

Abstract
Heart failure can be caused by pro-hypertrophic humoral factors such as angiotensin II (Ang II), which regulates protein kinase activities. The intermingled responses of these kinases lead to the early compensated cardiac hypertrophy, but later to the uncompensated phase of heart failure. We have shown that although beneficial, cardiac hypertrophy is associated with modifications in ion channels that are mainly mediated through mitogen-activated protein (MAP) kinase and phosphatidylinositol 3-kinase (PI3K) activation. This study evaluates the control of L-type Ca2+ current (ICa,L) by the Ang II/PI3K pathway in hypertrophied ventricular myocytes from volume-overload rats using the perforated patch-clamp technique. To assess activation of the ICa,L in cardiomyocytes, voltages of 350 ms in 10 mV increments from a holding potential of −85 mV were applied to cardiocytes, with a pre-pulse to −45 mV for 300 ms. Volume overload-induced hypertrophy reduces ICa,L, whereas addition of Ang II alleviates the hypertrophic-induced decrease in a PI3K-dependent manner. Acute administration of Ang II (10−6 mol/L) to normal adult cardiomyocytes had no effect; however, captopril reduced their basal ICa,L. In parallel, captopril regressed the hypertrophy and inverted the Ang II effect on ICa,L seemingly through a PI3K upstream effector. Thus, it seems that regression of cardiac hypertrophy by captopril improved ICa,L partly through PI3K.
Keywords: cardiac hypertrophy, Ang II, L-type calcium channel, captopril, PI 3-kinase
Introduction
Cardiac hypertrophy is the compensatory enlargement of the heart aimed at reducing stress induced by volume overload (VO) (Juric et al. 2010). Arteriovenous shunt or fistula has long been used as a model for inducing volume overload similar to that seen in conditions such as mitral valve regurgitation, aortic insufficiency, renal failure, obesity, hyperthyroidism, anemia, and bundle branch block (Wang et al. 2003). Although compensated hypertrophy may represent an adaptive response, in the long term, it is usually associated with high morbidity and mortality, often evolving to heart failure, in which altered cell Ca+2 cycling usually accompanies deterioration of contractile function (Carvalho et al. 2006). As a result of careful characterization of the volume overload-induced remodeling process, 3 distinct phases of remodeling have been identified: (i) an initial 2-week phase of chronic volume overload, (ii) a compensatory phase with normal systolic pressure and ejection fraction, and (iii) a decompensated phase in which hypertrophic mechanisms can no longer maintain an adequate mass-to-volume ratio, leading to heart failure (Ding et al. 2008). Recently, several studies have demonstrated that the magnitude of ICa,L is considerably altered in response to a number of physiological and pathophysiological influences, like α-adrenergic activation, cardiac hypertrophy, heart failure, hypo- or hyperthyroidism, diabetes, anoxia, and development. This suggests that changes in the inward L-type Ca+2 channels constitute a pathway for altering Ca+2 influx and cardiac contractility (Volk et al. 1999). While the L-type Ca+2 channels’ role in heart failure is not well established, Chen et. al (2002) unmask important changes in the density as well as regulation of L-type Ca+2 channels in failing human ventricular myocytes.
Electrophysiological remodeling is associated with the progression of hypertrophy and heart failure (Tsuji et al. 2000). During excitation-contraction coupling, Ca+2 is rapidly cycled between the cytosol (where it activates the myofilaments) and the sarcoplasmic reticulum, the Ca+2 stores (Prestle et al. 2003). These fluxes occur through the transient activity of Ca+2 pumps and channels. In end-stage heart failure, cardiac contractility is depressed due to alterations in the structure and function of proteins or protein complexes. Ding et al. (2008) demonstrated progressive ventricular hypertrophy, dilation, and contractile depression in response to chronic volume overload, as well as reduced protein expression of sarcoplasmic reticulum (Ca+2)-ATPase, and ryanodine receptors in myocytes of 10-week-old fistula rats. In addition, a reduction in the intracellular Ca+2 transient and the peak currents in VO induced hypertrophied cardiocytes was present, even though the L-type Ca+2 channel activity was normal (Ding et al. 2008). Under some conditions, such as stretch, renin gene expression is enhanced (Malhotra et al. 1999), and overexpression of the angiotensinogen gene in normal mice leads to hypertrophy of the right and left ventricles and to an increase of angiotensin II (Ang II) levels in both ventricles without any change in arterial blood pressure (Mazzolai et al. 1998, 2000). Ang II evokes positive inotropic responses in various species. However, the effects of this peptide on the cardiac L-type Ca+2 current (ICa,L) are still controversial, especially in the VO hypertrophied model. Early studies using multicellular preparations described an increase in ICa,L after Ang II treatment (Freer et al. 1976; Kass and Blair 1981). However, more recent observations using isolated myocytes reported contradictory results: increase (Allen et al. 1988; De Mello 1998; De Mello and Monterrubio 2004; Kaibara et al. 1994; Kass and Blair 1981), no effect (Ai et al. 1998; Ikenouchi et al. 1994), and even decrease (Habuchi et al. 1995) in ICa,L induced by Ang II. In the failing heart, Ang II administration enhances ICa,L; however, only the intracellular Ang II increased the rate of ICa,L inactivation (De Mello and Monterrubio 2004). These findings contrast with those obtained in normal controls, in which intracellular dialysis of the same dose of Ang II was unable to change the rate of ICa,L inactivation (De Mello and Monterrubio 2004).
