

Abstract
The HIV-1 accessory protein Nef is N-terminally myristoylated and this posttranslational modification is essential for Nef function in AIDS progression. Transfer of a myristate group from myristoyl coenzyme A to Nef occurs cotranslationally and is catalyzed by human N-myristoyl transferase-1 (NMT). To investigate the conformational effects of myristoylation on Nef structure as well as to probe the nature of the Nef: NMT complex, we investigated various forms of Nef with hydrogen exchange mass spectrometry. Conformational changes in Nef were not detected as a result of myristoylation and NMT had no effect on deuterium uptake by Nef in a myrNef:NMT complex. However, myrNef binding did have an effect on NMT deuterium uptake. Major HX differences in NMT were primarily located around the active site, with more subtle differences, at the longer timepoints, across the structure. At the shortest timepoint, significant differences between the two states were observed in two regions which interact strongly with the phosphate groups of coenzyme A. Based on our results, we propose a model of the Nef:NMT complex in which only the myristoyl moiety holds the two proteins together in complex and speculate that perhaps NMT chaperones Nef to the membrane and thereby protect the myristic acid group from the cytosol rather than Nef operating through a myristic acid switch mechanism.
Keywords: Hydrogen exchange, mass spectrometry, protein dynamics, myristoyl coenzyme A, myristoylation
A number of proteins live at the cytoplasmic face of biological membranes (1). These peripheral membrane proteins associate with membranes due to hydrophobic patches in the protein, and/or various types of anchors that keep the proteins at or near the membrane (2). Unlike integral membrane proteins, which are physically embedded in the membrane, most of the structure of peripheral membrane proteins is free to associate with other proteins at or near the membrane surface. Such interactions are important for many types of biological processes, including cellular signaling (3-4). One specific mechanism for targeting and anchoring proteins to biological membranes is the addition of myristic acid.
Myristic acid (C14:0) can be attached to the N-terminal glycine of both cellular and viral peripheral membrane protein substrates via an amide bond [reviewed in (5-6)]. The enzymes that carryout the attachment, N-myristoyl transferases (NMTs), add the myristic acid co-translationally. There are two mammalian isoforms of N-myristoyl transferases (NMT-1 and NMT-2) which have different specificities and are encoded by separate genes (7-8). Several NMT-1 homologs have been crystallized, including that from S. cervisiae (PDB 2NMT, 1IIC) (9-10), C. albicans (PDB 1NMT) (11) and H. sapiens (PDB 1RXT). The structural homology of NMT-1 between these diverse organisms has been maintained, however their substrate specificity (myristoylation consensus sequence) has diverged, making them an attractive target for fungicidal drugs (5).
Myristoylation is a weak membrane anchor and often membrane association requires additional forces such as charged sequences nearby or other proteins [reviewed in (6)]. In some proteins [e.g. Arf, (12)], a so-called myristoyl switch may occur in which the myr group is hidden from solvent or otherwise associated with the protein until it is necessary for anchoring in the membrane. The myr group is then exposed, a conformational change in the protein may/may not occur, and the protein becomes membrane associated via insertion of the myr group into the lipids of the membrane.
In this study, we wished to better understand the myristoylated form of the ~25 kDa HIV-1 Nef protein. HIV Nef is an essential factor for AIDS progression (13-16). Nef has no known catalytic activity but rather binds to a number of host-cell proteins (13, 17-19) to enhance viral replication, downregulate several endogenous cell surface receptors (MHC-1 and CD4) and activate resting T-cells. The N-terminal myristoylation sequence [Met1-Gly2-X3-X4-X5-(S/T)6], where X represents most amino acids, is 100% conserved in all functional nef alleles (20). In the current model, the myristate group is thought to act as a weak membrane anchor (21), which along with a basic cluster and several tryptophan residues in the N-terminal arm of Nef, localizes it to cellular membranes (22-23). Electrostatic attraction between the many basic residues in the N-terminus of Nef and an acidic lipid membrane are thought to drive Nef to first interact with the inner leaflet of the plasma membrane and then the myristate and several tryptophan residues insert into the membrane. These first two interactions comprise the fast association rate process. A much slower formation and insertion of an alpha helix into the membrane is believed to complete the process (24).
Two primary questions were addressed in the study presented here: can evidence of a myristoyl switch in Nef be found and what is the nature of the interaction between Nef and NMT. Several models (17-18) and some experimental evidence (25) have supported a myristoyl switch system for Nef but it remains unclear if conformational changes play a role in any potential switch. Using solution based hydrogen/deuterium exchange methods, we were unable to detect any conformational differences between a non-myristoylated form of SF2 Nef (hereafter referred to as nonMyrNef) when compared to the myristoylated form of SF2 Nef (MyrNef). Human NMT-1 (h-NMT-1, hereafter referred to as NMT) specifically myristoylates Nef and the two proteins are able to remain associated through immunoprecipitation experiments (26) and purification steps (see below). We characterized the NMT:MyrNef complex and compared the deuterium exchange into the proteins in the complex versus the protein when alone. The presence of NMT did very little to the conformation of MyrNef but NMT, on the other hand, was significantly affected by MyrNef.
Materials and Methods
Materials
Escherichia coli strain Rosetta2(DE-)pLysS was purchased from Merck Biosciences (Darmstadt, Germany); strain Codon Plus (DE-)-RIL was purchased from Agilent Technologies (Santa Clara, CA). Myristic acid, myristoyl coenzyme A, Bovine Serum Albumin (BSA), NaCl, Trizma HCl, glycerol and imidazole were purchased from Sigma-Aldrich (St. Louis, MO). Isopropylthioglactopyranoside (IPTG), ampicillin and chloramphenicol were purchased from Research Products International (Mt. Prospect, IL). TCEP was purchased from Thermo Scientific (Rockford, IL). Ni-NTA beads were purchased from QIAGEN (Hilden, Germany). Deuterium oxide (>99%), sodium deuteroxide and deuterium chloride were purchased from Cambridge Isotope Laboratories (Andover, MA).
Summary of protein expression and purification
Myristoylated SF2 Nef (MyrNef) and human N-myristoyl transferase-1 (h-NMT-1, referred to as NMT) were expressed using two different protocols. Since bacteria lack the necessary cellular enzymes to perform myristoylation, NMT was introduced into the expression host to accomplish the modification (27-29). At first, NMT was introduced using a two-vector system where DNA coding for Nef and NMT were on separate vectors each with different antibiotic selection (28-29). All proteins used in the hydrogen exchange MS experiments were made with this two-vector system. We also characterized expression and purification from a single Duet vector (30). In either expression method, ten minutes before inducting protein expression, the culture was supplemented with myristic acid in order for myristoylation to occur.
