

Summary
von Willebrand factor (VWF) is a multimeric glycoprotein that mediates platelet adhesion and is decreased in von Willebrand disease (VWD). 1-8 deamino-d-arginine vasopressin (DDAVP), the most common treatment for VWD, is limited by tachyphylaxis and inconvenience, and in 20% of the patients, unresponsiveness. Recombinant human interleukin-11 (rhIL-11), a gp-130 signalling cytokine with haematopoietic and anti-inflammatory activity, increases VWF antigen and its activity in heterozygous VWF+/− mice and dogs. To determine the biological efficacy and safety of rhIL-11 in non-bleeding human subjects with mild VWD, we conducted a phase II prospective open-label trial of rhIL-11 at 10, 25 and 50 μg kg−1 subcutaneously (s.c.), given daily for 7 days in nine subjects with mild VWD. VWF and factor VIII (FVIII) levels increased gradually and progressively after s.c. rhIL-11, which was sustained through 7 days of dosing to 1.5- to 3-fold over baseline. Following intravenous DDAVP, 0.3 μg kg−1, on day 7 there was a further boost in VWF and FVIII levels, suggesting that the mechanism of rhIL-11 differs from that of DDAVP. Platelet VWF mRNA expression measured by quantitative PCR increased from two- to eightfold over baseline, suggesting that the mechanism of rhIL-11 effect may be upregulation of VWF mRNA. VWF and FVIII levels returned to baseline by day 14. rhIL-11 was well tolerated with less than grade-1 hypertension, hypokalaemia and fluid retention. Recombinant IL-11 increases VWF levels in humans with mild VWD, justifying future clinical trials to determine its potential in preventing or reducing bleeding in this patient population.
Keywords: recombinant interleukin-11, von Willebrand disease, von Willebrand factor
Introduction
von Willebrand factor (VWF) is a multimeric plasma glycoprotein that mediates platelet adhesion to vascular endothelium and serves as a carrier molecule for FVIII [1,2]. Decreased VWF results in von Willebrand disease (VWD), a congenital bleeding disorder affecting 1% of the population [3], characterized by mucosal bleeding, including epistaxis, menorrhagia, postoperative and excessive post-traumatic bleeding [4,5]. Current treatment with DDAVP, which acts by stimulating release of VWF from endothelial stores in Weibel-Palade bodies, is efficacious but limited by tachyphylaxis, inconvenience of intravenous (i.v.) infusion especially in women with menorrhagia, the potential to induce hyponatraemia in children and unresponsiveness in 15–20% of patients [5-7]. Plasma-derived VWF concentrate, an alternative to DDAVP, is limited by higher cost and transmissible infectious agent risk [8]. Thus, development of safer, more effective products and alternative treatment strategies are goals for improving treatment of VWD [9,10]. Recombinant human interleukin 11 (rhIL-11, Neumega®; Wyeth Pharmaceuticals, Collegeville, PA, USA), a gp-130 signalling cytokine with haematopoietic and anti-inflammatory activity, currently FDA-approved for chemotherapy treatment of thrombocytopenia [11,12], has recently been shown to increase VWF antigen and activity with good tolerance in mice, dogs and healthy volunteers, and in heterozygous VWF+/− mice and dogs [13-15]. In the latter, the VWF increase was sustained over a range of doses (25, 50 and 75 μg kg day−1), for as long as the drug was given [14,15], suggesting the existence of a mechanism other than endothelial release [14,15], and the potential of rhIL-11 as an alternative agent to treat VWD. Taken together, these data suggest that rhIL-11 is a candidate as an alternative agent to treat human VWD. We therefore, conducted a prospective open-label Phase II study of rhIL-11 in non-bleeding subjects with mild VWD to test its biological efficacy and safety.
Materials and methods
Study subjects
Following signed informed consent, nine subjects 18 years of age or older with mild VWD, classified as type 1 VWD by recently published NHLBI criteria [10], were enroled in and completed the trial. Type 1 VWD was defined by the presence of a bleeding history; an abnormal bleeding severity score (BS) [8], low VWF:RCo, and normal distribution of VWF multimers [10]. All subjects had previous evidence of biological response to DDAVP, with correction by VWF tests into the normal range. One subject, Pt 7, with von Willebrand ristocetin cofactor (VWF:RCo) above 0.50 U mL−1, had a bleeding severity score of 6 and belonged to a large VWD kindred with affected siblings and children, all with significant bleeding, and thus was included, consistent with NHLBI criteria [10] as well as those in the genotype-phenotype studies in Canada [16] and Europe [17]. The reduced VWF:RCo/VWF:Ag ratios in Pt. 1, 2, 3, 7, 8 and 9 are also consistent with, but not diagnostic of type 2M VWD [10,16,17]. The presence of any of the other bleeding disorders, cardiac disease, congestive failure, arrhythmia, hypertension, myocardial infarction, stroke, thrombosis or past allergic reaction to rhIL-11 or DDAVP, or surgery within 8 weeks prior to study constituted the exclusion criteria. Concomitant use of immunomodulatory or experimental drugs, diuretics, antiplatelet drugs, anticoagulants, dextran, aspirin and non-steroidal anti-inflammatory drugs were not allowed, nor was treatment with DDAVP, cryoprecipitate, whole blood, plasma or plasma derivatives containing FVIII or VWF, within 5 days of study. Females who were pregnant or lactating were excluded from the study, and no hormone, oral contraceptive or other oestrogen use in the past 8 weeks was allowed. Subjects recruited for this study included the first nine subjects who met all entry criteria were willing to participate in this time-intensive study, and who in the judgement of the investigators were able to comply with all study requirements. All subjects provided signed informed consent in accordance with the Declaration of Helsinki. The protocol and informed consent documents were approved by the Clinical Translational Research Center (CTRC) Advisory Committee [formerly General Clinical Research Center (GCRC)] and the Institutional Review Board (IRB) at the University of Pittsburgh.
Study design and treatment schedule
This was a Phase II prospective open-label, escalating dose trial of rhIL-11. Three consecutive subjects each were scheduled to receive rhIL-11 at 25, 50 and 75 μg kg−1 subcutaneously (s.c.) daily for 7 days (days 1–7), followed by DDAVP at 0.3 μg kg−1 intravenously 30 min after rhIL-11 on day 7. Because coagulation levels, including VWF:RCo, VWF antigen (VWF:Ag), FVIII clotting activity (FVIII:C) and FVIII antigen (FVIII:Ag), increased up to fourfold over baseline at the 50 μg kg−1 dose level, with no toxicity, the 75 μg kg−1 dose level was removed; after discussion with the Data Safety Monitoring Board, a 10 μg kg−1 dose level was added to establish a dose–response relationship and avoid any potential toxicity at higher dose levels. Thus, the final three subjects received rhIL-11 at 10 μg kg−1 according to the schedule above.
