

Abstract
Caspase-6 is an apoptotic protease that also plays important roles in neurodegenerative disorders including Huntington’s and Alzheimer’s diseases. Caspase-6 is the only caspase known to form a latent state in which two extended helices block access to the active site. These helices must convert to strands for binding substrate. We probed the inter-converting region and found that the absence of helix-breaking residues is more critical than is a helix-bridging, hydrogen-bond network for formation of the extended conformation. In addition, our results suggest that caspase-6 must transition through a low-stability intermediate in order to bind active-site ligand.
Keywords: apoptosis, neurodegeneration, conformational switch, helix-to-strand, protease, high-energy intermediate
Caspases are a class of cysteine-aspartate proteases that play important roles in apoptosis, inflammation and development. Caspase-6 was originally classified primarily as an apoptotic executioner protease, as cleavage of the procaspase-6 zymogen by upstream caspases appeared to be essential for activation. During programmed cell death, caspase-6, along with caspase-3 and -7, cleaves critical factors, including nuclear and cytoskeletal proteins, leading to the demise of the cell. It has become clear in the recent past that caspase-6 also performs several non-apoptotic roles including axonal pruning during development (1) and B cell activation and differentiation (2). Currently, caspase-6 is most widely recognized for its role in cleaving neuronal substrates involved in neurodegeneration (3). It has become clear that cas-pase-6 plays important roles in Huntington’s (4), Alzheimer’s (5) and Parkinson’s diseases (6), where cleavage of substrates by caspase-6 is a causative event in development of each of these pathologies. As a result, despite historical difficulties in developing caspase-directed drugs, caspase-6 is now viewed as a promising drug target for neurodegeneration. The structure and regulation of caspase-6 is clearly unique among caspases, so a better understanding of caspase-6 structure and function is essential for developing caspase-6 specific therapies, which may prove important in treatment of several neurodegenerative diseases.
The structures of caspase-6 in the immature zymogen form (7), mature unliganded (apo) form (8, 9) and with active-site bound by a peptide inhibitor (7) have recently been determined. The active-site-bound and zymogen forms of caspase-6 are similar to all other caspases, but in apo mature caspase-6 the 60’s helix (amino acids 57–70) and the 130’s region (amino acids 128–142) are in extended helices that have not been observed previously (Fig. 1A). The formation of these helices causes the outward rotation of the 90’s helix and movement of the L1 loop. In the apo state, the loop above the 130’s helix occupies the substrate binding groove and the catalytic dyad is mispositioned for catalysis, suggesting that the apo enzyme is in a latent state (8). Thus in order to bind substrate, caspase-6 must undergo a dramatic reorganization of the 60’s, 90’s and 130’s helices and the L1 loop. Currently, perhaps the most compelling questions about caspase-6 are why and how it forms the extended helical state and what biological roles of caspase-6 require this unique conformation.
FIGURE 1.
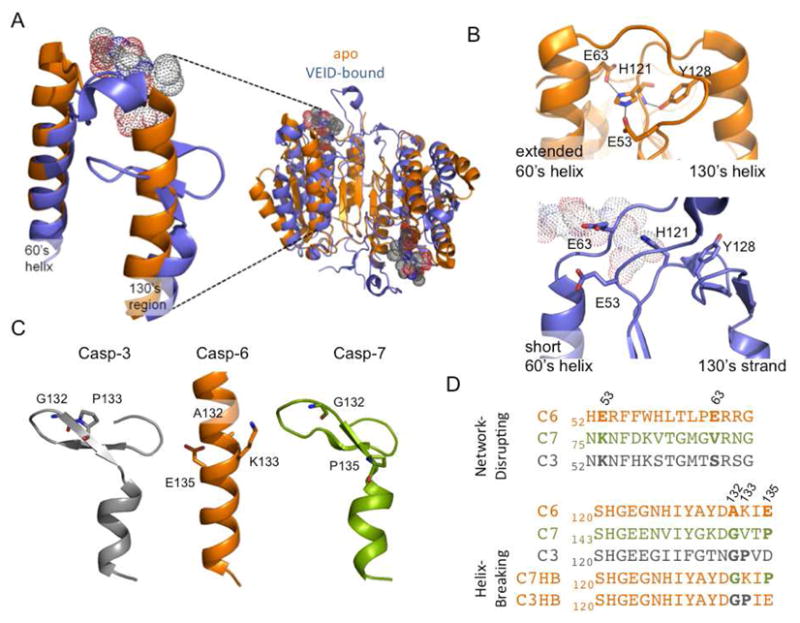
The caspase-6 latent state extended helices and helical network interrogated by mutations. A. Overlay of apo mature cas-pase-6 (3NKF, orange) and active-site bound caspase-6 (3OD5, blue). Two extended helices, the 130’s and 60’s helices, are lost upon ligand binding. The ligand VEID (dots) bound in the substrate-binding groove is incompatible with the extended helical conformation. B. The helix-bridging network (residues E63-H121-E53-Y128), which is only present in apo mature caspase-6 (orange), is broken in caspase-6 (blue) bound to the VEID (dots). C. 130’s region in apo caspase-3, -6 and -7 is helical only in caspase-6. Helix breaking residues at positions 132, 133, 135 in caspase -3, and -7 and the corresponding residues in caspase-6 (sticks) are located in the segment that undergoes a conformational change. D. Sequence alignment of 60’s and 130’s region in caspase-3, -6, and -7 inspired the design of the network-disrupting and helix-breaking (C3HB, C7HB) variants.