Expression and purification: Two vector system
A pET-14b vector containing codon-optimized, N-terminally 6xHis-tagged Nef SF2 [as in (28)] was obtained from T. Smithgall, University of Pittsburgh. Antibiotic resistance in this vector was afforded by ampicillin. For the expression of the C-terminally 6xHis-tagged Nef SF2, the Nef gene from the pET-14b Nef SF2 plasmid was amplified by PCR with NcoI and BamHI restriction sites at 5′ and 3′ end respectively. The reverse primer was designed to express a C-terminal thrombin cleavage site and 6xHis-tag for affinity chromatography. The PCR product was cloned into a pET-14b vector using NcoI and BamHI. The sequence of Nef in the final expressed protein had LVPRGSHHHHHH on the C-terminal end. A pET-30a vector containing DNA coding for residues 81-496 of human NMT-1 was also obtained from T. Smithgall. Antibiotic resistance in this vector was afforded by kanamycin. Both the SF2 Nef and h-NMT-1 vectors were cotransformed into Rosetta2 (DE-)pLysS and were maintained by dual ampicillin and kanamycin antibiotic selection.
The expression and Ni-affinity purification of Nef were carried out as previously reported (28, 31) with minor modifications. For expression, a fresh colony was selected and grown overnight in 50 mL Luria broth (LB) containing both ampicillin and kanamycin at 37 °C with shaking at 250 rpm. The following day, this starter culture was diluted to 500 mL LB with the same antibiotics and allowed to grow at 37 °C and shaking at 250 rpm until OD600 = 0.6-0.8. Ten minutes prior to induction with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), 12 mL of myristic acid solution (5 mM myristic acid, 0.6 mM BSA, pH 9, 60 °C) were added to the culture. After 4-6 hours of induction, cells were collected by centrifugation and stored at −80 °C until purification. For purification, a frozen cell pellet was resuspended in lysis buffer A (20 mM Tris, 100 mM NaCl, 20 mM imidazole, 10% glycerol, 3 mM DTT, pH 8.3) and lysed by sonication in the presence of phenylmethylsulphonyl fluoride (PMSF) and lysozyme. The lysate was clarified by centrifugation for 60 min at 16,000 rpm and 4 °C. Ni-NTA Agarose (QIAGEN) was added to the clarified lysate and mixed end-over-end at 4 °C for at least one hour. The bead slurry was loaded into a gravity flow column equipped with a UV detector (Pharmacia Biotech) and the beads washed extensively with lysis buffer A. The protein was eluted from the beads with elution buffer A (20 mM Tris, 100 mM NaCl, 200 mM imidazole, 10% glycerol, 3 mM DTT, pH 8.3).
The Ni-affinity eluate was immediately loaded onto a Superose 12 10/300 GL size exclusion column (GE Healthcare) connected to an AKTA FPLC (GE Healthcare/Amersham Pharmacia). The running buffer was 20 mM Tris, 100 mM NaCl, 3 mM DTT, pH 8.3. Fractions were collected across all peaks and analyzed with both SDS-PAGE and electrospray mass spectrometry to identify and characterize the purified protein. Although they could not be distinguished with electrophoresis, four versions of Nef could be seen with mass spectrometry (Supporting Information Figure S1). The myristoylated form (25553.3 Da) was typically the most intense peak. Some unmodified protein (25343.2 Da) was always present. Two other modifications, gluconoylation (+178 Da) and phosphogluconoylation (+258 Da), were also always seen in any culture where myristoylation was successful. N-terminal gluconoylation and phosphogluconoylation are known modifications in some N-terminally 6xHis tagged proteins expressed in E. coli (32). For this protein, however, the tag was moved to the C-terminus to allow for N-terminal myristoylation, and yet the chemical modifications persisted under the expression conditions used. Although the modification was not localized, we hypothesize that it was on the N-terminus, effectively blocking myristoylation on those proteins with gluconoylation and reducing the overall yield of myristoylated Nef. A fifth protein, later identified by mass spectrometry (see Supporting Information Figure S2) as h-NMT-1, was always present in the Ni-affinity eluate of any myristoylated Nef purification. Expression of myristoylated protein with the dual vector system resulted in approximately 10-fold less protein compared to the expression of the non-myristoylated form of the same protein.
In our hands, despite multiple attempts and contrary to what has been reported elsewhere (24, 28, 33), MyrNef was never purified without NMT being present (Figure 1A). High resolution mass spectrometry confirmed this fact in multiple purifications. Because MyrNef was always in complex with h-NMT-1, gel filtration chromatography was used to separate the MyrNef:NMT complex from the other Nef variants (unmodified, gluconoylated, and phosphogluconoylated) (Supporting Information Figure S2). In the gel filtration chromatogram, a small peak was present at the exclusion volume (MyrNef aggregate) followed by the MyrNef:NMT complex. Immediately after that the other Nef proteins eluted as one peak. The MyrNef:NMT complex (Figure 1A) was quite stable and could be concentrated in spin concentration devices and stored at −80 °C for up to four weeks.
Figure 1.

Purification of myristoylated SF2 Nef and h-NMT-1. (A) The MyrNef:NMT complex copurified in gel filtration as both species were seen in the mass spectrum (see also Supporting Information Figures 1, 2). (B) After the MyrNef:NMT complex was rebound to Ni-affinity beads, NMT was eluted with the addition of 10 μM myrCoA. (C) MyrNef was eluted from the Ni-affinity beads with imidazole. In these electrospray mass spectra, the masses of MyrNef (25553.5 Da) and NMT (48140.4 Da) match their theoretical mass values of 25554.0 Da and 48140.8 Da, respectively.