A medical history, physical examination, vital signs and fluid status assessment (height, weight, cardiac-, lung-and fundoscopic exam) were performed at baseline and before and 30 min and 4 h after daily rhIL-11. The screening visit (day 0) and final visit (day 14) were carried out at the Hemophilia Center of Western PA, and the biological effects study was carried out at the Montefiore University Hospital Clinical & Translational Research Center (MUH-CTRC), formerly the GCRC. Laboratory studies drawn at baseline and on day 1 (first day of rhIL-11), day 4, day 7 and on day 14 included the following coagulation tests: VWF:RCo, VWF:Ag, FVIII:C, FVIII:Ag, activated partial thromboplastin time (APTT), VWF multimers and closure times with epinephrine (CEPI) or adenosine diphosphate (CADP); clinical blood counts: haemoglobin, haematocrit and platelet count; and safety tests: electrolytes, electrocardiogram (EKG) rhythm strip. Platelet-rich plasma for platelet VWF mRNA was drawn at baseline, day 7 and day 14.
Dose escalation
The dose-limiting toxicity was defined as any grade 3 or higher treatment-related toxicity possibly or probably related to the investigational drug. Specific dose-limiting toxicities included persistent (>24 h) hyponatraemia (Na+ <130 mEq L−1) or hypokalaemia (K+ <3.5 mEq L−1), any platelet count >900,000, any grade 3 or higher treatment-related toxicity, or recurrence of any of these criteria. Dose escalation was permitted only if the occurrence of dose-limiting toxicity resolved within 24 h.
Laboratory assays
Plasma was assayed for VWF activity using ristocetin-induced platelet agglutination kits and a Chronolog aggregometer (Chronolog Corp., Havertown, PA, USA). VWF:Ag was determined by ‘sandwich’ ELISA as described [18-21]. The multimer distribution of VWF:Ag was analysed by sodium dodecyl sulfate gel electrophoresis using 1.5% or 0.65% agarose gels [18-20]. Anti-VWF antibodies used for VWF:Ag ELISA and multimer gels were purchased from Dako (A0082; Carpinteria, CA, USA). The VWF:Ag concentration and activity values are expressed in U mL−1, based on normal reference human plasma pool (George King Biomedical, Inc., Overland Park, KS, USA) that was assigned a value of 1.00 U mL−1. Human severe type 3 VWD plasma, VWF:RCo 15 U mL−1 and VWF:Ag 1 U mL−1 from one type 3 VWD patient (George King Bio-Medical, Inc) served as negative control. FVIII:C was determined by chromogenic substrate assay (Coamatic Factor VIII; DiaPharma Group, Westchester, OH, USA), as specified by the manufacturer. FVIII:C activity is expressed in U mL−1 based on normal human reference plasma (George King). FVIII:Ag was determined by immunoassay per the manufacturer’s instructions (Asserochrome FVIII:Ag; Diagnostica Stago, Gennevilliers, France). Human haemophilia A plasma served as negative control (George King Bio-Medical, Inc).
Platelet-poor plasma for the above assays was prepared from citrated blood by centrifugation at 1300 g for 15 min. The plasma was spun a second time at 1300 g for 7 min and stored at −80°C for batch testing. The APTT was performed by commercial reagents on an ST4 analyzer (Diagnostic Stago), and closure times, CEPI and CADP were performed by PFA-100 analyzer (Dade Behring, Marburg, Germany). VWF mRNA was performed on a lysed platelet pellet (prepared from platelet-rich plasma), the RNA was reverse transcribed into cDNA and amplified by quantitative PCR for VWF mRNA expression. Platelet-rich plasma was prepared from EDTA plasma by centrifugation at 250 g for 15 min at room temperature. Platelets were pelleted at 1500 g for 10 min, the supernatant discarded and the platelet pellet was stored at −80°C for batch testing. RNA isolation from the platelet pellet was performed using Tri-Reagent, bromochloropropane and DNAse I (Sigma, St Louis, MO, USA), and RNA quality was assessed by Nanodrop ND-1000 Spectrophotometer (Applied Biosystems, Foster City, CA, USA) with all RNA analysed at 260/280 nm >1.8. Real-time PCR was performed using High-Capacity cDNA Archive Kit and Taqman Universal PCR Master Mix (Applied Biosystems) with the gene-specific validated Taqman assays for VWF (Hs00169795_m1) and GPIBb (Hs02579226_s1). The VWF Taqman assay utilized a primer set that produces a 79-bp amplicon spanning exons 17 and 18. The GP1Bb Taqman assay produces a 178 bp amplicon spanning an exon 2 boundary. Both assays amplify only the intact mRNAs. Samples were reverse transcribed for 10 min at 25°C and 2 h at 37°C, followed by 43 rounds of amplification for 15 s at 95°C and 1 min at 60°C using the ABI Prism 7700 sequence detection system (Applied Biosystems). Quantification of the gene-specific VWF message level was based on a relative quantification comparison of the fluorescence intensity in the baseline (untreated) total RNA sample to the fluorescence intensity from day 7 and day 14 (7 days following treatment) total RNA samples. Amplification of the gene for GP1Bb, which is a constitutively expressed platelet-specific gene, was performed on all samples tested to control for variations in the RNA amounts. The VWF mRNA levels were subsequently normalized against the GP1Bb mRNA levels.
Endpoints and statistical methods
A minimum of nine evaluable subjects in this Phase II study was considered adequate to provide sufficient power to evaluate biological efficacy of the study [22], specifically with 95% confidence to show an increase of 0.2 VWF U mL−1, with σ of 0.1 VWF U mL−1 at α = 0.05 and β = 0.90 (one-sided). Because of the small sample size, descriptive statistics were used to evaluate VWF and related factors. Statistical analyses for this study were based on assessment of the primary endpoint, biological efficacy and secondary endpoint and safety. Assessment of biological efficacy response to rhIL-11 was based on an increase in VWF:RCo, VWF:Ag, FVIII:C and FVIII:Ag, assessed at baseline, day 1, 4 and 7, using descriptive statistics. Assessment of safety was based on incidence rates of adverse events, which were calculated as the number of subjects with an event divided by the number of subjects exposed to drug. Incidence rates for adverse event episodes were calculated by the number of adverse event cases over the number of doses. Adverse event incidence rates were categorized by body system, relationship to study drug, severity of adverse event, age, gender, time of onset of adverse event and duration of adverse event. Safety laboratory variables were analysed by descriptive statistics. Comparisons between VWF and FVIII activity and antigen levels by dose group were performed by t-test, comparing absolute values to baseline after rhIL-11 and after DDAVP.