The maintenance of Ca+2 homeostasis is essential to normal cardiac function. In ventricular cardiocytes, voltage-gated L-type Ca+2 channels represent the major pathway for Ca+2 entry and play a crucial role in excitation-contraction coupling. The regulatory role of L-type channels is enhanced by the modulation of this Ca+2 current by a large variety of hormones and mediators, mainly through protein kinase activation (Dolphin 1996; Dzhura et al. 2000; Kimura et al. 2000; Taylor et al. 2000). Angiotensin II activates Ca+2 entry by stimulating L-type Ca+2 channels through Gβγ-sensitive phosphoinositide 3-kinase (PI3K) and protein kinase C in venous myocytes (Macrez et al. 1997, 2001; Viard et al. 1999). The results of Quignard et al. (2001) demonstrate the specific involvement of the γ isoform of PI3K in the transduction pathway leading to calcium channel stimulation and the rise of Ca+2 induced by Ang II in rat portal vein myocytes. In addition, Ang II stimulates PI3K through the modulation of Gβγ (Viard et al. 1999). The pharmacological inhibition of PI3K strongly attenuated Ang II-induced calcium mobilization and vascular smooth muscle cell contraction (Do et al. 2009). The objective of this study is to delineate the role of PI3K in the regulation of the slow calcium channel by Ang II in the volume overload-induced cardiac hypertrophy model.
Materials and methods
Animal preparation
Conformity statement
As discussed in the following respective sections, all the procedures conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) publication No. 85–23, revised 1996. Male Sprague–Dawley rats of 200–250 g body weight were purchased from Charles River Laboratories (Wilmington, Mass., USA). The rats were allowed to recover and acquaint themselves with their new environment upon arrival at the animal house of the College of Medicine, Howard University, for 1 week. The animals were kept under secure, clean and controlled room temperature (21–23 °C) with a 6 h dark : 18 h light cycle and were fed food and water ad libitum.
Eccentric cardiac hypertrophy
Adult male Sprague–Dawley rats (200–250 g) were anesthetized with sodium pentobarbital (30 mg/kg body weight, i.p.). The abdominal aorta was punctured at the union of the segment two-third caudal to the renal artery and the one-third cephalic to the aortic bifurcation with an 18-gauge disposable needle. The needle was advanced into the abdominal aorta and vena cava at the point of anastomosis, shunting arterial blood into the venous system. A drop of cyanoacrylate glue was used to seal the aorta-punctured point. The patency of the shunt was verified visually by swelling of the vena cava and the mixing of arterial and venous blood. As a postoperative care measure, the rat was administered flunixin 2.5 mg/kg. The same procedure was performed on the age-matched sham rats, except for the insertion of the 18-gauge needle in the abdominal aorta and vena cava. One week after surgery, shunted animals were treated with angiotensin-converting enzyme inhibitor (ACEI) and 0.5 g/L captopril per day in drinking water, and the second group was given no drugs; 3 weeks were allowed for the cardiac hypertrophy to develop. On the experimentation day, visual inspection of the lungs did not show any signs or symptoms of pulmonary edema or pulmonary blood clots in all shunted animals used. This is relevant to the fact that these shunted rats are still in the compensated eccentric cardiac hypertrophy phase and eliminate any decompensatory process to heart failure.
