total assay volume of 0.5 mL). The pH of 5.5 was chosen based on measurements of the activity as a function of pH using NMN as substrate, which showed a plateau of maximal activity at pH 5.0 – 6.0 (data not shown). The enzyme concentration was 0.80 µM when using NMN as the substrate, 1.2 µM for p-nitrophenyl phosphate (pNPP), 4.0 µM for 5´-AMP and 3´-AMP, and 6.0 µM for and 2´-AMP. For each substrate concentration, the reaction was stopped by adding 1 mL of a quenching/detection solution (0.045 % (w/v) malachite green and 4.2% (w/v) ammonium molybdate in 4N HCl) after reaction times of 15 s, 75 s, 135 s and 195 s, and the citrate color development reagent (34 % (w/v) sodium citrate) was added 70 s after stopping each reaction. After 30 minutes, the inorganic phosphate concentrations were determined spectrophotometrically at 625 nm by reference to a standard curve constructed from solutions of known phosphate concentration. The initial rate was estimated by fitting data from the four time points to a line. Apparent values of Km and Vmax were estimated by fitting the initial rate data to the Michaelis-Menten equation using Origin 8.5 software. For each substrate tested, a preliminary data set was collected in order to estimate Km, and then a second data set was collected using a substrate range that spanned from less than 0.3Km to at least 3Km. The reported kinetic constants were determined from the second data set.
Analytical ultracentrifugation
Equilibrium sedimentation data were acquired at 20 °C using a Beckman XL-I Optima analytical ultracentrifuge equipped with an An50Ti rotor. Prior to centrifugation, the protein sample was dialyzed into a reference buffer of 50 mM acetate buffer at pH 6.0. Absorbance data (280 nm) were collected at three protein concentrations and three rotor speeds (11,000, 15,000, and 22,000 rpm). The 9 sets of data were fit globally using Origin 8 to an ideal single-species model (equation 9 of [26]). A solvent density of 1.00 g/cm3 was employed for the calculations, and the partial specific volume was assumed to be 0.73 cm3/g.
Immunological methods
Rabbit anti-rPmCCAP antiserum was obtained using standard immunological methods. Laboratory animals were utilized with approval of the University of Missouri Animal Care and Use Committee. Purified rPmCCAP (~250 µg) was dialyzed against 0.9 % NaCl, filter-sterilized, and emulsified in Freund’s complete adjuvant. The immunogen was then injected subcutaneously at multiple sites into a New Zealand white rabbit. Approximately 24 days after primary immunization, the response was boosted by single subcutaneous injection with similarly treated purified enzyme. Serum was collected by ear puncture 14 days following secondary immunization.
For Western blot detection of purified rPmCCAP and rP4, the protein was subjected to SDS-PAGE and then transferred to a Nitrocellulose (Bio Trace™ Pall Life Science) membrane in G-Biosciences 1X Tris-Glycine transfer buffer at 100 mA for 4 hr in an Owl Bandit Transblot apparatus. Rabbit anti-rPmCCAP polyclonal antibodies were used as primary antibody and detected with goat anti-rabbit or anti-mouse IgG (H+L) conjugated to alkaline phosphatase. Purified rP4 was obtained as previously described [8].
Detection of CCAP in clinical Pasteurella strains
Two P. multocida strains and one M. haemolytica strain were cultured from samples submitted to the Veterinary Medical Diagnostic Laboratory at the University Of Missouri College Of Veterinary Medicine. The two P. multocida samples originated from avian and porcine sources. The M. haemolytica strain originated from a bovine source. The strains were plated onto blood agar plates and incubated at 37 °C. After incubation, a colony was picked, resuspended in 10 mL of BHI broth and incubated overnight at 37°C under aerated conditions. The cells were harvested, disrupted using freeze/thaw cycles, and centrifuged. The supernatant was discarded, and the pellet was suspended in buffer and analyzed with Western blotting using rabbit anti-rPmCCAP IgG.
Results and discussion
Overall fold and active site structure
The fold of rPmCCAP comprises an α/β core domain situated below an α-helical cap domain and is almost identical to that of rP4 (Fig. 1A). The two enzymes superimpose with a root mean square deviation of 0.66 Å for Cα atoms.
