

Abstract
The bioactive signaling molecule D-erythro-sphingosine 1-phosphate (S1P) is irreversibly degraded by the enzyme S1P lyase (SPL). The reaction of SPL with C18-S1P generates ethanolamine phosphate and a long chain fatty aldehyde, trans-2-hexadecenal. Modulation of SPL expression in cells and organisms produces significant phenotypes, most of which have been attributed to corresponding changes in S1P-dependent signaling. However, the physiological functions of SPL products are not well understood. In the present study, we explored the biological activities of trans-2-hexadecenal in human and murine cells. We demonstrate that trans-2-hexadecenal causes cytoskeletal reorganization leading to cell rounding, detachment and eventual cell death by apoptosis in multiple cell types, including HEK293T, NIH3T3 and HeLa cells. Trans-2-hexadecenal stimulated a signaling pathway involving MLK3 and the respective phosphorylation of MKK4/7 and JNK, whereas ERK, AKT and p38 were unaffected. Trans-2-hexadecenal-induced apoptosis was accompanied by activation of downstream targets of JNK including c-Jun phosphorylation, cytochrome c release, Bax activation, Bid cleavage and increased translocation of Bim into mitochondria. The antioxidant N-acetylcysteine prevented JNK activation by trans-2-hexadecenal. Further, inhibition of JNK abrogated the cytoskeletal changes and apoptosis caused by trans-2-hexadecenal, whereas Rac1 and RhoA were not involved. In conclusion, our studies provide a new paradigm of sphingolipid signaling by demonstrating for the first time that S1P metabolism generates a bioactive product that induces cellular effects through oxidant stress-dependent MAP kinase cell signaling.
Keywords: sphingosine 1-phosphate, S1P lyase, sphingolipid, hexadecenal, JNK
1. Introduction
Sphingosine 1-phosphate lyase (SPL) catalyzes the final and irreversible step in the degradation of sphingolipids. The reaction catalyzed by SPL results in cleavage of the bioactive molecule sphingosine-1-phosphate (S1P) at the C2-3 carbon-carbon bond, generating two products, trans-2-hexadecenal (hereafter referred to as hexadecenal) and ethanolamine phosphate [1,2]. Other long chain base phosphates such as dihydrosphingosine 1-phosphate and phytosphingosine 1-phosphate can serve as substrates for the enzyme, resulting in the formation of ethanolamine phosphate and the corresponding fatty aldehyde. Modulation of SPL expression in cells and organisms leads to profound cellular effects and phenotypes. These effects implicate SPL function in the regulation of programmed cell death pathways, cell cycle progression, cell migration, vertebrate and invertebrate development, and innate and adaptive immune functions [3-10]. In most of these cases, the effects of SPL modulation have been attributed to the impact upon S1P pools available for intracellular and extracellular signaling through well-characterized pathways or to the accumulation of cytotoxic sphingolipid intermediates. In relatively few instances, the products of SPL have been implicated in biological activities, such as their suspected role in promoting murine embryonic carcinoma cell proliferation and the requirement for ethanolamine phosphate in Leishmania infectivity [11,12]. However, the physiological functions of these molecules remain poorly understood.
Free fatty aldehydes of the general formula CH3(CH2)nCHO with n = 6 to 20 or greater are generally unstable molecules that can react with amino groups found on proteins, carbohydrates, lipids and nucleic acids [13]; therefore, they are not easily detected in cells or tissues. They are known to function as pheromones in insects, contribute to bacterial films and waxes, and have signaling functions in plants and algae, triggering calcium transients from intracellular stores and inducing cell death and autophagy [13-16]. Some aldehydes inhibit proliferation and induce apoptosis, whereas short and long-chain aldehydes released in biomembranes during lipid peroxidation have been shown to form DNA lesions and strand breaks that are suspected to contribute to carcinogenesis [17,18].
In this study, we explored the effects of hexadecenal on various cellular activities. Our findings demonstrate that treatment of human and murine cells with an exogenous solution of hexadecenal leads to alteration of cellular cytoskeletal organization and adhesion, induction of apoptotic cell death and activation of a mixed lineage kinase 3 (MLK3)/MAP kinase kinase 4/7 (MKK4/7)/c-Jun N-terminal kinase (JNK) signaling pathway. Importantly, inhibition of JNK activation prevents hexadecenal-mediated apoptosis and cytoskeletal reorganization. We show for the first time that hexadecenal, which may be generated either through the actions of SPL or potentially through other biochemical or chemical reactions may exert significant and biologically relevant effects on cells and tissues through a specific JNK-dependent signal transduction pathway.
2. Materials and methods
2.1 Materials
Fatty acid free BSA, 4′, 6-diamidino-2-phenylindole dihydrochloride hydrate (DAPI), N-acetyl cysteine (NAC), palmitic acid and propidium iodide were from Sigma (St. Louis, MO). JNK inhibitor V was obtained from EMD Biosciences (Gibbstown, NJ). Rhodamine-conjugated phalloidin was from Cytoskeleton, Inc. (Denver, CO). Buffered formalin was from Fisher Scientific (Pittsburgh, PA). CEP11004, a pan-MLK inhibitor was a generous gift from Cephalon Inc. (Frazer, PA).
2.2 Hexadecenal synthesis
(2E)-Hexadecanal was synthesized by the reaction sequence of olefination of myristaldehyde with triethyl phosphonoacetate in the presence of potassium carbonate in aqueous 2-propanol, then reduction of the α,β-unsaturated ester with alane, and finally oxidation of the resulting alcohol with pyridinium chlorochromate, as described previously [19]. The structure and purity of the product were characterized by 1H- and 13C-NMR spectroscopy and high-resolution mass spectrometry [HRMS (M + Na)+ m/z calculated for C16H30NaO+ 261.2189, found 261.2192].