Safety analysis
Adverse events were assessed according to the NCI Common Terminology Criteria of Adverse Events (CTCAE) Version 3.0 [23]. As part of the safety assessment, specific focus was given to fluid retention and arrhythmias.
Results
A total of nine subjects with mild VWD, a past bleeding history and past DDAVP responsiveness were enroled on study between October 2004 and December 2006. Median age was 35 years (range 21 to 49 years) (Table 1).
Table 1.
Baseline characteristics of study subjects.
| Pt No. | Age/Gender | Bleeding severity score | History family | VWF:RCo (U mL−1) | VWF:Ag (U mL−1) | FVIII:C (U mL−1) | Multimers |
|---|---|---|---|---|---|---|---|
| Dose I: 10 μg kg−1 | |||||||
| Pt 1 | 38 (F) | 7 c, d, e, g, m, p | Yes | 0.22 | 0.72 | 0.82 | Normal |
| Pt 2 | 28 (F) | 5 c, e, g, m | No | 0.34 | 1.23 | 1.32 | Normal |
| Pt 3 | 35 (F) | 5 c, e, m, p | Yes | 0.23 | 0.47 | 0.25 | Normal |
| Dose II: 25 μg kg−1 | |||||||
| Pt 4 | 34 (F) | 6 c, d, m, mu | Yes | 0.40 | 0.46 | 0.81 | Normal |
| Pt 5 | 43 (M) | 4 c, d, e, mu | Yes | 0.44 | 0.60 | 0.70 | Normal |
| Pt 6 | 21 (M) | 5 c, mu, o | Yes | 0.39 | 0.30 | 0.16 | Normal |
| Dose III: 50 μg kg−1 | |||||||
| Pt 7 | 49 (M) | 6 c, d, g, mu, po | Yes | 0.57 | 1.18 | 0.16 | Normal |
| Pt 8 | 35 (M) | 3 c, mu, w | Yes | 0.40 | 0.64 | 0.21 | Normal |
| Pt 9 | 22 (F) | 5 c, m, mu, p | Yes | 0.47 | 0.78 | 0.66 | Normal |
DDAVP, 1-8 deamino-d-arginine vasopressin; VWF, von Willebrand factor; c, cutaneous; d, dental; e, epistaxis; g, gastrointestinal; m, menorrhagia; mu, musculoskeletal; o, oral; p, postpartum; pr, preoperative; w, wound bleeding; VWF:RCo, von Willebrand ristocetin cofactor; FVIII:C, clotting factor VIII; F, female; M, male; Ag, antigen.
Increase in VWF and FVIII Antigen and Activity in Response to IL-11 and DDAVP
There was an increase in VWF:RCo, VWF:Ag, FVIII:C and FVIII:Ag levels beginning at 24 h after rhIL-11, and persisting for the 7 days the drug was given. This increase was further boosted on day 7 after DDAVP infusion. By day 4 at 30 min after rhIL-11 injection, mean VWF:RCo was 1.49- to 1.62-fold higher than baseline, VWF:Ag was 1.56 to 2.19-fold higher, FVIII:C was 1.26 to 1.93-fold higher and FVIII:Ag was 1.25 to 2.23-fold higher than baseline levels, with significant increases in individual values (Tables 2 and 3). The effect persisted at 4 h after rhIL-11 injection and through day 7 and was further boosted on day 7 by i.v. DDAVP at 0.3 μg kg−1 given 30 min after rhIL-11, with an increase in VWF:RCo, 1.58 to 3.15-fold higher than baseline; VWF:Ag, 1.91 to 2.96-fold higher; FVIII:C, 2.08 to 3.16-fold higher and FVIII:Ag, 2.92 to 3.82-fold higher than baseline. By day 14, 7 days after the last dose of rhIL-11, VWF and FVIII levels returned to baseline. An example of the temporal changes in VWF:RCo, VWF:Ag and FVIII:C in one subject after rhIL-11, after DDAVP, and following cessation of the drug, is shown in Fig. 1
Table 2.
VWF and FVIII Activity and Antigen Levels After Recombinant IL-11.
| Pt ID screening | Day 1 |
Day 4 |
Day 7 |
Day 14 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre | 30 min | 4 h | 30 min | 4 h | 30 min | post-DDAVP† | 4 h | 7 day post-IL-11 | ||
| VWF:RCo, U mL−1 | ||||||||||
| Dose I: 10 μg kg−1 | ||||||||||
| Pt 1 | 0.92 | 0.86 | 1.08 | 1.40 | 1.54 | 1.16 | 2.32 | 3.52 | 3.12 | 0.90 |
| Pt 2 | 1.00 | 0.98 | 1.44 | 1.62 | 1.90 | 1.90 | NA | 2.88 | 1.38 | NA |
| Pt 3 | 0.81 | 0.68 | 0.61 | 0.84 | 0.71 | 0.52 | 0.72 | 1.60 | 0.94 | 0.68 |
| Mean | 0.91 | 0.84 | 1.04 | 1.29 | 1.38 | 1.19 | 1.52 | 2.67* | 1.81 | 0.79 |
| Dose II: 25 μg kg−1 | ||||||||||
| Pt 4 | 0.72 | 1.00 | 0.91 | 1.00 | 0.92 | 1.20 | 0.58 | 1.50 | 1.40 | 0.91 |
| Pt 5 | 0.61 | 0.45 | 0.87 | 0.83 | 1.24 | 1.10 | 1.40 | 1.72 | 1.00 | 0.47 |
| Pt 6 | 0.39 | 0.72 | 0.64 | 0.66 | 1.04 | 0.84 | 0.46 | 0.19 | 0.62 | 0.27 |