Fig. 1.

Structure of rPmCCAP. (A) Ribbon drawing of the protomer highlighting domain structure and similarity to rP4. rPmCCAP and rP4 (PDB code 3ET4) are shown in silver and green, respectively. Side chains of the four conserved Asp residues (DDDD motif) of rPmCCAP are shown in yellow. (B) Ribbon drawing of a dimer of rPmCCAP. The two chains are colored blue and pink. Asp residues of the DDDD motif are colored yellow. The dashed curve represents a disordered loop (residues 52–56) of chain A. The two insets show close-up views of intersubunit interactions.
The structural similarity of rPmCCAP to rP4 extends to the details of the active site, except for the Mg2+ ion, which is replaced by a water molecule in the rPmCCAP structure. Structures of rP4 have shown that the lower part of the active site contains the substrate phosphoryl binding site, which includes an essential Mg2+ ion surrounded by the four Asp residues of the DDDD motif [9, 15]. The roles of these Asp residues in rPmCCAP can be inferred from structures of rP4 complexed with substrates [15] and more generally, studies of HAD superfamily enzymes [27]. The first Asp (Asp63 in rPmCCAP, Fig. 2A) is the nucleophile that attacks the substrate phosphoryl group. The second Asp, aka Asp+2, is positioned two residues upstream from the nucleophile (Asp65, Fig. 2A), and functions as a general acid-base that protonates the substrate leaving group and deprotonates the water molecule that attacks the phosphoenzyme intermediate. The other two Asp residues of the DDDD motif (Asp180, Asp184 in rPmCCAP) are expected to bind the Mg2+ ion. Electron density maps indeed displayed a peak at the location expected for the metal ion (Fig. 2A, feature denoted "wa"). Consistent with the known metal ion site of rP4, the electron density peak is surrounded by the carboxylate groups of Asp63 and Asp180 as well as the carbonyl of Asp65. However, when a Mg2+ ion was modelled at this location, the distances between the metal ion and these coordinating oxygen ligands refined to values in the range of 2.7 – 2.9 Å, which are significantly longer than the Mg2+-O distance of 2.2 Å observed in the rP4 structure. Furthermore, the electron density map did not have features representing the three water molecules that complete the octahedral coordination sphere of the metal ion in rP4. It was therefore concluded that the electron density feature in rPmCCAP represents a water molecule (wa in Fig. 2A) rather than a metal ion. This assignment is consistent with the fact that the crystallization and cryoprotection solutions contained 0.2 M citrate [16], a Mg2+ chelator.
Fig. 2.

Close-up views of the phosphoryl binding site of rPmCCAP. (A) Electron density near the phosphoryl binding site. The cage represents a simulated annealing σA-weighted Fo - Fc omit map contoured at 3.0 σ. Prior to map calculation, the side chains and water molecules shown were removed, and simulated annealing refinement was performed using PHENIX. The water molecule denoted wa occupies the expected Mg2+ site. (B) Comparison of rPmCCAP (silver) and rP4 (green, PDB code 3ET4). Water molecules of rPmCCAP are colored red, while those of rP4 are colored blue. The Mg2+ ion of rP4 is shown in yellow. Black and green dashed lines denote electrostatic interactions in rPmCCAP and rP4, respectively.
Aside from the absence of a metal ion and its three coordinating water molecules, the active site of metal-free rPmCCAP is remarkably similar to that of Mg2+-bound rP4 (Fig. 2B). The only perceptible difference in the protein conformation involves Asp63, which is the enzyme nucleophile. This side chain is rotated around χ2 by 45° compared to Asp64 of rP4. The other three Asp residues display essentially the same conformations as in rP4 despite the absence of the metal ion. The structural integrity of the metal-free active site suggests that the metal ion plays a minimal role in organizing the active site residues for catalysis, and that its major role is to neutralize the negative charge of the substrate.
Oligomeric state
The oligomeric state of rPmCCAP in solution was determined with equilibrium analytical ultracentrifugation. Data from nine sedimentation experiments corresponding to three rotor speeds and three loading concentrations were fit globally to a single species model (Fig. 3). The best-fit value of the molecular weight is 60.1 ± 0.2 kDa, which is within 3 % of the theoretical molecular weight of a dimer (58.1 kDa). This result is consistent with sedimentation and size exclusion chromatography data showing that rP4 and the CCAPs from Helicobacter pylori and Clostridium perfringens are dimeric in solution [6, 12, 13].