Inspection of the caspase-6 apo structure reveals a helix-bridging hydrogen-bond network between the 130’s and 60’s helices which can only form in caspase-6 but not in other caspases due to both sequence and conformation. The interactions in this network position caspase-6 in the latent (catalytically inactive) conformation. In apo caspase-6, the side chain of the catalytic base, His121, which is located in the loop at the top of 130’s helix, is held 9Å apart from the catalytic nucleophile Cys163, preventing catalysis. His121 is held in this inactive position by Tyr128, Glu63 (60’s helix) and Glu53 (L1 loop above 60’s helix). Glu63 and Glu53 make salt bridges with the side chain of His121. Tyr128 (130’s helix) makes a salt bridge with the backbone amide of His121 (Fig. 1B). Position 53 is a glutamate residue only in caspase-1 and -6 and position 63 is a glutamate only in caspase-6, underscoring the uniqueness of this bridging network. Upon binding substrate, the top of the 130’s helix (containing Tyr128) transforms into a strand, the top of the 60’s helix (containing Glu63) transforms into a loop, the L1 loop (containing Glu53) changes conformation and the helix-bridging network is broken (Fig. 1B).
Comparison of the 130’s helix sequence in caspase-6 with that from other executioner caspases provides a second insight into why only caspase-6 can form the extended, helical conformation. Prolines and glycines are typically considered to be helix-breaking residues, although they have been observed in helices and their removal does not always lead to change in the helical conformation (10, 11). The most closely related caspases, cas-pase-3 and -7, contain helix-breaking residues in the 130’s region, whereas caspase-6 has none. Gly and Pro are seen in caspase-3 (residues 132 and 133) and in caspase-7 (133 and 135), but there are no glycines and prolines in this region of caspase-6 (Fig. 1C, D). Pairs of helix breaking residues are also present in the 130’s region of all other caspases, suggesting their importance in preventing the helical conformation. Based on sequence alone it appears that the 130’s region of caspase-6 has a higher propensity to adopt a helical conformation than other caspases.
The central motive behind this work is to understand the contributions of the helix-bridging network (Fig. 1B) and the helix-breaking residues in the 130’s region (Fig. 1C, D) in maintaining the helical conformation of mature caspase-6. To this end we introduced helix-network disrupting residues to mimic the residues present in caspase-7 and prevent formation of the hydrogen bond network: E53K, E63V, and E53K/E63V. We also introduced helix-breaking residues in the 130’s helix to generate the Caspase-7-like Helix Breaking variant (C7HB, containing the A132G and E135P substitutions) and the Caspase-3-like Helix Breaking variant (C3HB, containing the A132G and K133P substitutions) (Fig. 1D). All of the variants are active, as assessed by cleavage of fluorescent substrate, and have kcat and KM values similar to wild-type protein (Table 1) suggesting that these unique sequences are not required for caspase-6 catalytic properties.
Table 1.