| Mean | 0.57 | 0.72 | 0.81 | 0.83 | 1.07 | 1.05 | 0.81 | 1.14 | 1.01 | 0.55 |
| Dose III: 50 μg kg−1 | ||||||||||
| Pt 7 | 1.88 | 1.60 | 1.40 | 0.92 | 2.28 | 1.78 | 1.88 | 3.20 | 2.78 | 0.92 |
| Pt 8 | 0.59 | 1.24 | 1.22 | 1.40 | 2.40 | 2.36 | 2.56 | 6.40 | 4.16 | 1.00 |
| Pt 9 | 0.54 | 0.57 | 0.54 | 0.61 | 0.68 | 0.73 | 0.92 | 1.18 | 1.40 | 0.46 |
| Mean | 1.00 | 1.14 | 1.05 | 0.98 | 1.79 | 1.62 | 1.79 | 3.59 | 2.78 | 0.79 |
| VWF:Ag (U mL−1) | ||||||||||
| Dose I: 10 μg kg−1 | ||||||||||
| Pt 1 | 0.96 | 0.99 | 1.13 | 1.29 | 1.40 | 1.83 | 1.48 | 3.58 | 3.05 | 1.60 |
| Pt 2 | 0.76 | 1.39 | 1.72 | 1.83 | 2.30 | 1.58 | 2.39 | 3.86 | 2.42 | 1.00 |
| Pt 3 | 0.95 | 0.67 | 0.75 | 0.95 | 1.06 | 0.81 | 0.76 | 1.60 | 2.42 | 0.90 |
| Mean | 0.89 | 1.02 | 1.20 | 1.36 | 1.59 | 1.41 | 1.54 | 3.01* | 2.63* | 1.17 |
| Dose II: 25 μg kg−1 | ||||||||||
| Pt 4 | 0.42 | 0.49 | 0.08 | 0.26 | 0.29 | 0.57 | 0.24 | 1.08 | 0.58 | 1.41 |
| Pt 5 | 0.14 | 0.75 | 0.26 | 0.29 | 1.57 | 1.83 | 2.50 | 1.73 | 2.88 | 0.09 |
| Pt 6 | 0.33 | 0.53 | 0.62 | 0.41 | 1.20 | 1.67 | 0.41 | 0.58 | 0.60 | 0.12 |
| Mean | 0.30 | 0.59 | 0.32 | 0.32 | 1.02 | 1.36 | 1.05 | 1.13 | 1.35 | 0.54 |
| Dose III: 50 μg kg−1 | ||||||||||
| Pt 7 | 2.00 | 2.04 | 1.98 | 1.98 | 4.80 | 4.11 | 3.74 | 2.55 | 4.47 | 2.19 |
| Pt 8 | 0.57 | 0.53 | 0.95 | 0.68 | 1.23 | 1.63 | 1.55 | 2.57 | 2.45 | 0.71 |
| Pt 9 | 0.60 | 0.71 | 0.55 | 0.63 | 1.17 | 1.06 | 0.90 | 3.18 | 2.97 | 0.28 |
| Mean | 1.06 | 1.09 | 1.16 | 1.10 | 2.40 | 2.27 | 2.06 | 2.77* | 3.30* | 1.06 |
| FVIII:C (U mL−1) | ||||||||||
| Dose I: 10 μg kg−1 | ||||||||||
| Pt 1 | 0.74 | 1.26 | 1.19 | 0.91 | 1.67 | 2.06 | 2.39 | 3.05 | 1.81 | 1.08 |
| Pt 2 | 1.33 | 2.02 | 2.21 | 2.25 | 2.57 | 2.64 | 2.76 | 4.55 | 2.16 | 0.54 |
| Pt 3 | 1.33 | 1.16 | 1.26 | 1.19 | 1.35 | 1.50 | 0.83 | 1.64 | 1.95 | 1.47 |
| Mean | 1.13 | 1.48 | 1.55 | 1.45 | 1.86 | 2.07 | 1.99 | 3.08 | 1.97 | 1.03 |
| Dose II: 25 μg kg−1 | ||||||||||
| Pt 4 | 0.76 | 0.63 | 0.51 | 0.35 | 1.07 | 1.50 | 0.35 | 2.00 | 1.14 | 0.64 |
| Pt 5 | 0.98 | 0.99 | 0.79 | 0.78 | 2.05 | 1.94 | 1.50 | 2.52 | 2.35 | 1.12 |
| Pt 6 | 0.14 | 0.16 | 0.11 | 0.16 | 0.33 | 0.32 | 0.19 | 0.41 | 0.28 | 0.12 |
| Mean | 0.63 | 0.59 | 0.47 | 0.43 | 1.15 | 1.25 | 0.68 | 1.64 | 1.26 | 0.63 |
| Dose III: 50 μg kg−1 | ||||||||||
| Pt 7 | 0.24 | 0.28 | 0.24 | 0.27 | 0.48 | 0.40 | 0.50 | 0.90 | 0.80 | 0.33 |
| Pt 8 | 0.17 | 0.11 | 1.04 | 0.16 | 0.26 | 0.26 | 0.32 | 1.10 | 0.81 | 0.15 |
| Pt 9 | 0.71 | 1.01 | 0.93 | 0.95 | 1.21 | 1.18 | 1.24 | 2.43 | 2.05 | 0.81 |
| Mean | 0.37 | 0.47 | 0.43 | 0.46 | 0.65 | 0.61 | 0.69 | 1.48 | 1.22 | 0.43 |
| FVIII:Ag (U mL−1) | ||||||||||
| Dose I: 10 μg kg−1 | ||||||||||
| Pt 1 | 0.78 | 0.89 | 0.90 | 0.95 | 1.44 | 1.14 | 0.65 | 3.45 | 1.33 | 0.95 |
| Pt 2 | 1.24 | 1.30 | 1.24 | 1.33 | 2.18 | 2.23 | 3.27 | 4.22 | 4.55 | 0.88 |
| Pt 3 | 0.76 | 0.74 | 0.68 | 0.70 | 0.78 | 0.86 | 0.40 | 1.19 | 1.54 | 0.92 |
| Mean | 0.93 | 0.98 | 0.94 | 0.99 | 1.47 | 1.41 | 1.44 | 2.95** | 2.47 | 0.92 |
| Dose II: 25 μg kg−1 | ||||||||||
| Pt 4 | 0.71 | 0.80 | 0.60 | 0.72 | 1.18 | 0.70 | 0.89 | 2.39 | 1.79 | 0.66 |
| Pt 5 | 0.89 | 0.76 | 0.60 | 0.78 | 2.27 | 2.26 | 2.90 | 3.97 | 3.66 | 0.70 |
| Pt 6 | 0.07 | 0.18 | 0.08 | 0.10 | 0.41 | 0.22 | 0.14 | 0.27 | 0.26 | 0.07 |
| Mean | 0.56 | 0.58 | 0.43 | 0.53 | 1.29 | 1.06 | 1.31 | 2.21 | 1.90 | 0.48 |
| Dose III: 50 μg kg−1 | ||||||||||
| Pt 7 | 0.15 | 0.38 | 0.17 | 0.18 | 0.25 | 0.25 | 0.25 | 0.46 | 0.43 | 0.20 |
| Pt 8 | 0.07 | 0.08 | 0.08 | 0.10 | 0.17 | 0.17 | 0.21 | 0.50 | 0.39 | 0.10 |
| Pt 9 | 0.52 | 0.62 | 0.63 | 0.65 | 0.93 | 0.94 | 0.99 | 2.20 | 1.77 | 0.63 |
| Mean | 0.25 | 0.36 | 0.29 | 0.31 | 0.45 | 0.45 | 0.48 | 1.05 | 0.86 | 0.31 |
DDAVP, 1-8 deamino-d-arginine vasopressin; VWF, von Willebrand factor; IL, interleukin; VWF:RCo, von Willebrand ristocetin cofactor; rhIL-11, recombinant human interleukin 11; FVIII:C, clotting factor VIII; Ag, antigen.