2.3 Cell culture and hexadecenal treatment
Human embryonic kidney cells (HEK293T), human cervical carcinoma cells (HeLa) and mouse fibroblasts (NIH3T3) were procured from ATCC (Manassas, VA). Cells were propagated in Dulbecco’s modified Eagle’s medium with high glucose (Cell Culture Facility, UCSF, San Francisco, CA) containing 10% fetal bovine serum (JR Scientific, Woodland, CA), penicillin (100 units/ml) and streptomycin (100 μg/ml), at 37°C with 5% CO2. Hexadecenal was dissolved in chloroform:methanol (2:1 v/v). A thin film of hexadecenal was obtained by drying the organic solvent under a nitrogen stream. A 10 mM working stock solution of hexadecenal was prepared fresh by adding the vehicle (18% fatty acid free BSA, 5% ethanol and 150 mM sodium chloride) drop-by-drop while vortexing vigorously.
2.4 Phalloidin staining
NIH3T3 cells were grown for 24 h on 8-well chamber slides (BD Biosciences). After hexadecenal treatment, cells were washed in cold PBS and fixed for 15 min in 10% buffered formalin. After washing, cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature, washed and incubated with 100 nM Rhodamine phalloidin for 30 min at room temperature in the dark. Cells were washed 3 times, counterstained with 100 nM DAPI and mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Images were captured using a Zeiss Axioskop fluorescence microscope with a 100x oil objective (Carl Zeiss, Inc., Thornwood, NY). Images were processed using Adobe Photoshop CS4 (Adobe, San Jose, CA).
2.5 Plasmid constructs and transfection
Plasmid encoding enhanced green fluorescent protein, pcDNA3-EGFP plasmid (Addgene plasmid 13031) was a kind gift from Dr. Douglas Golenbock (U. Mass). Plasmid encoding dominant negative MLK3 pRK5-Flag-MLK3 K144A [20] was kindly provided by Dr. Kathleen Gallo, Michigan State University, East Lansing, MI. HEK293T cells were transfected with either pcDNA3-EGFP or pRK5-Flag-MLK3 K144A plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions.
2.6 Immunoblotting
Immunoblotting was performed as described [21]. Antibodies to Poly (ADP-ribose) polymerase (PARP), phospho-JNK (Thr183/Tyr185), phospho-MKK4 (Ser257/Thr261), phospho-MKK7 (Ser171/Thr275), phospho-c-Jun (Ser73), AKT, phospho-ERK (Thr202/Tyr204), ERK, phospho-p38 (Thr180/Tyr182), Bid, Bim and HRP-conjugated anti-rabbit antibodies were from Cell Signaling Technologies (Beverly, MA). Antibody to actin was from Sigma. Antibodies to MLK3 and cytochrome oxidase were from Epitomics (Burlingame, CA). Antibodies to JNK1/3, MKK4 and MKK7 were from GenScript Inc. (Piscataway, NJ). Antibody to phospho-AKT (Ser473) was from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to cytochrome c was from BD Pharmingen (San Diego, CA).
2.7 Flow cytometry
Annexin V binding assays were performed using the ApoAlert Annexin V Apoptosis Kit (Clontech Laboratories, Palo Alto, CA). Briefly, after hexadecenal treatment cells were trypsinized and washed once with PBS. Cells were rinsed once with binding buffer. Cells were resuspended in 200 μl of binding buffer and incubated with 5 μl of FITC-labeled Annexin V for 10 min in the dark. Cells were acquired using a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using CellQuest Pro software (BD Biosciences). Dead cells were excluded from analysis by propidium iodide staining.
2.8 Preparation of mitochondrial and cytosolic cell fractions
HEK293T cells were plated in 10 cm culture dishes. Cells were collected by centrifugation at 200 g for 5 min and at 4°C. The cells were then washed twice with ice-cold PBS, pH 7.4, followed by centrifugation at 200 g for 5 min. The cell pellet was resuspended in 200 μl of extraction buffer, containing 250 mM sucrose, 10 mM Tris-Cl, pH 7.4, 1 mM EDTA, 1 mM PMSF and protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN). After 30 min incubation on ice, cells were homogenized with a glass dounce and a B pestle (40 strokes). Cell homogenates were spun at 14,000 g for 30 min, then supernatants (cytosolic fraction) were collected and stored at −80°C until analysis. The mitochondrial pellet was washed once with extraction buffer and then lysed on ice for 30 min in lysis buffer containing 50 mM MOPS, pH 7.4, 1 mM EDTA, 1% Triton-X 100, 10% glycerol, 1 mM PMSF and protease inhibitor cocktail. The purity of the cytosolic and mitochondrial fractions was determined by immunoblotting using antibodies against actin and cytochrome oxidase, respectively.
2.9 Confocal microscopy
HeLa cells were grown for 24h on 8-well chamber slides (BD Biosciences). To detect activated Bax, cells were washed in cold PBS and fixed for 15 min in freshly prepared 4% paraformaldehyde. After washing, cells were permeabilized with 0.3% Triton-X-100 in PBS for 10 min at room temperature. Then, cells were blocked with 5% goat serum and 0.1% Triton X-100 in PBS for 1 h at room temperature. Cells were stained with a conformation specific Bax antibody (6A7) that detects the activated form of Bax (BD Pharmingen, San Diego, CA) at 1:150 dilution overnight at 4°C, washed 3 times with PBS, and stained with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA) for 1 h at room temperature. Nonspecific fluorescence was excluded by performing a control without primary antibody. Cells were washed 3 times, stained with DAPI and mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Images were captured using a laser scanning microscope and software (LSM 710, Carl Zeiss, Inc.). Images were processed using Adobe Photoshop CS4 software (Adobe, San Jose, CA).