Kinetic parameters and melting temperatures for wild-type (WT) caspases, the network-disrupters and helix-breakers.
| KM (μM) | kcat (s−1) | Apo Tm (°C) | Active site bound Tm (°C) | ΔTm (°) | |
|---|---|---|---|---|---|
| WT casp-6a | 66 ±7 | 0.81 ±0.01 | 77 ± 0.5 | 80.3 ± 0.4 | 3.3 |
| WT casp-3 | 5a | 9.1b | 83 | >90 | >7 |
| WT casp-7c | 23 ±2 | 0.35 ±3 ×l0−3 | 59.8 ±0 | 77.7 ± 3.8 | 17.9 |
| E53K | 100 ±28 | 0.7 ±0.07 | 77 ±0.1 | 77.2 ±0.4 | 0.2 |
| E63V | 121 ±24 | 0.78 ±0.08 | 74.6 ±1.5 | 77.5 ±0.1 | 2.9 |
| E53K+E63V | 68 ±16 | 0.74 ±0.06 | 75.5 ±0.2 | 78.3 ± 0.8 | 2.8 |
| C3HB (A132G, K133P) | 46 ± 3.5 | 0.84 ± 0.06 | 70.7 ±1.0 | 76.5 ±0.7 | 5.8 |
| C7HB (A132G, E135P) | 79 ±13 | 1.2 ±0.03 | 73.1 ±0.5 | 77.8 ±0.3 | 4.7 |
We have previously shown that caspase-6 exhibits a unique signature in the circular dichroism (CD) spectra at 222 nm resulting from the loss of the 130’s and 60’s helices upon substrate binding (Fig. 2) (7, 9). We have observed that in caspase-3 and caspase-7, caspases for which there is no evidence for the extended helical conformation, there is a very small change or no change in the 222 nm CD signal upon active-site ligand binding (Fig. 2). We use this CD signature to monitor the effect of the helical network-disrupting and helix-breaking substitutions on the structure of the 130’s and 60’s region. The spectra of each of the apo (unliganded) variants were first compared to wild-type cas-pase-6. Only C3HB variant showed weaker CD signal at 222 nm (Fig. S1) indicating decrease in helical content upon helix-breaking relative to wild-type capase-6. Overlay of the CD spectra of apo and active-site ligand (VEID, a covalent aldehyde-based peptide inhibitor) bound caspase-6 reveals that the 130’s helix-breaking mutants, C3HB and C7HB have a smaller difference in CD signal at 222 nm between apo and bound forms than in wild-type caspase-6 (Fig. 2). This smaller difference suggests that the apo state of C3HB and C7HB is more similar to the ligand-bound state (like caspase-3 and -7) than is the case for wild-type. Conversely, the network-disrupting variants exhibit a similar difference in CD signal as wild-type caspase-6. This suggests that the network-disrupting variants are as helical as wild-type in the apo state. These data suggest that the absence of helix-breaking residues is more critical to the formation of the caspase-6 extended helices than the bridging hydrogen-bond network.
FIGURE 2.

CD spectra of apo and ligand-bound wild-type caspase-6 reflect the change in structure of the 60’s and 130’s helix upon ligand binding. In contrast the CD spectra of caspase-7 and -3 do not reflect a change in helical content. The network-disrupting variant (E53K/E63V) shows a similar change in helical content upon ligand binding to wild-type caspase-6. The helix breaking variants C7HB and C3HB show a much decreased difference in CD spectra between apo and ligand bound, suggesting that the apo state is less helical in solution than wild-type caspase-6.
The melting temperature of a protein reflects the stability of that protein and the equilibria between folded and unfolded states. To assess the role of the helix-network and the lack of helix-breaking residues on stability, the apo and VEID-bound forms of all the variants were also thermally denatured as monitored by CD. Amongst the variants, with the exception of E53K, all of the helix-breaking and network-disrupting variants have somewhat lower melting temperatures (Tm) than wild-type caspase-6 in both the apo and ligand-bound states (Table 1). The E53K and E63V mutations do not affect the Tm of apo and bound form significantly. However, the Tm of apo C3HB is 6.3°C lower than apo wild-type caspase-6. The Tm of C7HB is 3.9°C lower than apo wild-type caspase-6. Thus introducing helix-breaking mutations in the 130’s helix destabilizes the apo form and underscores the importance of the 130’s helix in the stability of apo caspase-6.