Fig. 3.
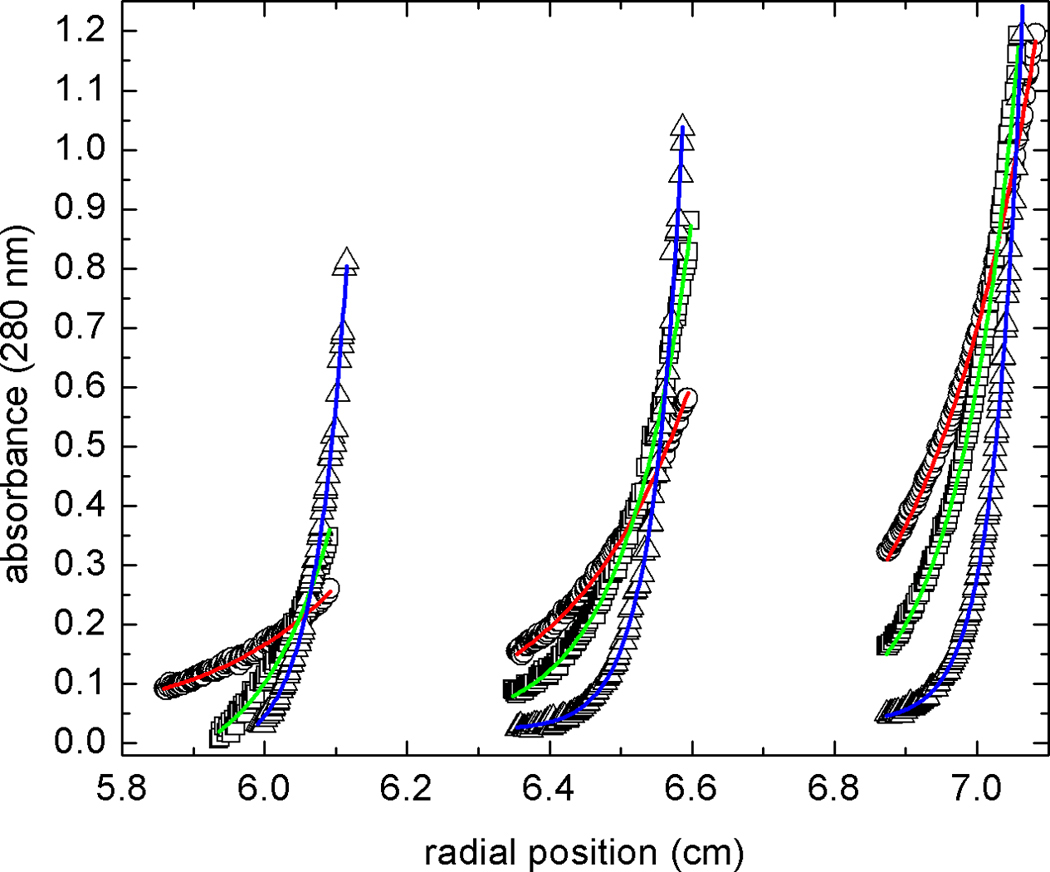
Global fit of sedimentation equilibrium data to an ideal single-species model. Data were acquired at 11,000 (circles), 15,000 (squares), and 22,000 (triangles) rpm at three different loading concentrations. The 9 data sets were subjected to non-linear least squares minimization as described in the text. The solid curves indicate the best fit to the data (red, 11,000 rpm; green, 15,000 rpm; blue, 22,000 rpm).