3. Results
3.1 Hexadecenal causes cytoskeletal reorganization and apoptosis
To explore the biological effects of hexadecenal, HEK293T cell cultures were observed over a period of hours to days after treatment with micromolar concentrations of exogenous synthetic hexadecenal. Hexadecenal applied at 25-50 μM was found to cause rounding and detachment of HEK293T cells. Similar effects were observed when human cervical carcinoma HeLa cells and mouse fibroblast NIH3T3 cells were treated with hexadecenal. The effect of hexadecenal on cytoskeletal organization was evaluated by labeling F-actin with phalloidin-Rhodamine in fixed NIH3T3 cells after treatment with hexadecenal or vehicle control. As shown in Figure 1, hexadecenal treatment reduced the number of stress fibers and induced lamellipodia formation.
Fig. 1. Hexadecenal causes cytoskeletal reorganization.
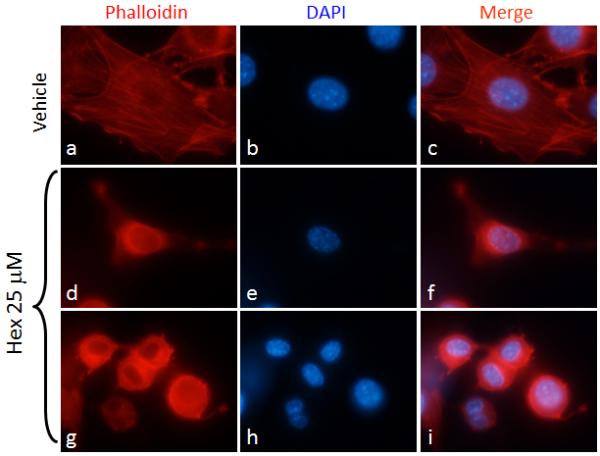
NIH3T3 cells were cultured in 8-chamber slides for 24 h. Cells were serum deprived for 3 h, then cells were treated with vehicle (a-c) or hexadecenal (25 μM) (d-i) for 30 min. F-actin was labeled with Phalloidin-Rhodamine and cells were counter stained with DAPI.
Detachment of anchorage-dependent cells from the surrounding extracellular matrix can induce apoptotic programmed cell death, or “anoikis”. Therefore, we investigated whether cell detachment after hexadecenal treatment induced apoptotic programmed cell death by measuring Annexin V labeling followed by flow cytometry. Our results revealed that treatment of NIH3T3, HeLa and HEK293T cells with 25 μM hexadecenal induces apoptotic cell death after detachment (Fig. 2A, B). In contrast to hexadecenal, the fatty acid palmitate at 25 μM had no effect on apoptosis in any of the cell types. Hexadecenal-induced apoptosis was confirmed in HEK293T and HeLa cells by demonstration of PARP cleavage as an indication of caspase-3 activation (Fig. 2C).
Fig. 2. Hexadecenal induces apoptosis in human and murine cells.
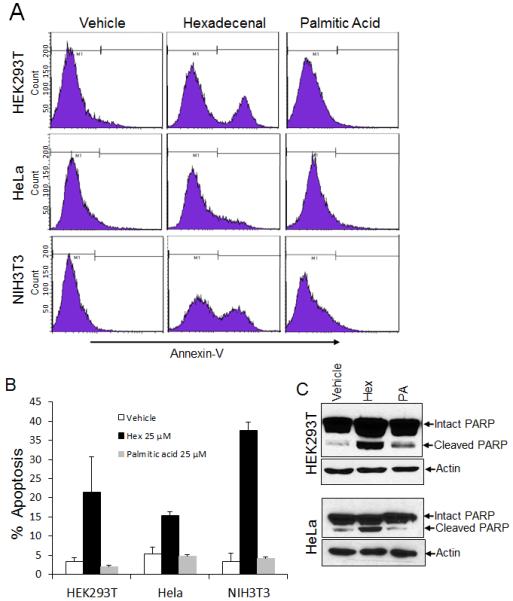
A-C, Cells were cultured in 10-cm culture dishes for 24 h. Cells were serum deprived for 3 h, followed by treatment with vehicle, hexadecenal (25 μM) or palmitic acid (25 μM) for 3 h. A, Apoptosis was determined by flow cytometry after Annexin V-FITC staining. B, Percentage of apoptotic cells (Annexin V positive) is shown in the histogram. Data are shown as mean ± SD. C, Cells were harvested and whole cell extracts were immunoblotted with PARP or actin antibodies. Immunoblots are representative of at least three independent experiments.
3.2 Hexadecenal causes robust activation of JNK
To investigate the mechanism by which hexadecenal induced cytoskeletal changes and apoptosis, we evaluated the effect of hexadecenal on the activation of the stress-induced signaling pathways mediated by p38, ERK and JNK, as well as on the PI3K/AKT signaling pathway known to regulate apoptotic cell death. Towards that end, immunoblotting was performed to measure the cellular levels of phosphorylated forms of AKT, p38, ERK and JNK. As shown in Figure 3A, JNK activation was observed within 15 min of hexadecenal treatment in HEK293T cells. JNK activation was further increased at later time points, reaching maximum activation 3 h after hexadecenal treatment and waning thereafter, whereas total JNK levels remained unchanged (Fig. 3A). Hexadecenal activated JNK and its downstream target c-Jun in a dose-dependent manner, with the lowest effective concentration tested being 25 μM, and higher doses producing more profound JNK activation (Fig. 3B). Hexadecenal is metabolized to palmitic acid [22]. Free fatty acids have also been shown to activate JNK pathway, albeit at a high concentration [23]. Therefore, to rule out the possibility that palmitic acid is the active molecule inducing the observed effects, cells were treated with 50 μM hexadecenal or palmitic acid. As shown in Figure 3C, hexadecenal showed a robust activation of JNK, whereas palmitic acid had no effect. Even at 100 μM concentration, palmitic acid did not elicit apoptosis or cell rounding (data not shown). To confirm whether JNK activation is correlated with cell detachment and apoptosis phenotypes observed in HEK293T, HeLa and NIH3T3 cells, each cell line was treated with hexadecenal and JNK activation was assessed. As shown in Figure 3D, hexadecenal treatment resulted in robust activation of JNK in HeLa and NIH3T3 cells, whereas palmitic acid had no effect on JNK activation (Fig. 3D). Similar effects were observed in HEK293T cells (data not shown). In contrast to our findings with JNK activation, hexadecenal treatment did not influence the activation of AKT, p38 or ERK signaling pathways (Fig. 3E, F). These findings confirm that hexadecenal specifically induces the JNK stress-activated signaling pathway.