The values of ΔTm (apo vs. ligand bound) showed gain in stability upon ligand binding for both caspase-7 (18°C) and caspase-3 (>7°C) with no significant conformational changes in the 130’s region (Table 1). The increase in the Tm for caspase-3 may be as large as that of caspase-7 but cannot be measured since ligand-bound caspase-3 melts above 90°C (the detection limit in water). In both caspase-3 and -7 no conformational changes in the 130’s region are observed upon ligand binding, but a number of new interactions are formed. These interactions between the substrate binding loops and across the dimer interface can only occur when ligand binds and thereby orders the loops. These interactions contribute significantly to the high stability of the ligand-bound state of caspase-3 and -7. In contrast, the change in Tm is only 2–3°C for WT caspase-6 upon substrate binding, denoting that the apo helical state and the strand-containing ligand-bound states of cas-pase-6 have similar stabilities, despite their large conformational differences. The C3HB and C7HB variants show ΔTm of 5.8°C and 4.7°C, which are greater than the wild-type ΔTm. A change in the magnitude of the ΔTm for C3HB and C7HB suggests that these variants populate the extended helical conformation and ligand bound (strand) conformation to different extents in the apo state than does wild-type caspase-6. Based on the CD spectra and the Tm values, the sequence of the 130’s helix, particularly the absence of helix-breaking residues, appears to be more critical for maintaining the helical conformation in apo caspase-6 than the helix-bridging network.
We used data from caspase-6 and caspase-7 to construct an energy diagram (Fig. 3) with points representing: the fully β-strand conformation of the 130’s region with disordered substrate-binding loops (apo caspase-7); the fully β-strand conformation of the 130’s region with ordered substrate binding loops (bound caspase-6 or -7) and the fully helical conformation of the 130’s region (apo caspase-6). Each of these states has unique stabilities (Tm). Using these endpoints we can then assess the contribution of each state to the conformation of the helix-breaking variants.
FIGURE 3.
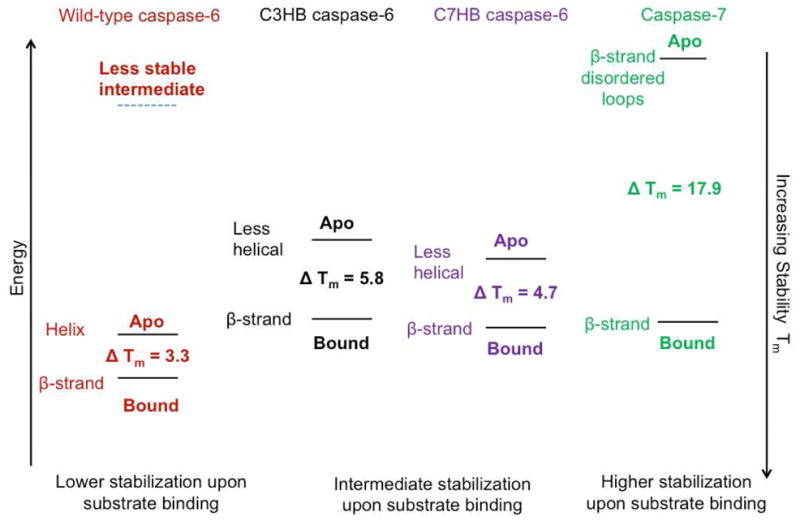
The relative stabilities of the apo- and active-site ligand-bound forms of caspase-6 and -7 and the helix breaking, C3HB and C7HB variants and of the predicted caspase-6 intermediate.
Caspase-6 undergoes a conformational change in the 130’s helix in order to bind ligand but it has a much lower gain in stability upon ligand binding than other caspases. In addition it is clear from crystal structures of caspase-6 that the extended conformation is incompatible with substrate binding. Thus it appears that the change in conformation of the 130’s helix causes apo-caspase-6 to transition through a less stable (high energy) intermediate in order to attain the strand conformation which is compatible with binding ligand (Fig 3). This hypothesis is supported by the properties of the C3HB and C7HB variants, which, as assessed by CD, populate the helical conformation in the apo state to a lower degree than wild-type caspase-6. They also show greater stabilization (greater ΔTm) upon ligand binding than wild-type caspase-6. This is because a larger fraction of the equilibrium apo population of these variants is in the strand conformation, like that seen in caspase-7. The high stability of the apo state of caspase-6 afforded by the extension of two helices may play a protective role, prolonging the caspase-6 lifetime.
The conformational switch in caspase-6 upon substrate binding is unique amongst proteases to caspase-6 but is not unprecedented in proteins generally. In the elongation factor, Ef-Tu, the effector C-terminus forms an α-helix when bound to GTP but converts to a β-hairpin upon GTP hydrolysis (12). The evolution of Cro family proteins is another excellent paradigm for secondary structural α-to-β switches (13). While the C-terminus of bacteriophage P22 Cro forms an α-helix, the homologous λ Cro forms a β-hairpin. α-to-β conformational switches are likewise critical for the propagation of prion infections (14). Thus, structural switches regulate function and facilitate new biological roles.