30-min post-DDAVP on day 7.
Statistical significance,
P < 0.01,
P < 0.05; as compared with Day 1, pre rhIL-11, at each dose.
Table 3.
Per cent increase in VWF and FVIII activity and antigen after recombinant IL-11.
| Dose I (10 μg kg−1) | Dose II (25 μg kg−1) | Dose III (50 μg kg−1) | |
|---|---|---|---|
| Mean level (per cent over baseline) | |||
| VWF:RCo | |||
| Day 1, pre | 85 (68–98)† | 72 (45–100) | 114 (57–160) |
| Day 1, 30 min | 104 (122%) | 81 (112%) | 105 (92%) |
| Day 4, 30 min | 138 (162%) | 107 (149%) | 179 (157) |
| Day 7, 30 min | 152 (179%) | 81 (144%) | 179 (157) |
| Day 7, after DDAVP | 267 (314%)* | 114 (158%) | 359 (315) |
| VWF:Ag | |||
| Day 1, pre | 101.8(67.6–139.2) | 59.1 (48.7–75.1) | 109.5 (53.0–204.3) |
| Day 1, 30 min | 120.0 (118%) | 32.0 (54%) | 116.3 (106%) |
| Day 4, 30 min | 158.6 (156%) | 101.8 (172%) | 240.3 (219%) |
| Day 7, 30 min | 154.5 (152%) | 104.9 (177%) | 206.3 (188%) |
| Day 7, after DDAVP | 301.5 (296%)* | 113.2 (191%) | 276.8 (253%)* |
| FVIII:C | |||
| Day 1, pre | 147.9 (115.8–202.2) | 59.4 (16.2–99.5) | 46.7 (10.9–101.1) |
| Day 1, 30 min | 155.2 (105%) | 47.2 (79%) | 42.7 (91%) |
| Day 4, 30 min | 186.2 (126%) | 114.8 (193%) | 64.9 (139%) |
| Day 7, 30 min | 199.5 (135%) | 67.9 (114%) | 68.7 (147%) |
| Day 7, after DDAVP | 307.9 (208%) | 164.4 (277%) | 147.8 (316%) |
| FVIII:Ag | |||
| Day 1, pre | 97.8 (74.0–129.8) | 57.8 (18.1–79.6) | 36.1 (8.2–62.2) |
| Day 1, 30 min | 93.9 (96%) | 42.9 (74%) | 29.1 (81%) |
| Day 4, 30 min | 146.9 (150%) | 129.0 (223%) | 45.1 (125%) |
| Day 7, 30 min | 143.9 (147%) | 131.1 (227%) | 48.5 (134%) |
| Day 7, after DDAVP | 295.5 (302%)** | 221.1 (382%) | 105.4 (292%) |
DDAVP, 1-8 deamino-d-arginine vasopressin; VWF, von Willebrand factor; VWF:RCo, von Willebrand ristocetin cofactor; IL, interleukin; FVIII:C, clotting factor VIII; Ag, antigen.
Range in parentheses. Statistical significance is indicated by
for P < 0.01 and
for P < 0.05.
Fig. 1.
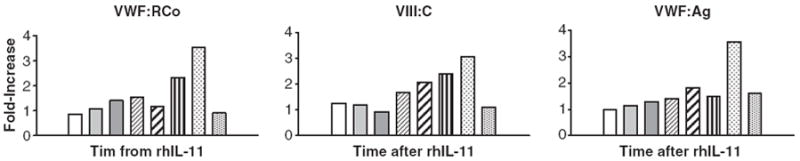
Temporal Change in von Willebrand factor (VWF) levels after rhIL-11 and 1-8 deamino-d-arginine vasopressin (DDAVP). The fold-increase in VWF:RCo, FVIII:C, and VWF:Ag are shown in one subject treated at dose I, the low dose level (10 μg kg−1), at baseline (white); on day 1 at 30 min (pale grey) at 4 h (dark grey); day 4 at 30 min (pale diagonal stripe) at 4 h (dark diagonal stripe); on day 7 at 30 min after rhIL-11 (before DDAVP) (vertical stripe) and 30 min after DDAVP (low density dots); and on day 14, 7 days after the last dose of rhIL-11 (high density dots). vWF:RCo, von Willebrand ristocetin cofactor; VWF:Ag, von Willebrand antigen, FVIII:C, clotting factor VIII; rhIL-11, recombinant human interleukin-11.
VWF Multimers following IL-11 and DDAVP
All patients had normal VWF multimer patterns at baseline, as analysed in 1.5% and 0.65% agarose gels. On day 7, very high-molecular-weight VWF multimers appeared after DDAVP, in parallel with the increase in plasma VWF antigen levels in all subjects and at all three doses of rhIL-11, as shown in one subject (Fig. 2).
Fig. 2.
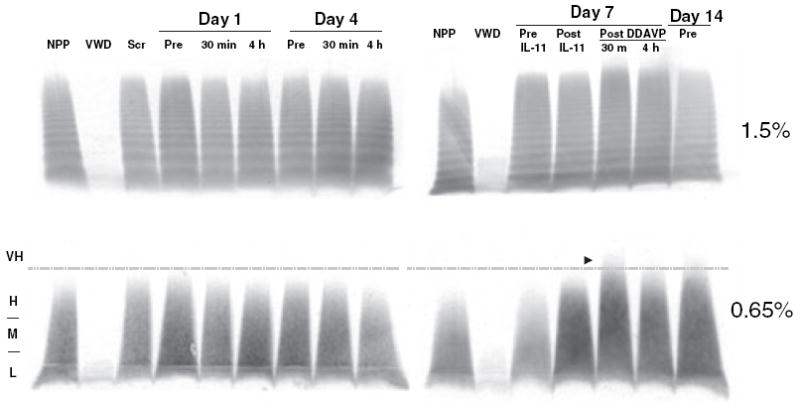
von Willebrand factor (VWF) multimers after rhIL-11 and 1-8 deamino-d-arginine vasopressin (DDAVP). VWF multimer bands are shown from one subject treated at dose III, the high dose level (50 μg kg−1), in 1.5% agarose (top panel) and in 0.65% agarose (lower panel), with separation of low (L), medium (M), high (H), and very high (VH) multimers. The first lane is normal pooled plasma (NPP), the positive control. The second lane is plasma from a type 3 severe patient with von Willebrand disease (VWF:RCo 15 U mL−1, VWF:Ag U mL−1), the negative control. In the next lanes are screening; day 1 and day 4, pre and 30 min and 4 h post-rhIL-11; and day 7 pre-rhIL-11, post-IL-11, and 30 min and 4 h post-DDAVP. Very high-molecular-weight multimers were detected on day 7 after DDAVP treatment (indicated by arrowhead). In the last lane on day 14, very high-molecular-weight multimers returned to the baseline. vWF:RCo, von Willebrand ristocetin cofactor; VWF:Ag, von Willebrand antigen; VWF:Ag, von Willebrand antigen; rhIL-11, recombinant human interleukin-11.