Fig. 3. Hexadecenal activates the JNK signaling pathway.
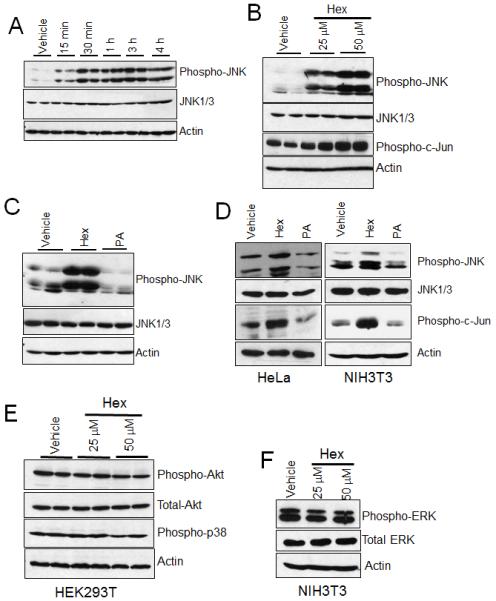
A, HEK293T cells were plated in 10-cm plates and cultured for 24 h. Cells were serum starved for 3 h and then treated with either vehicle or hexadecenal (50 μM). Cells were harvested at the indicated time period and whole cell extracts were immunoblotted with phospho-JNK, JNK1/3 or actin antibodies. B, HEK293T cells were treated with either vehicle or the indicated dose of hexadecenal (hex) for 1 h. Cell lysates were immunoblotted with phospho-JNK, JNK1/3, phospho-C-Jun or actin antibodies. C, HEK293T cells were treated with vehicle, hexadecenal (hex, 50 μM) or palmitic acid (PA 50 μM) for 1 h. Cell lysates were immunoblotted with phospho-JNK, JNK1/3 or actin antibodies. D, HeLa and NIH3T3 cells were cultured in 10-cm plates for 24 h. Cells were treated with either vehicle, hexadecenal (hex, 25 μM) or palmitic acid (PA 25 μM) for 1 h. Cell lysates were immunoblotted with phospho-JNK, JNK1/3, phospho-C-Jun or actin antibodies. E and F, HEK293T and NIH3T3 cells were cultured in 10-cm plates for 24 h. Cells were treated with either vehicle, or indicated dose of hexadecenal (hex) for 1 h. Cell lysates were immunoblotted with phospho-AKT, total AKT, phospho-p38, phospho-ERK, total ERK or actin antibodies. Immunoblots are representative of three independent experiments.
3.3 Hexadecenal activates JNK through a MLK3-dependent pathway
Activation of JNK occurs through its phosphorylation by dual-specificity kinases MKK4 or MKK7, which in turn become phosphorylated and activated by upstream serine/threonine kinases including apoptosis-signal regulating kinase 1 and MLK3 [24]. Therefore, we assessed the effect of hexadecenal on MKK4 and MKK7 activation. As shown in Figure 4A, hexadecenal activates both MKK4 and MKK7 in a dose-dependent manner. To examine whether activation of JNK occurs through MLK3 activation, HEK293T cells were pre-treated with a pan-MLK inhibitor prior to hexadecenal treatment. Inhibition of MLK3 completely prevented the hexadecenal-induced JNK activation (Fig. 4B). To rule out the possibility of a nonspecific effect of the MLK3 inhibitor, HEK293T cells were transfected with either a GFP construct or a dominant negative construct of MLK3 preceding hexadecenal treatment. As shown in Figure 4C, the dominant negative construct of MLK3 abrogated the JNK activation induced by hexadecenal. These findings suggest that MLK3 is required for hexadecenal-induced JNK activation.
Fig. 4. Hexadecenal activates JNK signaling through a Mixed Lineage Kinase 3 (MLK3)-dependent pathway.
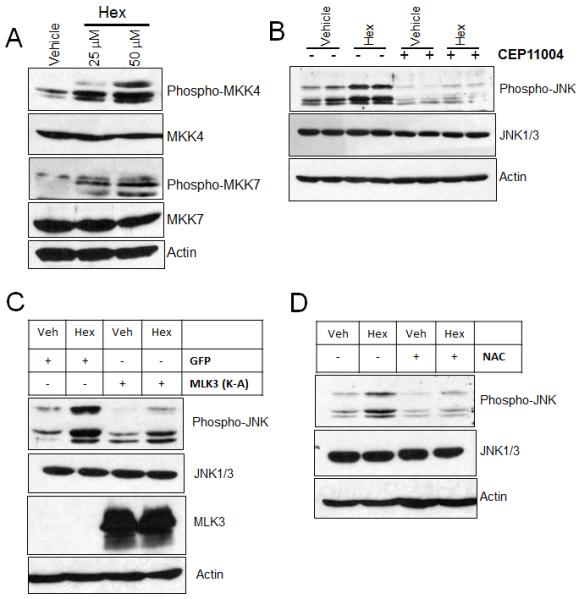
A, HEK293T cells were plated in 10-cm plates and cultured for 24 h. Cells were serum starved for 3 h and then were treated with either vehicle or with the indicated dose of hexadecenal for 1 h. Whole cell extracts were immunoblotted with phospho-MKK4, MKK4, phospho-MKK7, MKK7 or actin antibodies. B and D, HEK293T cells were plated in 10-cm plates and cultured for 24 h. Cells were serum deprived for 3 h in the presence or absence of a MLK3 inhibitor (CEP11004, 500 nM) or an antioxidant N-acetylcysteine (NAC, 10 mM) for 3 h. Then, cells were treated with either vehicle or hexadecenal (hex, 25 μM) for 1 h and cell lysates were immunoblotted with phospho-JNK, JNK1/3 or actin antibodies. C, HEK293T cells were transfected with GFP or a dominant negative construct of MLK3 (K-A). Forty-eight hours later, cells were serum deprived for 3 h and then treated with vehicle (veh) or hexadecenal (hex, 25 μM) for 1 h. Cell lysates were immunoblotted with phospho-JNK, JNK1/3, MLK3 or actin antibodies.