Those fractions that contained the MyrNef:NMT complex were pooled and either stored at −80 °C for later purification steps to separate the MyrNef:NMT complex or immediately concentrated with YM-10 Centricon devices (for analysis of the complex itself). For separation of MyrNef from NMT, an additional purification step was needed. Purified MyrNef:NMT complex (1 mL, 10 μM) was thawed on ice and Ni-NTA bead slurry (100 μL) was added and mixed for 1 hr at 4 °C to rebind the complex to the beads. The mixture was centrifuged at 300 × g for 2 min at 4 °C and the supernatant removed. The beads were washed two times each with 1 mL of lysis buffer A. NMT was competed off of MyrNef with 250 μL of 10 μM myristoyl coenzyme A in lysis buffer A. The sample was briefly centrifuged and the supernatant containing NMT removed, concentrated using spin concentration devices, characterized (see Figure 1B) and the NMT stored at −80 °C for later experiments. The remaining beads, which still were bound to MyrNef, were washed twice each with 1 mL of lysis buffer A and resuspended in 250 μL of elution buffer A to release MyrNef (Figure 1C). The sample was centrifuged and the supernatant containing MyrNef immediately used in HX MS experiments. In our hands, MyrNef, when not in complex with NMT, was very unstable and could not be concentrated or frozen.
Expression and purification: Duet Vector System
All hydrogen exchange MS experiments were completed using material prepared from the two-vector system. A paper appeared (30) describing a pET-Duet-1 vector containing both h-NMT-1 and SF2 Nef (C-terminal histidine tag) which efficiently expressed high levels of MyrNef and no copurification with h-NMT-1 or gluconoylation was reported. The pET-Duet-1 vector containing both h-NMT-1 (MCS1) and SF2 Nef (MCS2) was obtained from D. Willbold (30). The Duet vector was transformed into Codon Plus (DE-)-RIL cells using the manufacturer’s instructions. Expression in rich media was carried out as described previously (30).
The purification protocol followed that of (34) with minor modification. The frozen pellet was thawed on ice and resuspended in 10 mL of lysis buffer B (20 mM Tris, 500 mM NaCl, 20 mM imidazole, 15 mM BME, 1% Triton X-100, pH 8.0). Cells were lysed by sonication on ice in the presence of lysozyme and PMSF and then centrifuged (20,000 rpm, 4 °C, 1 hr). The supernatant of the clarified lysate was added to 0.5 mL of Ni-NTA bead slurry mixed end-over-end for 1 hour at 4 °C. The clarified lysate/Ni bead mixture was loaded into the gravity flow column/UV detector system as described above and washed with 25 mLs of lysis buffer B. Triton X-100 was removed by a second wash with 40 mLs of cholate buffer (20 mM Tris, 500 mM NaCl, 20 mM imidazole, 15 mM BME, 50 mM cholate, pH 8.0). Cholate was removed by another wash of 40 mLs of 20 mM Tris, 500 mM NaCl, 20 mM imidazole, 15 mM BME, pH 8.0). A step gradient (30 mM, 50 mM, 100 mM, 200 mM imidazole) was used to elute the protein from the beads. Nef was found to elute at around 100 mM imidazole. The fractions were checked with SDS-PAGE and electrospray mass spectrometry for purity and to confirm myristoylation. In rich media, the same expression pattern as seen in the dual vector system (gluconoylated, phosphogluconoylated, and MyrNef; as well as NMT copurification) was observed, with the exception of reduced levels of unmodified protein (Supporting Information Figure S1B). This reduction was a significant improvement over the two-vector system. However, the unwanted chemical modifications remained an obstacle to acquiring pure, MyrNef in a single purification.
The Duet vector expression system was also used in combination with modified M9 media to attempt to limit the amount of 6-phospho-gluconolactone available to modify the N-terminus of Nef. For expression in minimal media (modified M9: 200 mL 5 × M9 salts, 100 mL HEPES pH 7.4, 2 mL 1 M MgSO4, 0.1 mL 1 M CaCl2, 10 mL 10% casamino acids, 0.05% glucose per 1000 mL culture), a single colony was picked to inoculate a 50 mL starter culture in modified M9, and was grown overnight at 37 °C, with shaking at 250 rpm. The following morning, the starter culture was diluted to 500 mL with modified M9 and grown at 37 °C to OD600 = 0.6-0.8. The culture was moved to 16 °C and supplemented with 10 mL of myristic acid solution (as described above). Ten minutes later, IPTG was added to a 1 mM working concentration and the culture was allowed to express protein for 20 hours at 16 °C with shaking at 150 rpm. Cells were collected by centrifugation (3000 rpm, 4 °C) and the pellet stored at −20 °C until purification.
The combination of the Duet vector and modified M9 media resulted in high levels of MyrNef with significantly reduced levels of unmodified, gluconoylated and phospho-gluconoylated Nef (Supporting Information Figure S1C). The use of Triton X-100 in lysis reduced the amount of Nef lost in the pellet after centrifugation (data not shown). Expression of MyrNef from the Duet vector produced significantly more protein than the dual vector system. There was only a 2-fold reduction in expression levels of MyrNef when compared to expression of Nef alone from the Duet vector.
Intact protein mass spectrometry
Confirmation of myristoylation and the presence of the other Nef variants (unmodified, gluconoylated and phospho-gluconoylated) were determined by mass spectrometry (see also Supporting Information Figure S1, S2) as they could not be visualized or separated by SDS-PAGE. For each analysis, approximately 200 pmol of protein were injected and trapped on a self-packed POROS 20R2 trap (ABI Sciex) and manually desalted with 1 mL of Buffer A (ddH2O, 0.05% TFA). The protein was eluted into the mass spectrometer with a short (4 min) gradient of 15-75% acetonitrile, 0.05% TFA. Mass analyses were performed with a Waters LCT Premier mass spectrometer, equipped with a standard electrospray interface. Tof calibration was maintained by an infusion of horse heart apo myoglobin at the end of each injection.