Platelet VWF mRNA by quantitative PCR
The increase in VWF was accompanied by a two- to eightfold increase in VWF mRNA expression over baseline, by quantitative PCR, as shown in one subject (Fig. 3).
Fig. 3.
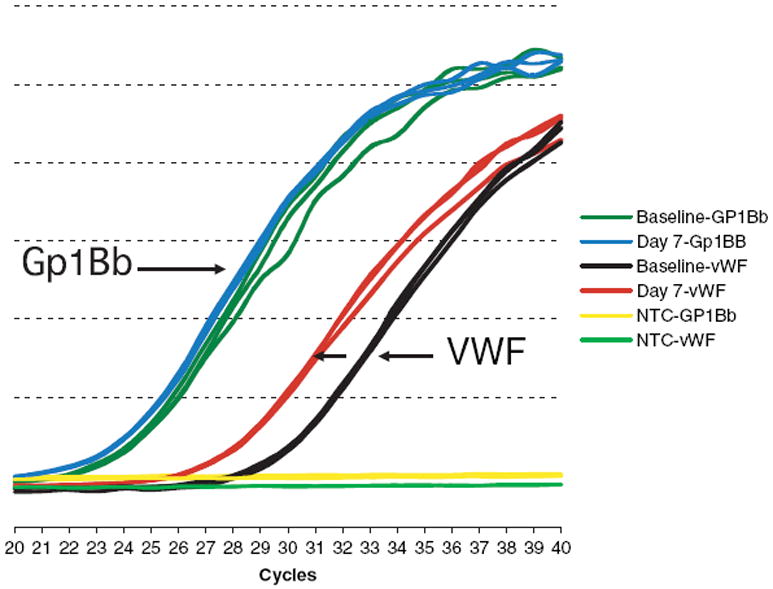
Platelet von Willebrand factor (VWF) mRNA expression after rhIL-11. Platelet VWF mRNA from one subject treated at dose I, the low dose level (10 μg kg−1), was isolated from platelet-rich plasma and reverse transcribed into VWF cDNA, and then amplified by quantitative PCR (see methods). When no RNA template was added, no reaction occurred and no message was detected, as seen in the yellow line. After rhIL-11, there was no change in GPIBb expression, as seen in the nearly superimposed green (baseline) and blue (after rhIL-11) lines. By contrast, after rhIL-11, there was a two- to eightfold increase in VWF mRNA expression as seen by the increase from brown (baseline) to red (after rhIL-11) lines (see arrow). NTC, no template control; rhIL-11, recombinant human interleukin-11.
Adverse effects and safety of IL-11 and DDAVP
Recombinant human IL-11 was well tolerated with minor adverse effects, all less than grade 1 toxicity (Table 4). Minor adverse events included hypertension, fluid retention and hypokalaemia, all less than grade 1 toxicity. Asymptomatic hypertension defined as diastolic blood pressure of 90 mmHg or higher, occurred in three subjects at the two higher dose levels, one of whom was found to have baseline labile hypertension. Fluid retention, including ring finger swelling in two subjects at the two higher dose levels was self-limited, and non-pitting oedema in a third subject on the 50 μg kg−1 dose arm resolved with oral Lasix (Furosemide, Mylan Pharmaceuticals, Morgantown, WV, USA). Hypokalaemia occurred in three subjects at the two high dose arms: the lowest potassium level, 3.1 mEq L−1, was asymptomatic, caused no changes on EKG and resolved with orange juice. One 38-year-old male subject on the 50 μg kg−1 dose arm developed anxiety and atypical chest pain, the evening before a scheduled visit. His chest X-ray, EKG, blood gases and spiral computerized tomography scan were all negative, and his symptoms, which resolved spontaneously without treatment, were judged to be unrelated to the study drug. There was a mean 2.2-fold increase (P < 0.01) in fibrinogen by day 4, mean 349 to 671 mg dL−1, but no thrombotic events occurred during the study.
Table 4.
Safety and tolerance of rhIL-11 in study subjects.
| Dose I (10 μg kg−1) | Dose II (25 μg kg−1) | Dose III (50 μg kg−1) | |
|---|---|---|---|
| Hypertension | |||
| No. abnormal | 0 | 2* | 1 |
| Median diastolic BP, mmHg | 84 | 91 | 81 |
| Hypokalaemia, No. | |||
| No. abnormal† | 0 | 1 | 2 |
| Median K+, mEq L−1 | 3.5 | 3.5 | 3.2 |
| Fluid retention‡ | |||
| No. abnormal† | 0 | 1 | 2 |
| Median weight gain, kg | 1.7 | 0.7 | 0.8 |
| Anaemia, dilutional | |||
| No. abnormal† | 3 | 3 | 3 |
| Median haemoglobin, g dL−1 | 10.2 | 12.1 | 12.6 |
| Secondary thrombocytosis§ | |||
| No. abnormal† | 1 | 0 | 0 |
| Median platelet no. × 103 μL−1 | 390 | 367 | 320¶ |
BP, blood pressure.
One subject (25 μg kg−1 dose) had baseline labile hypertension.
Abnormal tests in each category listed above were all less than grade 1 toxicity.
Fluid retention included ring finger swelling (one each, 25 and 50 μg kg−1 doses) and non-pitting pedal oedema (one, 50 μg kg−1 dose).
At 14 or 7 days after last rhIL-11 injection.
One subject (50 μg kg−1 dose) had baseline thrombocytopenia related to hepatitis C infection.