3.4 JNK activation by hexadecenal requires ROS generation
Both MLK3 and JNK are activated by reactive oxygen species (ROS) generation [25,26]. Therefore, we investigated whether JNK activation by hexadecenal requires ROS generation by pretreating cells with the antioxidant compound NAC. As shown in Figure 4D, pretreatment with NAC attenuated JNK activation by hexadecenal indicating that the latter acts by stimulating a MLK3/MKK4/7/JNK signaling pathway in a ROS-dependent manner.
3.5 Downstream targets of JNK signaling are modulated by hexadecenal
It became important to establish whether JNK signaling induced by hexadecenal could be responsible for cellular changes and apoptosis in hexadecenal-treated cells. JNK has an essential role in modulating the functions of pro- and antiapoptotic proteins located in the mitochondria [27]. Toward that end, we investigated the effects of hexadecenal on downstream targets of JNK signaling including cytochrome c release and expression/translocation of Bcl-2 family proteins. To examine the cytochrome c release, the cytosolic and mitochondrial fractions from vehicle- or hexadecenal-treated cells were prepared and immunoblotted with a mouse monoclonal cytochrome c antibody. As shown in Figure 5A, a dose-dependent cytochrome c release was observed from mitochondria of hexadecenal-treated cells. Bax and Bak activation is essential for the JNK-stimulated cytochrome c release and initiation of apoptosis [28]. Therefore we examined Bax activation after hexadecenal treatment in HeLa cells by confocal microscopy using an antibody that recognizes the activated form of Bax [29]. Hexadecenal treatment caused a substantial increase in punctate labeling for Bax, indicating the activation of Bax (Fig. 5B). The BH3-only proteins Bid and Bim are the primary activators of Bax [30]. Active Bid, also known as truncated Bid (tBid), induces the oligomerization of Bax [31]. We analyzed the protein levels of Bid and Bim by immunoblotting of whole cell lysates of hexadecenal-treated cells. As shown in Figure 5C, treatment of cells with increasing concentrations of hexadecenal was correlated with an increase in Bid cleavage, producing tBid. Bim exists in three splice forms: BimS, BimL and BimEL [32,33]. BimL and BimEL are normally found in the cytosol. In response to an apoptotic signal, phosphorylation of Bim by JNK allows it to translocate to the mitochondria, where Bim can activate Bax [32-34]. An increase in the levels of BimL and BimEL proteins was observed in the mitochondrial fraction of hexadecenal-treated cells (Fig. 5D), whereas the protein levels of all the Bim isoforms remained unaltered, as shown in the whole cell extracts (Fig. 5C). These findings suggest that hexadecenal induced-JNK activation induces apoptosis through cytochrome c release caused by tBid- and Bim-activated Bax.
Fig. 5. Hexadecenal induces apoptosis through Bid cleavage and Bax and Bim translocation.
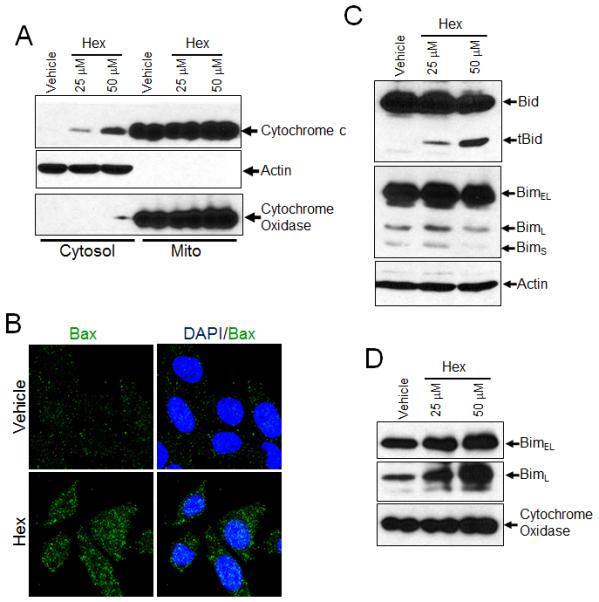
A, HEK293T cells treated with either vehicle or with the indicated dose of hexadecenal for 2 h. Cytosolic and mitochondrial fractions were immunoblotted with cytochrome c, cytochrome oxidase or actin antibody. B, HeLa cells were grown on chamber slides and treated with vehicle or hexadecenal (hex 25 μM) for 2 h. Cells were fixed and immunostained with a conformation-specific Bax antibody and counterstained with DAPI. Images were acquired using a laser scanning microscope 710 with a 63x oil objective. C, HEK293T cells treated with either vehicle or with the indicated dose of hexadecenal (hex) for 3 h. Whole cell extracts were immunoblotted with Bid, Bim or actin antibody. D, HEK293T cells treated with either vehicle or with indicated dose of hexadecenal for 2 h. Mitochondrial fractions were immunoblotted with Bim or cytochrome oxidase.