Isolation of adult rat cardiomyocytes
All the reagents were purchased from Sigma-Aldrich Chemicals (St. Louis, Mo., USA). Double-distilled water from the Milli-Q system (Millipore Corporation, Bedford, Mass., USA) was used to prepare all solutions. Stock buffer solution contained (mmol/L): 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, and 10 HEPES. Animals were injected with sodium heparin (1000 U/kg, i.p.) and anesthetized with pentobarbital sodium (40 mg/kg; i.p.), 20 min prior to removal of the heart. After the heart was excised, it was quickly transferred to a Langendorff setup for retrograde coronary perfusion through the aorta at 10 mL/min (37 °C) for an initial 5 min equilibration with a perfusion buffer (mmol/L): 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 HEPES, 1.1 d-glucose, and 10.2 butanedione monoxime. The experimental protocol consisted of continuing the retrograde perfusion of the hearts for 12 min with (30 mL) perfusion buffer solution (Digestion Buffer) containing (mg): 25 BSA (essentially fatty-acid free), 25 collagenase (type 2), and 3 protease (Type XIV). Then the heart was perfused for 5 min with perfusion buffer solution containing (5%) fetal calf serum and 50 μmol/L CaCl2. The ventricles were cut and minced into the stop buffer. Calcium was progressively reintroduced to the cells and the dissociated cardiomyocytes were kept in 1.5 mmol/L calcium containing buffer solution until later experimentation. Freshly isolated myocytes showing no signs of blebs or round edges were used for up to 12 h.
Electrophysiological studies
The perforated patch-clamp technique, using nystatin in the pipette solution (240 μg/mL), was performed on adult rat cardiomyocytes to avoid the known run-down phenomenon of the ICa,L. Patch pipettes (1 MΩ) were pulled from borosilicate glass capillary tubing with a 2-stage puller (David Kopf Instruments, Tujunga, Calif., USA). Ventricular myocytes were placed on the stage of an inverted microscope and perfused with an extracellular buffer (mmol/L): 5 KCl, 1 MgCl2, 140 NaCl, 10 HEPES, 10 d-glucose, 1.5 CaCl2, 20 TEA, 4-AP, and pH at 7.3. The intracellular solution contained (mmol/L): 130 CsOH-H2O, 130 l-glutamic acid, 10 HEPES, 10 d-glucose, and pH at 7.3. After the formation of a gigaohm seal, capacitance was estimated by integrating the area of the capacitance transient. The measured currents were divided by the cell capacitance to normalize for cell size changes between normal and hypertrophied cardiomyocytes. The cardiomyocytes were stimulated in voltage-clamp mode using pCLAMP 9.0 software (Molecular Devices, Sunnyvale, Calif., USA) connected to an Axopatch 200B amplifier through an A/D converter (Digidata 1320A; Molecular Devices). The resulting ionic currents were stored on a computer for analysis with pCLAMP 9.0. All patch-clamp experiments were performed at room temperature (20–22 °C). The holding potential was kept at −85 mV. A 300 ms pre-pulse to −45 mV was applied before the voltage protocol to inactivate all sodium channels. The current-voltage stimulus consisted of 350 ms of incremental 10mV voltage steps from −80 mV to +50 mV. Peak current levels were plotted as a function of the command potential. The action of Ang II in the presence and absence of specific blockers or inhibitors was analyzed for its effects on the current-to-voltage (I-V) relationship. The specific cell-permeable inhibitor for PI3K, LY 294002, was purchased from Cell Signaling Technology (Beverly, Mass., USA).
Western blotting
Activation of Akt was assessed using Western blot technique. Protein samples were prepared from perfused heart tissue using a lysis buffer containing (mmol/L): 20 β-glycerophosphate,1 EGTA, 0.5 NaVO3, 2 dithiothreitol, 10 benzamidine, 0.2 Na3VO4, 2 EDTA, 20 NaF, and 0.6% deoxycholate, 0.1% Triton X-100, and 1 tablet/10 mL of Complete protease inhibitors cocktail (Roche, Porterville, Calif., USA) (pH 7.5). Samples were matched for protein concentration, separated by SDS–PAGE, and transferred onto nitrocellulose membranes. After blocking in 5% nonfat milk in TBST (10 mmol/L Tris, 150 mmol/L NaCl, 0.1% Tween 20), the membranes were incubated with dual total and phospho-specific antibodies to Akt (Cell Signaling Technology) overnight at 4 °C. Afterward, membranes were washed 3 times in TBST, incubated with appropriate secondary antibody conjugated to horseradish peroxidase (Cell Signaling Technology) for 2 h, and then washed 3 times with TBST. Bands were visualized by Chemiluminescence (Renaissance, NEN Life Science Products, PerkinElmer, Waltham, Mass., USA). Films from at least 3 independent experiments were scanned and densities of the immunoreactive bands were evaluated using NIH Image software and normalized. Kinase activities were evaluated as the ratio of phosphorylated kinase over total protein kinase per experiment.