Platelet counts following rhIL-11
There was a delayed increase in platelet count following rhIL-11 treatment. By day 7, there was a mean 19 ± 5% increase (not significant) and, by day 14, a 125 ± 15% increase (P < 0.001) in platelet count. The increase in platelet number on day 14 was by 66 000 to 183 000 μL−1, with the highest absolute platelet count not exceeding 492 000 μL−1. This increase in platelets may have been, in part, reactive. Dilutional anaemia occurred in all subjects, with an average 2.7 g% decrease in haemoglobin (range, 1.8 to 3.5%), concomitant with mild fluid retention and, on average, a 0.9 kg weight gain (range, −0.5 to 1.9 kg). Both the weight gain and dilutional anaemia resolved by day 14 (7 days after cessation of rhIL-11), constituting less than grade 1 toxicity. There was no significant correlation between VWF levels and platelet count at baseline, r = −0.30, P > 0.40; at day 7, r = 0.371, P > 0.50; or at day 14, r = 0.099, P > 0.50.
Discussion and conclusion
This study demonstrates that rhIL-11 given s.c. causes a gradual and progressive increase in VWF and FVIII levels in subjects with mild VWD. The VWF:RCo and VWF:Ag levels increased 1.4 to 2.3-fold above baseline, sustained for the 7 days during which rhIL-11 was given. On day 7 after DDAVP, there was release of very high-molecular weight VWF multimers, confirmed on 0.65% agarose gel, which was not observed after rhIL-11 alone (Fig. 2a,b). These data suggest that the mechanism of the rhIL-11-induced VWF increase differs from that of DDAVP, and, specifically, that the rhIL-11-induced VWF increase occurs by a mechanism other than through release of VWF from endothelial stores [6]. The further boost in VWF and FVIII and release of very high-molecular weight VWF multimers by DDAVP implies that the DDAVP-releasable VWF pool persists despite rhIL-11 treatment, and that there is little, if any, effect of rhIL-11 on VWF trafficking to endothelial Weibel-Palade bodies, consistent with previous findings in dogs [15].
The two- to eightfold increase in VWF mRNA expression following rhIL-11 administration, as demonstrated by quantitative PCR, implies that the mechanism of rhIL-11 effect may be through upregulation of VWF mRNA. Previous studies in dogs have demonstrated a similar increase in VWF mRNA following rhIL-11 injection [15], although this was not the case in mice [14]. Whether rhIL-11 induces an increase in Weibel-Palade bodies or prevents loss of responsiveness to DDAVP is not known [15].
The findings of this study also show that when DDAVP is given after seven consecutive days of rhIL-11 treatment, VWF levels are increased above that induced by rhIL-11 alone, suggesting that rhIL-11 may be a useful agent in those refractory or unresponsive to DDAVP. It is unlikely, however, that the converse is true, i.e. that rhIL-11 boosts DDAVP response, given the difference in mechanisms and temporal separation in responses: DDAVP acts via a secretory pathway and induces an immediate release (within min) of substantial amounts of stored, very high-molecular-weight multimers that are cleared over the next 24 h, while rhIL-11 appears to induce a sustained increase in constitutive VWF release that progressively increases over days.
The rhIL-11-induced increase in FVIII:C suggests rhIL-11 may be a potentially effective haemostatic agent in mild haemophilia A. Further, the increase in FVIII:C after rhIL-11 injection appears to be to a lesser degree than the increase in VWF. Whether the increase in FVIII:C occurs by a separate mechanism from that of the VWF increase, or whether it involves upregulation of FVIII mRNA, however, remains unresolved.
While the site where FVIII and VWF first interact has not been established [24], recent data in haemophilia liver transplant recipients suggest that co-localization of FVIII and VWF are necessary for FVIII secretion after DDAVP [25]. DDAVP is known to be a specific agonist for the vasopressin V2 receptor (V2R) [26], and it is well established that with DDAVP binding to endothelial V2R, VWF is secreted from endothelial Weibel-Palade bodies [27]. The mechanism of FVIII release, however, is not known, and further studies are needed to clarify the mechanism of VWF and FVIII responses to rhIL-11 and DDAVP.
It should be noted that the baseline VWF:RCo and VWF:Ag levels measured at the start of the study (Table 2) were higher than those measured at diagnosis (Table 1). The reasons for this are not known but may relate to many factors. First, the study samples and diagnosis samples were drawn on widely different days and at widely different times and run in different laboratories. Day-to-day and lab-to-lab variability in VWF levels has been previously described and likely to contribute to these differences [2]. Second, the acute stress of an in-hospital research study of a new agent is also likely to contribute to these differences. Notably, this study was performed in the CTRC at the University of Pittsburgh which allowed all study blood samples to be drawn at the same time and all medications to be given at the same time on each study day. Thus, we believe that the results showing an increase in VWF activity, antigen and mRNA following rhIL-11 accurately reflect changes induced by rhIL-11. Third, it should be noted that the FVIII increase after rhIL-11 in subjects 6, 7 and 8 did not appear to increase sufficiently for potential surgical challenge. It is not clear why the FVIII increase was inferior in these subjects to the VWF increase, but could suggest potential increase in available VWF:Ag binding sites [2,10,28].
In summary, recombinant IL-11 is well tolerated and safely increases VWF and FVIII into the range associated with clinical haemostasis in humans with mild VWD while not bleeding. These data justify a clinical trial to determine whether rhIL-11 will reduce clinical bleeding when present in this population or when procedures or surgery would be expected to be associated with excessive bleeding. Those refractory to or with a limited response to DDAVP might also benefit, and those who desire the safety of treatment with other than plasma-derived concentrates would also be of interest to study. The potential target population may include those with type 1 VWD or type 2M VWD, although, as genotyping becomes more common, the full spectrum of those responsive to rhIL-11, just as with DDAVP, will likely become evident [2]. Clinical trials to determine clinical efficacy of rhIL-11 in VWD are underway.
Acknowledgments
M. Ragni and T Nichols designed the research; M. Ragni and R. Jankowitz performed the clinical trial; H. Chapman obtained and managed the clinical data and performed nursing assessments on study subjects; E. Merrick, M. Kloos and A. Dillow performed laboratory assays; M.Ragni and T. Nichols analysed the results and made figures and M.Ragni wrote the paper with contributions from R. Jankowitz and T. Nichols.
We also acknowledge nursing by Dana McDermott, BSN, RN and Kristen Jaworski, BSN, RN at the Hemophilia Center of Western PA and Jill Huey, BSN, RN at the Montefiore University Hospital Center for Translation Research, MUH-CTRC. We also acknowledge Ms. Robin A. Raymer, B.S. for technical support in performance of VWF assays.