Several new roles may be enabled by the helical conformation of the caspase-6 latent state. Both apoptotic and inflammatory caspases can perform functions independent of their catalytic activity. Caspase-1 and -2 can non-catalytically activate NF-κB, caspase-8 can be activated by c-FLIPL, a catalytically inactive caspase, and functional caspase-12 exists in two catalytically inactive forms (for review see (15)). We suspect that caspase-6 in the latent conformation may likewise perform non-catalytic functions. The absence of helix breaking residues and the stable latent state may explain caspase-6 resistance to binding inhibitors in the active site. Intriguingly, the inhibitors of apoptosis (IAPs) bind and inhibit the active sites of caspase-3 and -7 but not of caspase-6 (16). The conformation of IAPs bound to caspases appears to be structurally incompatible with the latent state of caspase-6 (Fig. S2). Thus it is likely that formation of the latent state prevents IAP binding. Lack of IAP binding potentially improves the response of caspase-6 to substrate or to other non-catalytic stimuli.
In conclusion, our two major findings address why caspase-6 can uniquely adopt the helical latent state. First, upon substrate binding, part of the extended 130’s helical region switches to a β-strand conformation through a high energy, substrate-compatible intermediate. Second, the absence of helix-breaking residues is critical to the extended conformation of the 130’s helix region in the latent state structure of caspase-6, whereas the hydrogen-bond network in this region is not essential. Given the important role caspase-6 plays in a number of neurodegenerative diseases, these two findings may be exploited to design small molecule inhibitors that are specific for caspase-6, either binding to the extended helical (latent) state or preventing the transition through the high energy intermediate required for substrate binding.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 GM080532. We thank Elih Velázquez for caspase-3 purification and Tm and Witold Witkowski for purified caspase-7.
Footnotes
SUPPORTING INFORMATION AVAILABLE. Protein expression and purification, circular dichroism spectroscopy, enzyme kinetics are described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nikolaev A, McLaughlin T, O’Leary D, Tessier-Lavigne M. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Watanabe C, Shu G, Zheng T, Flavell R, Clark E. The Journal of Immunology. 2008;181:6810–6809. doi: 10.4049/jimmunol.181.10.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klaiman G, Petzke T, Hammond J, LeBlanc A. Molecular & Cellular Proteomics. 2008;7:1541–1555. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galvan V, Gorostiza O, Banwait S, Ataie M, Logvinova A, et al. PNAS USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham R, Deng Y, Slow E, Haigh B, Bissada N, et al. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 6.Giaime E, Sunyach C, Druon C, Scarzello S, Robert, et al. Cell Death & Differentiation. 2009;17:158–169. doi: 10.1038/cdd.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang XJ, Cao Q, Liu X, Wang KT, Mi W, et al. EMBO Rep. 2010;11:841–847. doi: 10.1038/embor.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumgartner R, Meder G, Briand C, Decock A, D’Arcy A, et al. Biochem J. 2009;423:429–439. doi: 10.1042/BJ20090540. [DOI] [PubMed] [Google Scholar]
- 9.Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA. J Mol Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alber T, Bell JA, Sun DP, Nicholson H, Wozniak JA, Cook S, Matthews BW. Science. 1988;239:631–635. doi: 10.1126/science.3277275. [DOI] [PubMed] [Google Scholar]
- 11.Hardy JA, Nelson HC. Protein Sci. 2000;9:2128–2141. doi: 10.1110/ps.9.11.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polekhina G, Thirup S, Kjeldgaard M, Nissen P, Lippmann C, Nyborg J. Structure. 1996;4:1141–1151. doi: 10.1016/s0969-2126(96)00122-0. [DOI] [PubMed] [Google Scholar]
- 13.Newlove T, Konieczka J, Cordes M. Structure. 2004;12:569–581. doi: 10.1016/j.str.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 14.Pan K, Baldwin M, Nguyen J, Gasset M, Serban A, et al. PNAS USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 16.Deveraux Q, Takahashi R, Salvesen G, Reed J. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 17.Thornberry NA, Rano TA, Peterson EP, Rasper DM, et al. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 18.Witkowski WA, Hardy JA. Protein Sci. 2009;18:1459–1468. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.