Peptide level HX MS
Deuterium labeling methods were similar to those previously described for Nef (35). Briefly, single aliquots (10 μL, 50-100 pmol, equating to concentrations of 5-10 μM) of each protein sample were labeled by adding a 10-fold excess of 20 mM Tris, 100 mM NaCl, and 3 mM DTT in 99% deuterium oxide (pD 7.9). The labeling reactions were quenched at predetermined times by the addition of an equal volume of ice-cold 0.8 M guanidinium HCl, 0.8% formic acid, 100% H2O, pH 2.1 and the samples were placed on ice. Off-line pepsin digestion was used immediately after quenching to digest the protein into short peptides thereby allowing the localization of deuterium uptake. Pepsin (10 mg/mL in ddH2O, 0 °C) was added at a 1:1 ratio (w/w) pepsin:substrate to quenched samples (220 μL total volume) and allowed to incubate on ice for 5 min. The peptides were then injected and trapped on a self-packed POROS 20R2 trap (ABI Sciex) and manually desalted with 1 mL of ice cold Buffer A (ddH2O, 0.05% TFA, pH 2.5). The peptides were eluted from the trap at a flow rate of 50 μL/min onto a 100 mm × 0.25 mm (ID) reversed phase capillary perfusion HPLC column (POROS 10R2 media) or a 1.0 × 50 mm Zorbax 300SB-C18 reversed phase column (Agilent) and separated with a gradient of 5 to 40% acetonitrile, 0.05% TFA, pH 2.5 in 9 min. Valves, columns and tubing were submerged in an ice bath to maintain quench conditions throughout the injection, trapping and chromatographic separation (36). Mass analyses were performed with either a Waters Qtof API-US or LCT Classic mass spectrometer, both equipped with standard electrospray interfaces. Continuous lock-mass correction was carried out using Glu-fibrinogen peptide. Peptic peptides were identified on a Waters QTof premier mass spectrometer using MSE and Waters IdentityE software (37). Deuterium uptake for each peptide was calculated with HX-Express (38) software by subtracting the unlabeled centroid mass of each peptide from the centroid mass of the deuterium labeled peptides. The deuterium uptake was not corrected for back-exchange so the resulting plots are of the relative deuterium level (39).
HX MS data were interpreted on tertiary structures and/or models of each protein. For h-NMT-1, the yeast homolog structure [PDB 1IIC (10)] was chosen as the human crystal structure (PDB 1RXT) was lacking more of the N-terminus than the yeast structure. Overlaying these two structures showed very significant structural homology. The HIV Nef model (40) that was used is a combination of crystallographic data of the core domain of NL4-3 Nef [PDB 1EFN (41)], NMR of the N-terminus [PDB 1QA5 (42)] and modeling of the internal disordered loop; coordinates were provided by M. Geyer (40).
Results and Discussion
It has been proposed that MyrNef may adopt a different tertiary and quaternary structure than nonMyrNef (17, 28-29). Analytical gel filtration, ultracentrifugation, antibody binding and fluorescence spectroscopy indicated that MyrNef was smaller than nonMyrNef and had different accessibility in solution, leading to the conclusion that MyrNef had undergone a conformational change (28). Other studies including small angle x-ray scattering, dynamic light scattering and analytical ultracentrifugation indicated that MyrNef was more monomeric than nonMyrNef and that there were changes to the quaternary structure upon myristoylation (29). In a fluorescence assay, Nef was reported to bind to a peptide identical to the first six residues of its N-terminus only when that peptide was myristoylated, although the location of binding was not determined (25). The structural techniques (limited proteolysis, gel filtration, AUC, Trp fluorescence and fluorescence quenching) used to observe conformational changes upon myristoylation detect mainly global changes. To identify the location of the proposed conformational differences between MyrNef and nonMyrNef, we compared hydrogen exchange between both forms of the protein.
Utility of HX MS for analysis of Nef
HX MS has been reviewed previously (39, 43-45). Briefly, labile hydrogens in proteins are constantly in flux with hydrogens in solution; this exchange, however, is modulated by hydrogen bonding and solvent occlusion. As changes in protein structure alter both solvent accessibility and hydrogen bonding, hydrogen exchange is a very sensitive probe of protein structure. By measuring the deuterium uptake of different forms of the same protein with mass spectrometry, regions that undergo conformational changes, and hence have altered hydrogen exchange, can be determined. HX MS as a method has several characteristics that make it particularly well suited to study Nef: the sensitivity of the mass spectrometer allows for very little protein to be used per analysis (as little as 1.0-1.5 μgs) and the concentration can be quite dilute (< 1 μM). As Nef is prone to aggregation at higher concentrations, the use of dilute solutions ensured that the protein was monomeric (in all of the HX MS experiments in this work, the concentration of Nef never exceeded 10 μM). Preparation of large quantities of MyrNef is challenging but with such small sample requirements, a number of HX MS analyses could be performed with a small yield of purified material.
Myristoylated vs. non-myristoylated Nef
Previous HX MS work on Nef (35) used SF2 Nef with an N-terminal 6xHis purification tag. In order to study an N-terminally MyrNef, the purification tag first was moved to the C-terminus to allow for N-terminal myristoylation. Deuterium uptake in the intact protein, as well as at the peptide-level, was indistinguishable between the two variants (data not shown), demonstrating that the position of the purification tag had little or no effect on the conformation or dynamics of the Nef protein.
To determine the location of structural changes due to myristoylation, both myristoylated and non-myristoylated forms of Nef were overexpressed and purified. The proteins were labeled with deuterium, digested and analyzed by mass spectrometry as detailed in the Materials and Methods section. There were no detectable differences between the two forms, either at the C-terminus, the internal loops, or in the core of Nef (Supporting Information Figure S3). The bulk of the N-terminal arm also showed no differences in deuterium uptake. The only difference between nonMyrNef and MyrNef was manifested in the absolute N-terminus of Nef. The three myristoylated N-terminal peptides (encompassing residues 1-12, 1-14 and 1-17) acquired ~1 more deuterium than their non-myristoylated counterparts (Supporting Information Figure S3). We interpret this to mean the N-terminal myristoylation protected the N-terminal amide (on glycine) from back-exchange during analysis. It is understood (46) that the N-terminal amide of a polypeptide has a very fast exchange rate and that any deuterium incorporated in this position will be lost during HPLC. By myristoylating the N-terminus, the rate of exchange of this N-terminal amide was reduced such that deuterium exchange into this position was retained during analysis, resulting in the ~1 Da increase when compared to the non-myristoylated peptides. If the myristoylated N-terminus was interacting with another part of Nef (e.g., the core domain) those peptides would most likely exhibit significant protection from exchange and have less deuterium uptake. We did not observe any such protection. Based on these data, we conclude that in solution in the absence of lipid bilayers, and at concentrations less than 10 μM, there is no conformational difference between MyrNef and nonMyrNef. While conformational changes upon myristoylation seem to be consistent with the prior observations (17, 28-29), it remains possible, although unlikely in our view, that such changes could go undetected by HX MS. If the acyl chain is only making contact with hydrophobic sidechains of Nef residues, for example, it may not have changed the ability of the amide hydrogens in the N-terminal arm to exchange with deuterium in solution, although protection of the part of Nef interacting with the acyl chain would be expected. Another possible explanation is that conformational changes could still have occurred but the dynamics of the protein are sufficiently fast such that deuteration still takes place to the same extent by the first exchange timepoint (10 seconds). Alternatively, the changes observed by other techniques may be a result of the higher concentrations that were used in those other methods (generally in excess of 25 μM, and as high as 200 μM) and we are not able to see those differences below 10 μM. A way to test the effect of concentration would be to measure HX MS at several concentrations, starting at 10 μM and extending up to 200 μM. Although we have not done those experiments here, it would be interesting to determine if concentration plays a role in the ability of the myristoyl group to alter Nef conformation.