The dimeric assembly shown in Fig. 1B is most likely the one formed in solution. The asymmetric unit contains one such dimer, which is formed by chains A and B (AB dimer, Fig. 4A). There is a third molecule in the asymmetric unit (chain C), and rotation of it around the crystallographic 2-fold axis (−x, y, −z) followed by translation of (1, 0, 0) generates the CC´ dimer shown in Fig. 4A. Analysis of protein-protein interfaces using the PISA server [28] shows that this dimer has a buried interface area of 2900 Å2, which is the largest interface in the lattice. The estimated free energy of dissociation from PISA (42 kcal/mol) suggests that the dimer is stable in solution. Furthermore, an identical dimer is found in both the tetragonal and hexagonal rP4 crystal lattices [9, 15], and several intersubunit interactions are observed in both enzymes. These conserved interactions include an intersubunit Glu-Arg-Asn electrostatic network that involves an active site loop (Fig. 1B, lower inset) and a hydrophobic interaction in which a tryptophan near the C-terminus of one protomer (Trp250 in rPmCCAP) packs against three aromatic residues (Tyr74, Trp77, Trp93) of the cap domain of the other protomer (Fig. 1B, upper inset). Taken together, these results suggest that the dimer of Fig. 1B represents the solution form of rPmCCAP and confirms this assembly as the dimer of rP4.
Fig. 4.

Involvement of the C-terminal polyhistidine tag in crystal packing. (A) Section of the crystal lattice showing three dimers interacting via His-tags to form a hexameric assembly. The asymmetric unit consists of chains A, B, and C. Crystallographic symmetry was used to generate chains A´, B´, and C´. The dotted lines indicate disordered residues of chains B and C. The view is looking along the a–c diagonal with the b-axis vertical. (B) Close-up view of the binding of the tag of chain B (magenta) in the active site of chain C (yellow). 5´-AMP from the structure of a substrate-trapping mutant of rP4 complexed with 5´-AMP is shown in green (PDB code 3OCV).
Stabilization of the crystal lattice by the polyhistidine affinity tag
The crystal lattice exhibits an unusual case of the polyhistidine tag participating in crystal contacts (Fig. 4A). We note that the rPmCCAP gene was created so that the expressed protein has a non-cleavable hexahistidine tag following Lys253. Electron density was evident for the last five histidine residues of chain C (residues 255–259) but absent for Lys253 and His254. The connectivity of the C-terminus of chain B was better, with only His255 omitted from the final model. Electron density for the tag of chain A was not evident. As shown in Fig. 4A, the His-tag of chain B interacts with chain C (or equivalently, C´), while the tag of chain C (or C´) interacts with chain A (or A´), resulting in a hexameric assembly. The hexamer is an artifact of crystallization, since the sedimentation data showed that His-tagged rPmCCAP forms a dimer in solution.
The terminal residue His259 appears to mimic the base of nucleoside monophosphate substrates (Fig. 4B). Previous structures of a substrate-trapping mutant of rP4 complexed with nucleoside monophosphates showed that the base of the substrate binds in an aromatic box formed by Phe86, Trp91, and Tyr221 [15]. The analogous residues of rPmCCAP are Phe85, Trp90, and Tyr220, and they form a substructure identical to that of rP4. The imidazole ring of His259 binds in the aromatic box and is sandwiched between Phe85 and Tyr220. This arrangement is reminiscent of the adenine ring of 5´-AMP binding in the aromatic box of rP4. Thus, substrate mimicry appears to contribute to the stabilization of the polyhistidine affinity tag, and hence, crystal formation.
We note that ordered polyhistidine tags in crystal structures are common. A recent query of the PDB using PDBeMotif [29] revealed 15 structures with at least 5 histidine residues built at the N-terminus and 69 structures with at least 5 histidine residues built at the C-terminus.
Kinetic analysis
Preliminary activity assays showed that the enzyme exhibited activity with nucleoside monophosphate substrates as well as the synthetic aryl phosphate substrates pNPP and 4-methylumbelliferyl phosphate. The steady-state kinetic constants were measured at 37°C and pH 5.5 for the substrates NMN, 5´-AMP, 3´-AMP, 2´-AMP, and pNPP (Table 2). Among the nucleoside monophosphate substrates tested, the enzyme exhibits the highest catalytic efficiency (kcat/Km) for NMN, followed by 5´-AMP, 3´-AMP, and 2´-AMP. This rank ordering is the same as that of rP4 [15]. The lowest Km was obtained with 5´-AMP (0.08 mM), and the highest was obtained with 2´-AMP (1.4 mM). This trend was also observed for rP4. The observation that rPmCCAP catalyzes the hydrolysis of nucleoside 5´- and 3´-monophosphates is consistent with other kinetic analyses suggesting that CCAPs are nonspecific nucleotidases [5–14].