Statistical analysis
All statistical analysis was performed using SigmaStat software and verified using Microsoft Excel. The paired Student’s t test was used to compare data before and after drug treatment of the same animal group. The heteroscedastic 2-sample unpaired Student’s t test assuming unequal variances was used when comparing the drug effects between 2 different animal groups (sham vs. shunt). Using the null hypothesis, p ≤ 0.05 was considered significant.
Results
Structural parameters
The data on the structural parameters from sham and shunted adult rats confirmed the development of the eccentric cardiac hypertrophy within 3 weeks post-surgery, as seen in Table 1. The shunted rats had greater heart weights (shunts 2478 ± 91 mg vs. shams 1210 ± 22 mg), as well as relative heart weights (shunts 524 ± 4 mg/100 g body weight vs. shams 335 ± 6 mg/100 g body weight) when compared with the sham animals. Captopril-treated animals showed regression in the absolute (shams 1236 ± 15 mg vs. shunts 1908 ± 34 mg) and relative heart weights (sham 368 ± 9 mg/100 g body weight; shunt 530 ± 9 nmg/100 g body weight). In addition, the cellular membrane capacitance was significantly greater in the cardiomyocytes from hypertrophied hearts as compared with normal ones (273 ± 19 pF vs. 201 ± 6 pF).
Table 1.
Structural parameters of sham and volume-overloaded shunt adult rat hearts.
| Heart weight (mg) | Relative heart weight (mg/100 g body weight) | |
|---|---|---|
| Sham (25) | 1210±22 | 335±6 |
| Shunt (21) | 2478±91 | 524±4* |
| Sham + captopril (19) | 1236±15 | 368±9 |
| Shunt + captopril (17) | 1908±34 | 530±9*, † |
Note: Values are means ± SEM; number of rats is indicated within parentheses.
p < 0.05 vs. untreated sham.
p < 0.05 vs. captopril-treated sham.
PI3K inhibition and Ang II effects on ICa,L channels in sham and shunted hearts
Hypertrophied myocytes, as compared with normal myocytes, showed significant decreases in the peak basal current density levels of the L-type calcium current channels, ICa,L (Hypertrophied −6.4 ± 1.7 pA/pF vs. Normal −11.9 ± 1.1 pA/pF; p < 0.05, n = 8) (Fig. 1). Figure 2 shows that superfusion of normal cardiomyocytes with Ang II (10−6 mol/L) resulted in no effect on the ICa,L current density; however, the effect of Ang II on hypertrophied cardiomyocytes shows a significant increase in ICa,L densities (−13.4 ± 0.8 pA/pF; p < 0.05, n = 10) (Fig. 3). Similar to Ang II, the PI3K inhibitor, LY294002, had no effect on ICa,L channels in normal cardiomyocytes (Fig. 4). Interestingly, in hypertrophied cardiomyocytes, PI3K inhibition significantly increased current density for ICa,L (−13.1 ± 0.7 pA/pF; p < 0.05, n = 10) (Fig. 5).
Fig. 1.

L-type Ca+2 current (ICa,L) voltage relationship for normal cardiomyocytes and volume overload-induced hypertrophied cardiomyocytes, with the respective stimulation protocol shown as an inset. The inset in the graph shows the respective representative current responses at +0 mV for ICa,L. Data are presented as average current density ± SEM with n = 8. *, p < 0.05 vs. Control.
Fig. 2.