This study was supported by CTRC/CTSI grant: NIH/NCRR/CTSA UL-1 RR024153; Maternal and Child Health Bureau, HRSA, Contract #1-H-30-MC-00038-01; CDC Contract U24CCU318053; and the Pennsylvania State Department of Health Contract, SAP #04100000330. The study was also supported by Wyeth Pharmaceuticals, Inc. who supplied rhIL-11 (but provided no financial support). There were no competing financial interests by any authors. The study was registered at ClinicalTrials.Gov under NCT00151125.
Footnotes
Disclosures The authors declare no competing financial interests.
References
- 1.Ginsburg D, Sadler JE. Von Willebrand disease: a database of point mutations, insertions and deletions. Thromb Haemost. 1993;69:177–94. [PubMed] [Google Scholar]
- 2.Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 3.Rodeghiro F, Castaman G, Dini E. Epidemiologic investigation of the prevalence of Von Willebrand disease. Blood. 1987;69:454–9. [PubMed] [Google Scholar]
- 4.Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84:160–74. [PubMed] [Google Scholar]
- 5.Ragni MV, Bontempo FA, Cortese-Hassett AL. Von Willebrand disease and bleeding in women. Haemophilia. 1999;5:313–7. doi: 10.1046/j.1365-2516.1999.00342.x. [DOI] [PubMed] [Google Scholar]
- 6.Mannucci PM. Desmopressin in the treatment of bleeding disorders: the first 20 years. Blood. 1997;90:2515–21. [PubMed] [Google Scholar]
- 7.Rodeghiero F, Castaman G, Tosetto A, et al. The discriminant power of bleeding history for the diagnosis of type 1 von Willebrand disease: an international multi-center study. J Thromb Haemost. 2005;3:2619–26. doi: 10.1111/j.1538-7836.2005.01663.x. [DOI] [PubMed] [Google Scholar]
- 8.Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infection. N Engl J Med. 2006;355:1303–5. doi: 10.1056/NEJMp068178. [DOI] [PubMed] [Google Scholar]
- 9.Healthy People 2010. Medical Product Safety. Vol. 17. Rockville, MD: US Department Health and Human Services; 2003. pp. 3–19. Available at: http://www.healthypeople.gov. [Google Scholar]
- 10.Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart Lung and Blood Institute (NHLBI) Expert Panel Report (USA) Haemophilia. 2008;14:171–232. doi: 10.1111/j.1365-2516.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- 11.Issacs C, Robert NJ, Bailey FA, et al. Randomized placebo-controlled study of recombinant human interleukin-11 to prevent chemotherapy- induced thrombocytopenia in patients with breast cancer receiving dose-intensive cyclophosphamide and doxorubicin. J Clin Oncol. 1997;15:3368–77. doi: 10.1200/JCO.1997.15.11.3368. [DOI] [PubMed] [Google Scholar]
- 12.Physician’s Desk Reference. Neumega: product information. Montvale, NJ: Thomson PDR; 2007. pp. 3435–40. [Google Scholar]
- 13.Kaye J, Loewy J, Blume J, et al. Recombinant human interleukin eleven (Neumega, rhIL-11 growth factor) increases plasma von Willebrand factor and fibrinogen concentrations in normal human subjects. Blood. 1994;84:278a. [Google Scholar]
- 14.Denis CV, Kwack K, Saffaripour S, et al. Interleukin 11 significantly increases plasma von Willebrand factor and factor VIII in wild type and von Willebrand disease mouse models. Blood. 2001;97:465–72. doi: 10.1182/blood.v97.2.465. [DOI] [PubMed] [Google Scholar]
- 15.Olsen EHN, McCain AS, Merricks EP, et al. Comparative response of plasma VWF in dogs to up-regulation of VWF mRNA by interleukin-11 versus Weibel-Palade body release by desmopressin (DDAVP) Blood. 2003;102:436–41. doi: 10.1182/blood-2003-01-0290. [DOI] [PubMed] [Google Scholar]
- 16.James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 17.Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1 VWD) Blood. 2007;109:112–21. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- 18.Haberichter SL, Merricks EP, Fahs SA, Christopherson PA, Nichols TC, Montgomery RR. Re-establishment of VWF-dependent Weibel-Palade bodies in VWD endothelial cells. Blood. 2005;105:145–52. doi: 10.1182/blood-2004-02-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benson RE, Catalfamo JL, Dodds WJ. A multispecies enzyme-linked immunosorbent assay for von Willebrand’s factor. J Lab Clin Med. 1992;119:420–7. [PubMed] [Google Scholar]
- 20.Brouland JP, Egan T, Roussi J, et al. In vivo regulation of von Willebrand factor synthesis: von Willebrand factor production in endothelial cells after lung transplantation between normal pigs and von Willebrand factor-deficient pigs. Arterioscler Thromb Vasc Biol. 1999;19:3055–62. doi: 10.1161/01.atv.19.12.3055. [DOI] [PubMed] [Google Scholar]
- 21.Nichols TC, Bellinger DA, Reddick RL, et al. The roles of von Willebrand factor and factorVIII in arterial thrombosis: studies in canine von Willebrand disease and hemophilia A. Blood. 1993;81:2644–51. [PubMed] [Google Scholar]
- 22.Estey EH, Thall PF. New designs for phase 2 clinical trials. Blood. 2003;102:442–8. doi: 10.1182/blood-2002-09-2937. [DOI] [PubMed] [Google Scholar]
- 23.Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events, version 3.0, DCTD, NCI, NIH, DHHS. 2003 Available at: http://ctep.cancer.gov.
- 24.Kaufmann JE, Vischer UM. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP) J Thromb Haemost. 2003;1:682–9. doi: 10.1046/j.1538-7836.2003.00190.x. [DOI] [PubMed] [Google Scholar]
- 25.Lamont PA, Ragni MV. Lack of DDAVP response in men with hemophilia A following liver transplantation. J Thromb Haemost. 2005;3:2259–63. doi: 10.1111/j.1538-7836.2005.01553.x. [DOI] [PubMed] [Google Scholar]
- 26.Bernat A, Hoffman P, Dumas A, Gal SL, Raugaste D, Herbert JM. V2 receptors antagonism of DDAVP-induced release of hemostasis factors in conscious dogs. J Pharmacol Exp Ther. 1997;282:597–602. [PubMed] [Google Scholar]
- 27.Kaufmann JE, Oksche A, Wolheim CB, Gunther G, Rosenthal W, Vischer UM. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J Clin Invest. 2000;106:107–16. doi: 10.1172/JCI9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montgomery RR, Gill JC. Interactions between von Willebrand factor and factor VIII: where did they first meet? J Pediatr Hematol Oncol. 2000;20:2689–95. doi: 10.1097/00043426-200005000-00017. [DOI] [PubMed] [Google Scholar]