Conformational changes in Nef when bound to NMT
We next turned to characterization of the MyrNef:NMT complex. In order to determine the location of the interaction surface between MyrNef and NMT, and to elucidate any other conformational changes accompanying Nef binding to NMT, the deuterium exchange into MyrNef alone was compared to exchange into MyrNef when in complex with NMT. The hydrogen exchange rate is very sensitive to any changes in solvent accessibility and hydrogen bonding as result of complex formation and has been used to determine such information about many intra- and inter-protein associations (47). Surprisingly, there were no significant differences in deuterium uptake in MyrNef when bound to NMT versus when it was free in solution (Supporting Information Figure S4). All of the uptake curves were nearly on top of one another and the deuterium content at each individual timepoint was the same in both cases, within the error of the analysis. So although MyrNef binds very tightly to NMT and copurifies through cell lysis, affinity and gel filtration chromatography, the actual interaction between the two proteins does not have a detectable effect on the backbone amide hydrogen deuterium uptake of any regions in Nef, either right near the myristoylation site at the N-terminus or in any other regions of Nef. This result was somewhat surprising as in a prior study (26), N-terminal Nef fragments consisting of the first 30 or 68 amino acids were sufficient to precipitate NMT from cells, however less efficiently than precipitate of NMT with full-length Nef, suggesting that other elements in Nef besides the N-terminal arm also participate in interactions with NMT.
NMT conformation was affected by MyrNef binding
To be clear about the data that follow, it is useful to review the protein myristoylation pathway. The enzymatic reaction of protein myristoylation transfers the myristoyl moiety from myrCoA to the N-terminus of a protein in an ordered Bi-Bi reaction (48). The reaction begins when myrCoA binds NMT and makes NMT competent for binding the myristoylation consensus sequence of a target protein. The enzymatic reaction connecting myristic acid to the glycine at the N-terminus of the target protein occurs and CoA, no longer covalently attached to the acyl chain, is free to diffuse from the active site. Finally, in the rate limiting step, disassembly of the NMT-myrProtein complex occurs due to the loss of interactions from the pantetheine moiety of CoA with NMT’s Ab loop, resulting in structural changes in NMT (49).
In the HX MS experiments, MyrNef bound to NMT was compared to “free NMT” which was actually NMT in complex with myrCoA. MyrNef and NMT began their life in complex during translation and remained associated throughout cell lysis and purification. The acyl chain of myristic acid, now attached to Nef, was presumably still bound in the NMT active site while the CoA moiety had long ago diffused away. “Free NMT” was produced when high concentrations of myrCoA competed NMT off the complex and MyrNef remained bound via its 6xHis tag to the Ni-affinity resin.
A large number of NMT peptic peptides were produced and followed during the study of HX into NMT (see Supporting Information Figure S5). A summary of the differences in deuterium uptake between the two forms of NMT is shown in Figure 2 while all the deuterium uptake curves are shown in the Supporting Information Figure S6. As seen in Figure 2, there were specific regions where exchange in NMT was different between when it was bound to MyrNef versus myrCoA (see also Supporting Information Figure S7). The difference indicies (DI(1)/(2)) (50) for this comparison were 54/52. Several regions of NMT, encompassing residues 40-89, 170-185 and 360-413 (the carboxy-terminus of NMT), showed significant levels of deprotection when MyrNef was bound compared to when myrCoA was bound. The locations of the differences were plotted onto the crystal structure of NMT (Figure 3, see also Supporting Information Movie S1). Altered exchange was predominantly located in the region of the NMT active site. At the shortest labeling timepoint (10 sec), several peptides in the MyrNef-bound state showed increases in deuterium uptake greater than 2 Da when compared to the myrCoA-bound state. Such differences are likely the result of the loss of interactions between NMT and CoA. As CoA was no longer in the active site, the hydrogen bonds which were holding the CoA moiety in position were no longer present, allowing these positions to become deuterated much more quickly. At the longer periods in deuterium, there were increases in deuterium uptake in the MyrNef-bound state of NMT across the structure. By the 30 min timepoint, MyrNef-bound NMT saw increases in deuterium very distant from the myrCoA binding pocket. Either the binding of MyrNef, or the loss of hydrogen bonds to CoA were communicated through NMT resulting in increased flexibility throughout the NMT structure which ultimately led to increased deuterium uptake.
Figure 2.

Difference in deuterium uptake between NMT in complex with MyrNef versus NMT alone (NMT+myrCoA). The relative difference plot shows changes for five time points: 10 sec (orange), 5 min (red), 30 min (cyan), 2 hr (blue), and 4 hr (black) in D2O. The residues of each peptide (see also Supporting Information Figures S5-S7) are shown on the left. Data to the left of zero indicate MyrNef-induced protection, data to the right indicate MyrNef-induced deprotection. These data are an average of two independent experiments. The gray dashed lines indicate the range of significant different, i.e. data points within the dashed lines are not significantly different [see (50) for detailed explanation of this type of graphical comparison].
Figure 3.

The location of differences in deuterium exchange in NMT. The differences summarized in Figure 2 (see full dataset in Supporting Information Figure S6) were mapped onto the crystal structure of yeast NMT [PDB 1IIC (10)] for the four time points indicated. Differences in deuterium uptake >2.0 Da were classified as a major difference (red), between 0.5 and 2.0 Da as a minor difference (yellow), and differences <0.5 Da were classified as no difference (light blue). Regions where deuterium uptake could not be monitored (gray) and portions of the yeast NMT structure that is not present (green) in the human NMT-1 construct in these experiments are also indicated. Myristic acid is shown in purple. The four timepoints in this figure constitute the frames of Movie S1 in the Supporting Information.