3.6 JNK regulates cell detachment and apoptosis mediated by hexadecenal
The members of the Rho-GTPase subfamily, Rho, Rac1 and Cdc42 are known to regulate the organization of the cytoskeleton [35]. Therefore, we examined the activation of Rac1 and Cdc42 after hexadecenal treatment. We found that neither Rac1 nor Cdc42 are activated by hexadecenal (data not shown). Consistent with this finding, pre-treatment of cells with either Rac1 or Rho inhibitors had no effect on the ability of hexadecenal to elicit cytoskeletal changes, apoptosis or JNK activation. Importantly, inhibition of JNK using a pharmacological inhibitor attenuated the cytoskeletal changes caused by hexadecenal (Fig. 6A). Further, pre-treatment with a JNK inhibitor prevented hexadecenal-induced apoptosis (Fig. 6B). Taken together, these findings suggest that hexadecenal induces detachment and apoptosis through a JNK-dependent pathway that is independent of Rac, Rho and Cdc42.
Fig. 6. Inhibition of JNK prevents hexadecenal-induced cytoskeletal reorganization and apoptosis.
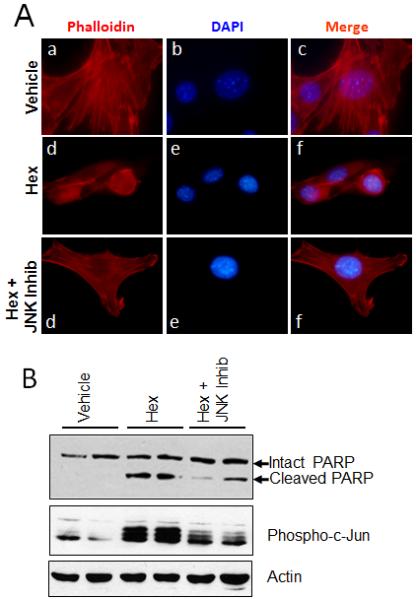
A, NIH3T3 cells were cultured in 8-chamber slides for 24 h. Cells were serum deprived for 3 h in the absence or presence of a JNK inhibitor V (JNK Inhib, 20 μM), then cells were treated with vehicle (a-c) or hexadecenal (25 μM) (d-i) for 30 min. F-actin was labeled with Phalloidin-Rhodamine and cells were counter stained with DAPI. B, Cells were cultured in 10-cm culture dishes for 24 h. Cells were serum deprived for 3 h in the presence or absence of a JNK inhibitor V (JNK inhib, 20 μM), then cells were treated with vehicle or hexadecenal (25 μM) for 3 h. Cells were harvested and whole cell extracts were immunoblotted with PARP, phospho-C-Jun or actin antibodies. Immunoblots are representative of at least three independent experiments.
4. Discussion
Our study provides evidence that the long chain aldehyde hexadecenal mediates cytoskeletal changes and induces apoptosis through a JNK-dependent signaling pathway. The JNK pathway is one of several MAPK signaling pathways that are activated in response to stressful cellular conditions including UV irradiation, ionizing radiation, osmotic shock or heat shock [24]. JNK is directly activated through phosphorylation by MKK4/7, which is itself activated by MAP kinase kinase kinases such as MLK3 [24,36]. Each step of this phosphorylation cascade appears to be induced in response to hexadecenal exposure. Disruption of microtubule structures by several anti-cancer agents induces apoptosis by activating MLK3 and JNK [37-39]. Thus, it is possible that the hexadecenal effect is mediated in this manner as well. However, we cannot exclude the possibility that hexadecenal might affect cell morphology via covalent reactions with cytoskeletal components, since fatty aldehydes are known to be highly reactive with amino groups present on proteins. In that regard, we note that the α,β-unsaturated long chain aldehyde 4-hydroxynonenal induces rapid disappearance of microtubule networks by causing adduct formation with tubulin [40,41]. Whether hexadecenal forms adducts with tubulin or other cytoskeleton proteins remains to be determined.
JNK can activate apoptotic signaling either through nuclear or mitochondrial signaling pathways [24]. Activated JNK translocates to the nucleus where it induces transcription of pro-apoptotic genes such as Bax and Bak, and decreases the expression of pro-survival genes including c-Jun [24]. Hexadecenal induces apoptosis within 3h of treatment, and it is unlikely that JNK induces the transcription of pro-apoptotic genes through c-Jun and other transcription factors within this time frame. Consistently, we did not observe any change in the expression levels of Bcl-2 family proteins such as Bcl-2, Bcl-xL, Mcl-1, Bax, Bik, Bok, Puma and Bad (data not shown). UV radiation-induced apoptosis requires Bax-mediated cytochrome c release [24,42]. Further, tBid and Bim promote Bax-mediated cytochrome c release [30]. BimL and BimEL normally remain sequestered in the cytosol by the dynein motor complex via an interaction with dynein light chain 1. In response to an apoptotic signal, phosphorylation of Bim by JNK dissociates Bim from the dynein motor-complex allowing Bim, along with LC8, to translocate to the mitochondria [32-34]. Their translocation induces the release of cytochrome c from the mitochondria by activating Bax. Our finding that hexadecenal treatment results in Bid cleavage and increased localization of Bim isforms BimL and BimEL in the mitochondria further suggests that hexadecenal induces apoptosis through a mechanism that depends upon JNK-mediated posttranslational protein modifications.