Table 2.
Kinetic parameters for rPmCCAP
| Substrate | Km (mM) | kcat (s−1) | kcat / Km (s−1M−1) |
|---|---|---|---|
| NMN | 0.29 ± 0.06 | 4.4 ± 0.3 | 15000 ± 3300 |
| pNPP | 0.5 ± 0.1 | 6.2 ± 0.5 | 12000 ± 2700 |
| 5′-AMP | 0.08 ± 0.01 | 0.18 ± 0.01 | 2300 ± 310 |
| 3′-AMP | 0.8 ± 0.2 | 0.19 ± 0.01 | 240 ± 61 |
| 2′-AMP | 1.4 ± 0.3 | 0.13 ± 0.01 | 90 ± 21 |
Detection of proteins using anti-rPmCCAP IgG
Since antibodies against outer membrane proteins are potentially useful in vaccine development, we raised polyclonal anti-rPmCCAP antibodies in a rabbit. Western blotting confirms that the anti-rPmCCAP antibodies react with rPmCCAP (Fig. 5A, lane 1). The high structural similarity between rPmCCAP and rP4 prompted us to test whether anti-rPmCCAP antibodies cross-react with rP4. As shown in Fig. 5A (lane 3), anti-rPmCCAP antibodies also recognize rP4.
Fig. 5.

Western blot analysis using anti-rPmCCAP IgG. (A) Detection of purified rPmCCAP (lane 1) and rP4 (lane 3) using a 1:80,000 antiserum dilution. Lane 2 contains molecular weight standards. (B) Analysis of clinical P. multocida and M. haemolytica clinical strains. Lane 1 contains molecular weight standards. Lanes 2 and 3 contain cell pellet fractions of P. multocida clinical strains cultured from avian and porcine sources, respectively. Lane 6 contains the cell pellet fraction from a M. haemolytica clinical strain (bovine source). The antiserum was diluted 1:160,000 in each case.
The rabbit anti-rPmCCAP IgG was used to detect PmCCAP in clinical strains. The strains of P. multocida tested were cultured from avian and porcine sources. The cells were disrupted, and the pellet obtained by centrifugation, which presumably includes membrane-anchored proteins, was assayed using Western blotting. As shown in Fig. 5B, PmCCAP is detected in both clinical strains (lanes 2 and 3). These results are consistent with the prediction that PmCCAP is lipidated and exported from the cytoplasm [16]. Further work will be needed to determine whether the enzyme is associated with the inner or outer membrane, but the high sequence identity to P4 implies that it is an outer membrane protein.
Anti-rPmCCAP was also used to detect the CCAP in M. haemolytica cultured from an infected bovine source. Formerly known as Pasteurella haemolytica, M. haemolytica is closely related to P. multocida. Comparison of the PmCCAP amino acid sequence with the non-redundant sequence database using BLAST [30] revealed a predicted M. haemolytica CCAP having 65 % global sequence identity with PmCCAP (RefSeq accession number ZP_05990345). As shown in Fig. 5B (lane 6), Western blot analysis of the M. haemolytica clinical strain using anti-rPmCCAP was positive, consistent with the prediction from sequence analysis.
Antibodies against rPmCCAP may have clinical utility. The use of rP4 as a component of a recombinant vaccine against nontypeable H. influenzae [2–4] suggests the potential for developing a vaccine to protect animals against P. multocida infection. Another possibility is to develop a diagnostic test for P. multocida and closely related pathogens using anti-rPmCCAP. The structure reported here should aid these efforts by providing a three-dimensional structural basis for the design of catalytically inactive mutant enzymes having enhanced immunogenicity.