The most dramatic changes in deuteration were found in the myristic acid binding pocket of NMT. The peptic peptide encompassing NMT residues 170-185 showed a major difference in deuterium uptake between the two states across all timepoints (Figure 4). Based on the crystal structure, this peptide has five amide hydrogens (green balls, Figure 4C) which actively bond to the phosphate groups in the pyrophosphate moiety of myrCoA (49). In the myrCoA-bound state these hydrogens would be protected from exchange resulting in less deuterium uptake in this peptide. After the enzymatic reaction coupling myristic acid to the Nef, CoA can diffuse out of the active site. In MyrNef, these same five amide hydrogens would now be exposed to solvent and could readily exchange with deuterium. Without accounting for back-exchange (see Materials and Methods), there was a ~4 Da increase in the MyrNef-bound state at the 10 sec timepoint, which when ~18-25% backexchange in this experimental HX MS system is considered, is in good agreement with the five amide hydrogens predicted to be available for exchange when CoA is not present.
Figure 4.

Deuterium uptake in the NMT myristoyl coenzyme A binding pocket. (A) Mass spectra of the peptide covering residues 170-185, m/z 616.39 (+3), for both the myrCoA- and MyrNef-bound states. (B) Mass spectra of the peptide covering residues 40-50, m/z 635.32 (+2), for both the myrCoA- and MyrNef-bound states. In panels A and B, the amount of time in D2O is indicated in the center and the dashed red line, meant as a visual reference, is drawn through the centroid value of the isotope distribution of each peptide at the myrCoA-bound 4 hour time point. (C) Structural location and deuterium uptake of the two peptides from panels A and B. Peptide 170-185 is shown in green and 40-50 in purple. Myristoyl coenzyme A and the myristoyl moiety of MyrNef are indicated. Backbone amide hydrogens which hydrogen bond to the phosphate groups of myrCoA are shown as space filling balls. The deuterium uptake curves for these two peptides, as measured from the spectra in panels A and B, are the average of two independent experiments (error bars are set to ±0.5 Da). The myrCoA-bound state is shown in blue and the MyrNef-bound state in red. See also Supporting Information Figures S5 and S6.
The peptide encompassing NMT residues 40-50 showed a change in dynamics between the two bound states (Figure 4). Based on the X-ray structure (10), in this peptide one amide hydrogen could be bonded to the adenosine 3′-phosphate and two more amide hydrogens adjacent to the N-terminus of this peptide (Q38 and F39, see also Supporting Information Figure S5) may form hydrogen bonds to the 3′-phosphate (49). With the CoA missing (the MyrNef-bound state), this peptide was no longer tethered to CoA through the hydrogen bonds of residues 38-40, was exposed to solvent, and therefore became quickly deuterated (to ~5.5 Da not accounting for back-exchange). When myrCoA was bound in the active site (“free NMT”), this peptide presumably associated with CoA, resulting in the protection seen in the uptake curve for residues 40-50 in Figure 4C. However, by 30 minutes in deuterium, both forms had the same amount of deuteration which indicates that this region of NMT is quite dynamic, that the bonds protecting this region from deuteration are transient, or both.
Conclusions
The hydrogen exchange data presented here correspond to a situation in which proteins in complex lack significant protection versus proteins alone. While HX MS is able to detect protein:protein interactions by defining the surface in which solvent has been occluded (47), no such interface was observed for the MyrNef-NMT complex. With two reasonably large proteins (25.5 kDa for MyrNef and 48.1 kDa for h-NMT-1), it is unlikely that a potential large interaction surface would go undetected by HX MS. The driving force that keeps these two proteins together may be the protection of the myristoyl moiety of Nef by the binding pocket of h-NMT-1. Considering the hydrophobic nature of the myristic acid acyl chain, high exposure to aqueous solvent would not be favored. Given the apparent lack of protection of the myristoylated N-terminus by Nef itself, the complex is maintained by the stabilization of the acyl chain in the binding pocket of NMT. Such a complex between MyrNef and NMT is shown in Figure 5. In this model the sole interaction maintaining the MyrNef-NMT complex is the binding of the myristoyl moiety of Nef in the binding pocket of h-NMT-1. The N-terminal arm of Nef acts as a tether connecting h-NMT-1 to the globular core domain of Nef.
Figure 5.
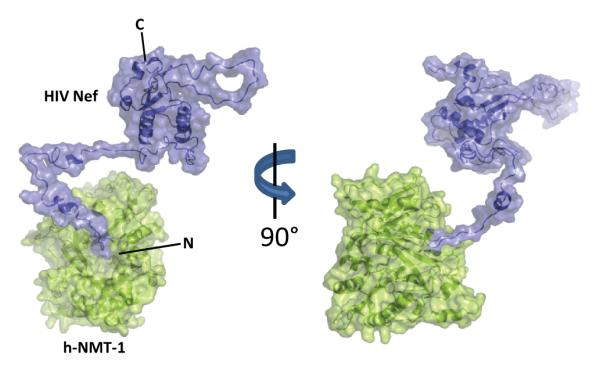
Model depicting h-NMT-1 (green) and myristoylated Nef (blue). PDB 1IIC (10) was used for NMT and a MyrNef model (40) was used for Nef. This model shows a complex which lacks a significant interaction surface and is driven primarily by the myristoyl moiety of Nef remaining in the active site of h-NMT-1. The N- and C-termini of Nef are shown.
Major differences in h-NMT-1 deuterium uptake were primarily located around the active site, with more subtle differences, at the longer timepoints, across the structure. At the shortest timepoint, significant differences between the two states were seen in two peptides which clearly interact strongly with the phosphate groups of CoA. This significant increase in deuterium uptake is directly related to the loss of protein-CoA interactions in the MyrNef-bound state. No differences were detected in Nef due to either myristoylation or binding to NMT. Our findings suggest that if there is a closed conformation of MyrNef as has been proposed (17, 28-29), the N-terminal arm is still highly solvent exposed, no structure forms in the absence of membranes, and the acyl chain of the myristic acid makes contact with a region on the body of Nef that does not cause backbone amide hydrogens exchange rates to change. Given that the interaction of the acyl chain with NMT caused very obvious and measurable changes in HX, we would expect to see a change in HX in parts of Nef where the myristic acid made contact. Perhaps the interaction is via a hydrophobic patch on the core of Nef and this hydrophobic interaction does not alter backbone HX rates. .