The routine recycling of complex sphingolipids and the activation of sphingomyelin metabolism by stressful conditions such as radiation and hypoxia are well-established phenomena [43,44]. Sphingolipid turnover generates bioactive metabolites that ultimately result in formation of S1P. Intracellular S1P catabolism by the enzyme SPL results in the formation of hexadecenal and ethanolamine phosphate [45]. Fatty aldehydes such as hexadecenal are metabolized by a microsomal enzyme, fatty aldehyde dehydrogenase (FALDH). Although under normal conditions hexadecenal levels are low, the toxic effects of fatty aldehydes are dramatically demonstrated in patients with Sjogren-Larsson syndrome. This heritable disorder is caused by mutations in the gene encoding FALDH [46,47]. Sjogren-Larsson patients lack FALDH activity and suffer from neurological and cognitive defects and ichthiosis thought to result from accumulation of fatty aldehydes and alcohols formed from metabolism of sphingolipids, fatty alcohols, leukotriene B4, ether glycerolipids and plasmalogens. Thus, we propose that FALDH deficiency may enhance the effects of hexadecenal on cells and tissues, and conversely that the symptoms of this disease could arise in part from accumulation of hexadecenal. Fatty aldehydes are also known to induce DNA damage in cells and are thereby thought to thereby contribute to the carcinogenic process [18]. Whether hexadecenal mediates DNA damage and/or other cellular effects associated with long chain aldehydes remains to be determined.
Hexadecenal can also be generated by non-enzymatic degradation of sphingolipids. For example, hexadecenal formation was noted after photolysis and radiolysis of sphingomyelin and lysosphingomyelin, respectively [48], which can result in high local concentrations of hexadecenal in the membrane. Lysosphingolipids such as d-erythro-sphingosylphosphorylcholine (SPC), sphingosine and S1P associate predominantly with high-density lipoproteins (HDL) in the plasma. HDL-associated lysosphingolipids have been suggested to mediate, in part, the atheroprotective effects of HDL [49]. Myeloperoxidase produced reactive chlorinating species have been shown to attack HDL-associated SPC and S1P, thereby generating hexadecenal [50]. Our findings elucidating the bioactivity of hexadecenal raise the possibility that degradation of circulating S1P to its fatty aldehyde metabolite may not only attenuate the S1P signal but may serve to antagonize the effects of S1P on vascular permeability, endothelial cell survival, and platelet functions. Thus, our findings represent a new paradigm in sphingolipid signaling in which the ultimate conversion step in the sphingolipid degradative pathway provides an additional opportunity for fine-tuning of the balance between cell survival and cell death.
5. Conclusion
Our studies demonstrate that trans-2-hexadecenal induces cellular detachment from substratum, cytoskeletal rearrangements and apoptosis in multiple mammalian cell types.
These effects are mediated through a JNK-dependent mechanism via generation of ROS and activation of MLK3.
Our cumulative results provide a new paradigm of sphingolipid signaling by demonstrating that S1P degradation generates a bioactive product that induces cell changes and programmed cell death through a stress-activated MAPK pathway.
Acknowledgements
This work was supported by NIH Grants CA77528 (to JDS) and NIH HL-083187 (to RB). Confocal images were acquired at the CHORI Microimaging Facility supported by NIH Grant S10RR025472 and the Children’s Hospital Branches, Inc.
Abbreviations
- DAPI
4′, 6-diamidino-2-phenylindole dihydrochloride hydrate
- FALDH
fatty aldehyde dehydrogenase
- HDL
high density lipoprotein
- HEK293T
human embryonic kidney cell line 293T
- JNK
c-Jun N-terminal kinase
- MKK4/7
MAP kinase kinase 4/7
- MLK3
mixed lineage kinase 3
- NAC
N-acetyl cysteine
- PARP
Poly (ADP-ribose) polymerase
- ROS
reactive oxygen species
- S1P
sphingosine-1-phosphate
- SPC
sphingosylphosphorylcholine
- SPL
sphingosine-1-phosphate lyase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Van Veldhoven PP. In: Sphingolipid Metabolism and Cell Signaling Part A. Merrill AH Jr., Hannun YA, editors. Academic Press; New York: 2000. pp. 244–254. [Google Scholar]
- [2].Serra M, Saba JD. Adv. Enzyme Regul. 2010;50:349–362. doi: 10.1016/j.advenzreg.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kumar A, Saba JD. Expert Opin. Ther. Tar. 2009;13:1013–1025. doi: 10.1517/14728220903039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Min J, Van Veldhoven PP, Zhang L, Hanigan MH, Alexander H, Alexander S. Mol Cancer Res. 2005;3:287–296. doi: 10.1158/1541-7786.MCR-04-0197. [DOI] [PubMed] [Google Scholar]
- [5].Li G, Foote C, Alexander S, Alexander H. Development. 2001;128:3473–3483. doi: 10.1242/dev.128.18.3473. [DOI] [PubMed] [Google Scholar]
- [6].Oskouian B, Sooriyakumaran P, Borowsky A, Crans A, Dillard-Telm L, Tam Y, Bandhuvula P, Saba J. Proc. Natl. Acad. Sci. U. S. A. 2006;103:17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schwab S, Pereira J, Matloubian M, Xu Y, Huang Y, Cyster J. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- [8].Oravecz T, Donoviel MS, Anderson SJ, Carson K, Sun W, Swaffield J, Liu Q, Kimball SD, Piggott JR, Main AJ, Zambrowicz BP, Sands AS, Turner CA, Augeri DJ. Annual Meeting of the American Society of Hematology; 2007. [Google Scholar]