Acknowledgements
We thank Dr. Jay Nix of ALS beamline 4.2.2 for help with data collection. This research was supported by the NIH grant U54 AI057160 to the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research (MRCE) and the University of Missouri Research Board. H.S. was supported by a pre-doctoral fellowship from National Institutes of Health grant DK071510 and a Chancellor’s Dissertation Completion Fellowship from the University of Missouri-Columbia. Part of this research was performed at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thaller MC, Schippa S, Rossolini GM. Protein Sci. 1998;7:1647–1652. doi: 10.1002/pro.5560070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotomi M, Ikeda Y, Suzumoto M, Yamauchi K, Green BA, Zlotnick G, Billal DS, Shimada J, Fujihara K, Yamanaka N. Vaccine. 2005;23:1294–1300. doi: 10.1016/j.vaccine.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 3.Mason KW, Zhu D, Scheuer CA, McMichael JC, Zlotnick GW, Green BA. Vaccine. 2004;22:3449–3456. doi: 10.1016/j.vaccine.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 4.Green BA, Baranyi E, Reilly TJ, Smith AL, Zlotnick GW. Infect. Immun. 2005;73:4454–4457. doi: 10.1128/IAI.73.7.4454-4457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reilly TJ, Chance DL, Smith AL. J. Bacteriol. 1999;181:6797–6805. doi: 10.1128/jb.181.21.6797-6805.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reilly TJ, Smith AL. Protein Expr. Purif. 1999;17:401–409. doi: 10.1006/prep.1999.1157. [DOI] [PubMed] [Google Scholar]
- 7.Reilly TJ, Green BA, Zlotnick GW, Smith AL. FEBS Lett. 2001;494:19–23. doi: 10.1016/s0014-5793(01)02294-3. [DOI] [PubMed] [Google Scholar]
- 8.Ou Z, Felts RL, Reilly TJ, Nix JC, Tanner JJ. Acta Cryst. 2006;F62:464–466. doi: 10.1107/S1744309106012358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felts RL, Ou Z, Reilly TJ, Tanner JJ. Biochemistry. 2007;46:11110–11119. doi: 10.1021/bi701016m. [DOI] [PubMed] [Google Scholar]
- 10.Passariello C, Schippa S, Iori P, Berlutti F, Thaller MC, Rossolini GM. Biochim. Biophys. Acta. 2003;1648:203–209. doi: 10.1016/s1570-9639(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 11.Malke H. Appl. Environ. Microbiol. 1998;64:2439–2442. doi: 10.1128/aem.64.7.2439-2442.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reilly TJ, Calcutt MJ. Protein Expr. Purif. 2004;33:48–56. doi: 10.1016/j.pep.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 13.Reilly TJ, Chance DL, Calcutt MJ, Tanner JJ, Felts RL, Waller SC, Henzl MT, Mawhinney TP, Ganjam IK, Fales WH. Appl. Environ. Microbiol. 2009;75:3745–3754. doi: 10.1128/AEM.01599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang R, Ohtani K, Wang Y, Yuan Y, Hassan S, Shimizu T. Microbiology. 2010;156:167–173. doi: 10.1099/mic.0.030395-0. [DOI] [PubMed] [Google Scholar]
- 15.Singh H, Schuermann JP, Reilly TJ, Calcutt MJ, Tanner JJ. J. Mol. Biol. 2010;404:639–649. doi: 10.1016/j.jmb.2010.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh H, Felts RL, Ma L, Malinski TJ, Calcutt MJ, Reilly TJ, Tanner JJ. Acta Crystallogr. 2009;F65:226–231. doi: 10.1107/S1744309109001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harper M, Boyce JD, Adler B. FEMS Microbiol. Lett. 2006;265:1–10. doi: 10.1111/j.1574-6968.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 18.Rice JA, Carrasco-Medina L, Hodgins DC, Shewen PE. Anim. Health Res. Rev. 2007;8:117–128. doi: 10.1017/S1466252307001375. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 20.Matthews BW. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 21.Kantardjieff KA, Rupp B. Protein Sci. 2003;12:1865–1871. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr., Sect. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris RJ, Perrakis A, Lamzin VS. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Acta Cryst. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. Anal. Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 26.Lebowitz J, Lewis MS, Schuck P. Protein Sci. 2002;11:2067–2079. doi: 10.1110/ps.0207702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen KN, Dunaway-Mariano D. Trends Biochem. Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Krissinel E, Henrick K. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 29.Golovin A, Henrick K. BMC Bioinformatics. 2008;9:312. doi: 10.1186/1471-2105-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Engh RA, Huber R. Acta Cryst. A. 1991;47:392–400. [Google Scholar]
- 32.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]