While Nef functions at the membrane, a significant fraction of Nef is localized in the cytoplasm (most likely a function of the membrane localization being driven by the weakest of membrane anchors, myristoylation). Transit from the ribosome to the inner side of the plasma membrane is required for Nef to function, as many of its binding partners are at the plasma membrane. Rather than utilizing a myristic acid switch in which the N-terminal myristic acid is protected from the cytoplasm by the protein itself, perhaps Nef utilizes another mechanism in which NMT participates in protecting the myristoyl moiety while accompanying Nef to the membrane. As we only were able to purify myrNef as complex with NMT, there is clearly a strong interaction between the two proteins that may likely persist in the cytoplasm. More detailed cellular assays will be required to validate this NMT-Nef chaperone theory.
Supplementary Material
Acknowledgements
We gratefully acknowledge P. Narute and T. Smithgall, University of Pittsburgh, for supplying the SF2 Nef and h-NMT-1 plasmids, J. Glück and D. Willbold, Forschungszentrum Jülich, for providing the pET Duet 1 Nef/NMT vector, and M. Geyer, MPI-Dortmund, for sharing the HIV Nef model coordinates. This is contribution 983 from the Barnett Institute.
Abbreviations
- NMT
Human N-myristoyl transferase
- nonMyrNef
non-myristoylated Nef
- myrNef
myristoylated Nef
- myrCoA
myristoyl coenzyme A
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- BSA
bovine serum albumin
- PMSF
phenylmethylsulphonyl fluoride
- HX
hydrogen exchange
- MS
mass spectrometry
- TFA
trifluoroacetic acid
- DTT
dithiothreitol
Footnotes
This work was supported by funding from the NIH (R01-GM086507) and a research collaboration with the Waters Corporation.
Supporting Information Available
Supporting information accompanies this paper in the form of seven supplemental figures (Figures S1-S7) and an animation (Movie S1). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Seaton BA, Roberts MF. Peripheral membrane proteins. In: Merz KM, Roux B, editors. Biological Membranes. Birkhauser; Boston, MA: 1996. pp. 355–403. [Google Scholar]
- 2.Walsh CT. Posttranslational modification of proteins. Roberts and Co.; Englewood, CO: 2006. [Google Scholar]
- 3.Patwardhan P, Resh MD. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol Cell Biol. 2010;30:4094–4107. doi: 10.1128/MCB.00246-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bijlmakers MJ. Protein acylation and localization in T cell signaling (Review) Mol Membr Biol. 2009;26:93–103. doi: 10.1080/09687680802650481. [DOI] [PubMed] [Google Scholar]
- 5.Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J Biol Chem. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- 6.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 7.Giang DK, Cravatt BF. A second mammalian N-myristoyltransferase. J Biol Chem. 1998;273:6595–6598. doi: 10.1074/jbc.273.12.6595. [DOI] [PubMed] [Google Scholar]
- 8.Glover CJ, Felsted RL. Identification and characterization of multiple forms of bovine brain N-myristoyltransferase. J Biol Chem. 1995;270:23226–23233. doi: 10.1074/jbc.270.39.23226. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar RS, Futterer K, Farazi TA, Korolev S, Murray CL, Jackson-Machelski E, Gokel GW, Gordon JI, Waksman G. Structure of N-myristoyltransferase with bound myristoylCoA and peptide substrate analogs. Nat Struct Biol. 1998;5:1091–1097. doi: 10.1038/4202. [DOI] [PubMed] [Google Scholar]
- 10.Farazi TA, Waksman G, Gordon JI. Structures of Saccharomyces cerevisiae N-myristoyltransferase with bound myristoylCoA and peptide provide insights about substrate recognition and catalysis. Biochemistry. 2001;40:6335–6343. doi: 10.1021/bi0101401. [DOI] [PubMed] [Google Scholar]
- 11.Weston SA, Camble R, Colls J, Rosenbrock G, Taylor I, Egerton M, Tucker AD, Tunnicliffe A, Mistry A, Mancia F, de la Fortelle E, Irwin J, Bricogne G, Pauptit RA. Crystal structure of the anti-fungal target N-myristoyl transferase. Nat Struct Biol. 1998;5:213–221. doi: 10.1038/nsb0398-213. [DOI] [PubMed] [Google Scholar]
- 12.Ames JB, Tanaka T, Stryer L, Ikura M. Secondary structure of myristoylated recoverin determined by three-dimensional heteronuclear NMR: implications for the calcium-myristoyl switch. Biochemistry. 1994;33:10743–10753. doi: 10.1021/bi00201a023. [DOI] [PubMed] [Google Scholar]
- 13.Arhel NJ, Kirchhoff F. Implications of Nef: host cell interactions in viral persistence and progression to AIDS. Curr Top Microbiol Immunol. 2009;339:147–175. doi: 10.1007/978-3-642-02175-6_8. [DOI] [PubMed] [Google Scholar]
- 14.Das SR, Jameel S. Biology of the HIV Nef protein. Indian J Med Res. 2005;121:315–332. [PubMed] [Google Scholar]
- 15.Baur A. Functions of the HIV-1 Nef protein. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:309–313. doi: 10.2174/1568008043339749. [DOI] [PubMed] [Google Scholar]
- 16.Trono D. HIV accessory proteins: leading roles for the supporting cast. Cell. 1995;82:189–192. doi: 10.1016/0092-8674(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 17.Arold ST, Baur AS. Dynamic Nef and Nef dynamics: how structure could explain the complex activities of this small HIV protein. Trends Biochem Sci. 2001;26:356–363. doi: 10.1016/s0968-0004(01)01846-1. [DOI] [PubMed] [Google Scholar]
- 18.Fackler OT, Baur AS. Live and let die: Nef functions beyond HIV replication. Immunity. 2002;16:493–497. doi: 10.1016/s1074-7613(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 19.Saksela K. Therapeutic targeting of interactions between Nef and host cell proteins. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:315–319. doi: 10.2174/1568008043339776. [DOI] [PubMed] [Google Scholar]
- 20.Harris M. The role of myristoylation in the interactions between human immunodeficiency virus type I Nef and cellular proteins. Biochem Soc Trans. 1995;23:557–561. doi: 10.1042/bst0230557. [DOI] [PubMed] [Google Scholar]