- [9].Allende M, Bektas M, Lee B, Bonifacino E, Kang J, Tuymetova G, Chen W, Saba J, Proia R. J. Biol. Chem. 2010 doi: 10.1074/jbc.M110.171819. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhao Y, Gorshkova I, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk P, Pendyala S, Oskouian B, Saba J, Garcia J, Natarajan V. Am. J. Respir. Cell Mol. Biol. 2011 doi: 10.1165/rcmb.2010-0422OC. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kariya Y, Kihara A, Ikeda M, Kikuchi F, Nakamura S, Hashimoto S, Choi CH, Lee YM, Igarashi Y. Genes Cells. 2005;10:605–615. doi: 10.1111/j.1365-2443.2005.00862.x. [DOI] [PubMed] [Google Scholar]
- [12].Zhang K, Pompey J, Hsu F, Turk J, Bandhuvula P, Saba J, Beverley S. EMBO J. 2007;26:1094–1104. doi: 10.1038/sj.emboj.7601565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Forman H. Ann. N. Y. Acad. Sci. 2010;1203:35–44. doi: 10.1111/j.1749-6632.2010.05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Millar J. Annu. Rev. Entomol. 2000;45:575–604. doi: 10.1146/annurev.ento.45.1.575. [DOI] [PubMed] [Google Scholar]
- [15].Vardi A, Formiggini F, Casotti R, De Martino A, Ribalet F, Miralto A, Bowler C. PLoS Biol. 2006;4:e60. doi: 10.1371/journal.pbio.0040060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thukkani A, McHowat J, Hsu F, Brennan M, Hazen S, Ford D. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A. [DOI] [PubMed] [Google Scholar]
- [17].Guéraud F, Atalay M, Bresgen N, Cipak A, Eckl P, Huc L, Jouanin I, Siems W, Uchida K. Free Radic. Res. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- [18].Huang H, Kozekov I, Kozekova A, Wang H, Lloyd R, Rizzo C, Stone M. Environ. Mol. Mutagen. 2010;51:625–634. doi: 10.1002/em.20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Berdyshev E, Goya J, Gorshkova I, Prestwich G, Byun H, Bittman R, Natarajan V. Anal. Biochem. 2011;408:12–18. doi: 10.1016/j.ab.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gallo KA, Mark MR, Scadden DT, Wang Z, Gu Q, Godowski PJ. J. Biol. Chem. 1994;269:15092–15100. [PubMed] [Google Scholar]
- [21].Kumar A, Oskouian B, Fyrst H, Zhang M, Paris F, Saba J. Cell Death Dis. 2011 doi: 10.1038/cddis.2011.3. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dobrosotskaya I, Seegmiller A, Brown M, Goldstein J, Rawson R. Science. 2002;296:879–883. doi: 10.1126/science.1071124. [DOI] [PubMed] [Google Scholar]
- [23].Jaeschke A, Davis RJ. Mol. Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dhanasekaran DN, Reddy EP. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lo YY, Wong JM, Cruz TF. J. Biol. Chem. 1996;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- [26].Hong HY, Kim BC. Biochem. Biophys. Res. Commun. 2007;362:307–312. doi: 10.1016/j.bbrc.2007.07.165. [DOI] [PubMed] [Google Scholar]
- [27].Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S. J. Biol. Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- [28].Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. Mol. Cell. Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Upton JP, Valentijn AJ, Zhang L, Gilmore AP. Cell Death Differ. 2007;14:932–942. doi: 10.1038/sj.cdd.4402092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Green DR. Cancer Cell. 2006;9:328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- [31].Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. J. Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ley R, Hadfield K, Howes E, Cook SJ. J. Biol. Chem. 2005;280:17657–17663. doi: 10.1074/jbc.M412342200. [DOI] [PubMed] [Google Scholar]
- [33].Ley R, Ewings KE, Hadfield K, Cook SJ. Cell Death Differ. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- [34].Lei K, Davis RJ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaibuchi K, Kuroda S, Amano M. Annu. Rev. Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- [36].Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. J. Biol. Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- [37].Kolomeichuk SN, Terrano DT, Lyle CS, Sabapathy K, Chambers TC. FEBS J. 2008;275:1889–1899. doi: 10.1111/j.1742-4658.2008.06349.x. [DOI] [PubMed] [Google Scholar]
- [38].Xiao D, Pinto JT, Soh JW, Deguchi A, Gundersen GG, Palazzo AF, Yoon JT, Shirin H, Weinstein IB. Cancer Res. 2003;63:6825–6837. [PubMed] [Google Scholar]
- [39].Muller GJ, Geist MA, Veng LM, Willesen MG, Johansen FF, Leist M, Vaudano E. J. Neurochem. 2006;96:1242–1252. doi: 10.1111/j.1471-4159.2005.03590.x. [DOI] [PubMed] [Google Scholar]
- [40].Stewart BJ, Doorn JA, Petersen DR. Chem. Res. Toxicol. 2007;20:1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- [41].Kokubo J, Nagatani N, Hiroki K, Kuroiwa K, Watanabe N, Arai T. Cell Struct. Funct. 2008;33:51–59. doi: 10.1247/csf.07038. [DOI] [PubMed] [Google Scholar]
- [42].Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- [43].Hannun YA, Obeid LM. Nat. Rev. Mol. Cell Bio. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- [44].Kolesnick R, Fuks Z. Oncogene. 2003;22:5897–5906. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- [45].Stoffel W, Sticht G, LeKim D. Hoppe. Seylers Z. Physiol. Chem. 1968;349:1149–1156. doi: 10.1515/bchm2.1968.349.2.1149. [DOI] [PubMed] [Google Scholar]
- [46].James P, Zoeller R. J. Biol. Chem. 1997;272:23532–23539. doi: 10.1074/jbc.272.38.23532. [DOI] [PubMed] [Google Scholar]
- [47].Rizzo W. Mol. Genet. Metab. 2007;90:1–9. doi: 10.1016/j.ymgme.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lisovskaya IL, Shcherbachenko IM, Volkova RI, Ataullakhanov FI. Chem. Biol. Interact. 2009;180:433–439. doi: 10.1016/j.cbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- [49].Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F. J. Biol. Chem. 2001;276:31780–31785. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- [50].Brahmbhatt VV, Hsu FF, Kao JL, Frank EC, Ford DA. Chem. Phys. Lipids. 2007;145:72–84. doi: 10.1016/j.chemphyslip.2006.10.006. [DOI] [PubMed] [Google Scholar]