- 21.Murray D, Hermida-Matsumoto L, Buser CA, Tsang J, Sigal CT, Ben-Tal N, Honig B, Resh MD, McLaughlin S. Electrostatics and the membrane association of Src: theory and experiment. Biochemistry. 1998;37:2145–2159. doi: 10.1021/bi972012b. [DOI] [PubMed] [Google Scholar]
- 22.Welker R, Harris M, Cardel B, Krausslich HG. Virion incorporation of human immunodeficiency virus type 1 Nef is mediated by a bipartite membrane-targeting signal: analysis of its role in enhancement of viral infectivity. J Virol. 1998;72:8833–8840. doi: 10.1128/jvi.72.11.8833-8840.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaminchik J, Margalit R, Yaish S, Drummer H, Amit B, Sarver N, Gorecki M, Panet A. Cellular distribution of HIV type 1 Nef protein: identification of domains in Nef required for association with membrane and detergent-insoluble cellular matrix. AIDS Res Hum Retroviruses. 1994;10:1003–1010. doi: 10.1089/aid.1994.10.1003. [DOI] [PubMed] [Google Scholar]
- 24.Gerlach H, Laumann V, Martens S, Becker CF, Goody RS, Geyer M. HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat Chem Biol. 2010;6:46–53. doi: 10.1038/nchembio.268. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann S, Jonas E, Konig S, Preusser-Kunze A, Willbold D. Nef protein of human immunodeficiency virus type 1 binds its own myristoylated N-terminus. Biol Chem. 2007;388:181–183. doi: 10.1515/BC.2007.020. [DOI] [PubMed] [Google Scholar]
- 26.Hill BT, Skowronski J. Human N-myristoyltransferases form stable complexes with lentiviral nef and other viral and cellular substrate proteins. J Virol. 2005;79:1133–1141. doi: 10.1128/JVI.79.2.1133-1141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duronio RJ, Jackson-Machelski E, Heuckeroth RO, Olins PO, Devine CS, Yonemoto W, Slice LW, Taylor SS, Gordon JI. Protein N-myristoylation in Escherichia coli: reconstitution of a eukaryotic protein modification in bacteria. Proc Natl Acad Sci U S A. 1990;87:1506–1510. doi: 10.1073/pnas.87.4.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breuer S, Gerlach H, Kolaric B, Urbanke C, Opitz N, Geyer M. Biochemical indication for myristoylation-dependent conformational changes in HIV-1 Nef. Biochemistry. 2006;45:2339–2349. doi: 10.1021/bi052052c. [DOI] [PubMed] [Google Scholar]
- 29.Dennis CA, Baron A, Grossmann JG, Mazaleyrat S, Harris M, Jaeger J. Co-translational myristoylation alters the quaternary structure of HIV-1 Nef in solution. Proteins. 2005;60:658–669. doi: 10.1002/prot.20544. [DOI] [PubMed] [Google Scholar]
- 30.Gluck JM, Hoffmann S, Koenig BW, Willbold D. Single vector system for efficient N-myristoylation of recombinant proteins in E. coli. PLoS One. 2010;5:e10081. doi: 10.1371/journal.pone.0010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trible RP, Emert-Sedlak L, Wales TE, Ayyavoo V, Engen JR, Smithgall TE. Allosteric loss-of-function mutations in HIV-1 Nef from a long-term non-progressor. J Mol Biol. 2007;374:121–129. doi: 10.1016/j.jmb.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geoghegan KF, Dixon HB, Rosner PJ, Hoth LR, Lanzetti AJ, Borzilleri KA, Marr ES, Pezzullo LH, Martin LB, LeMotte PK, McColl AS, Kamath AV, Stroh JG. Spontaneous alpha-N-6-phosphogluconoylation of a “His tag” in Escherichia coli: the cause of extra mass of 258 or 178 Da in fusion proteins. Anal Biochem. 1999;267:169–184. doi: 10.1006/abio.1998.2990. [DOI] [PubMed] [Google Scholar]
- 33.Szilluweit R, Boll A, Lukowski S, Gerlach H, Fackler OT, Geyer M, Steinem C. HIV-1 Nef perturbs artificial membranes: investigation of the contribution of the myristoyl anchor. Biophys J. 2009;96:3242–3250. doi: 10.1016/j.bpj.2008.12.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayburt TH, Grinkova YV, Sligar SG. Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Letters. 2002;2:853–856. [Google Scholar]
- 35.Hochrein JM, Wales TE, Lerner EC, Schiavone AP, Smithgall TE, Engen JR. Conformational features of the full-length HIV and SIV Nef proteins determined by mass spectrometry. Biochemistry. 2006;45:7733–7739. doi: 10.1021/bi060438x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol Cell Proteomics. 2006;5:589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 39.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 40.Geyer M, Fackler OT, Peterlin BM. Structure--function relationships in HIV-1 Nef. EMBO Rep. 2001;2:580–585. doi: 10.1093/embo-reports/kve141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH-domain. Cell. 1996;85:931–942. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- 42.Geyer M, Munte CE, Schorr J, Kellner R, Kalbitzer HR. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J Mol Biol. 1999;289:123–138. doi: 10.1006/jmbi.1999.2740. [DOI] [PubMed] [Google Scholar]
- 43.Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 44.Smith DL, Deng Y, Zhang Z. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 45.Tsutsui Y, Wintrode PL. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr Med Chem. 2007;14:2344–2358. doi: 10.2174/092986707781745596. [DOI] [PubMed] [Google Scholar]
- 46.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 47.Engen JR. Analysis of protein complexes with hydrogen exchange and mass spectrometry. Analyst. 2003;128:623–628. doi: 10.1039/b212800b. [DOI] [PubMed] [Google Scholar]
- 48.Farazi TA, Manchester JK, Waksman G, Gordon JI. Pre-steady-state kinetic studies of Saccharomyces cerevisiae myristoylCoA:protein N-myristoyltransferase mutants identify residues involved in catalysis. Biochemistry. 2001;40:9177–9186. doi: 10.1021/bi0107997. [DOI] [PubMed] [Google Scholar]
- 49.Bhatnagar RS, Futterer K, Waksman G, Gordon JI. The structure of myristoyl-CoA:protein N-myristoyltransferase. Biochim Biophys Acta. 1999;1441:162–172. doi: 10.1016/s1388-1981(99)00155-9. [DOI] [PubMed] [Google Scholar]
- 50.Houde D, Berkowitz SA, Engen JR. The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J Pharm Sci. 2011 doi: 10.1002/jps.22432. in press, DOI 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.