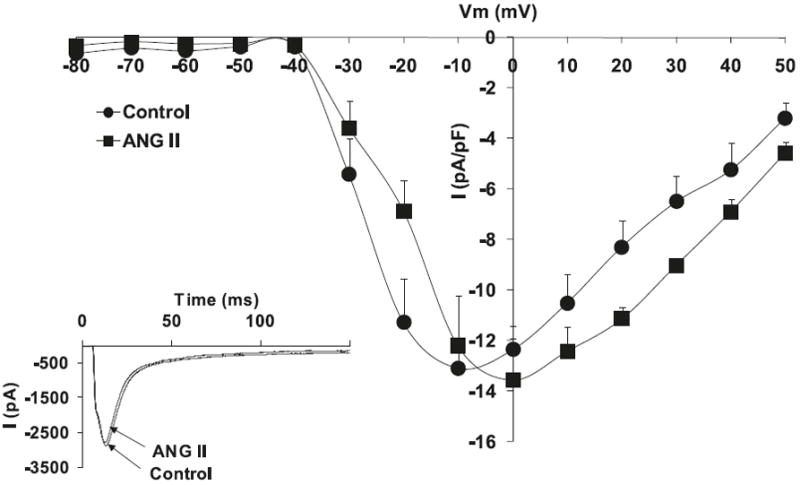
Effects of angiotensin II (Ang II) (10−6 mol/L) on L-type Ca+2 current (ICa,L) voltage relationship for normal cardiomyocytes, with the respective stimulation protocol shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa,L. Data are presented as average current density ± SEM with n = 10.
Fig. 3.
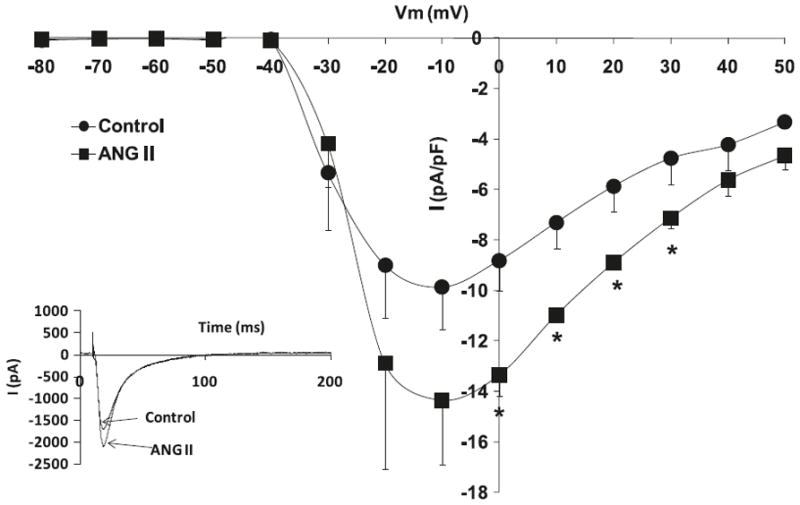
Effects of angiotensin II (Ang II) (10−6 mol/L) on L-type Ca+2 current (ICa,L) voltage relationship for volume overload-induced hypertrophied cardiomyocytes, with the respective stimulation protocol shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa,L. Data are presented as average current density ± SEM with n = 10. *, p < 0.05 vs. Control.
Fig. 4.
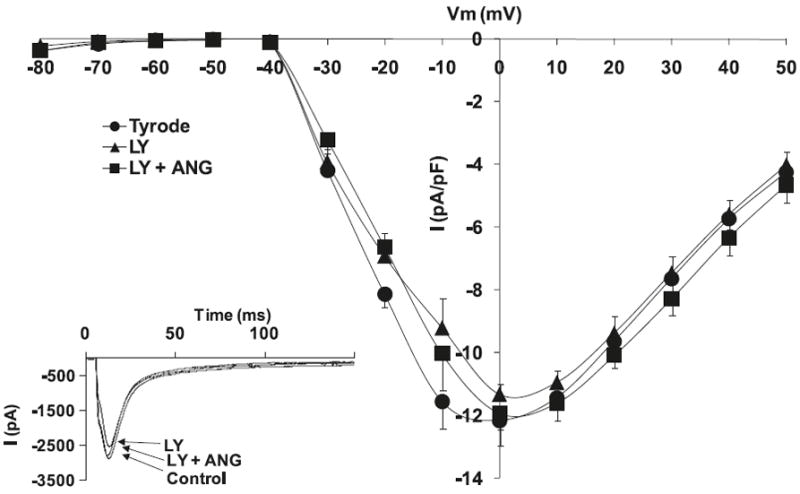
Effects of angiotensin II (Ang II) (10−6 mol/L) and phosphoinositide 3-kinase (PI3K) inhibitor, LY 294002 (1 μmol/L), on L-type Ca+2 current (ICa,L) voltage relationship for normal cardiomyocytes, with the respective stimulation protocol shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa,L. Data are presented as average current density ± SEM with n = 8.
Fig. 5.
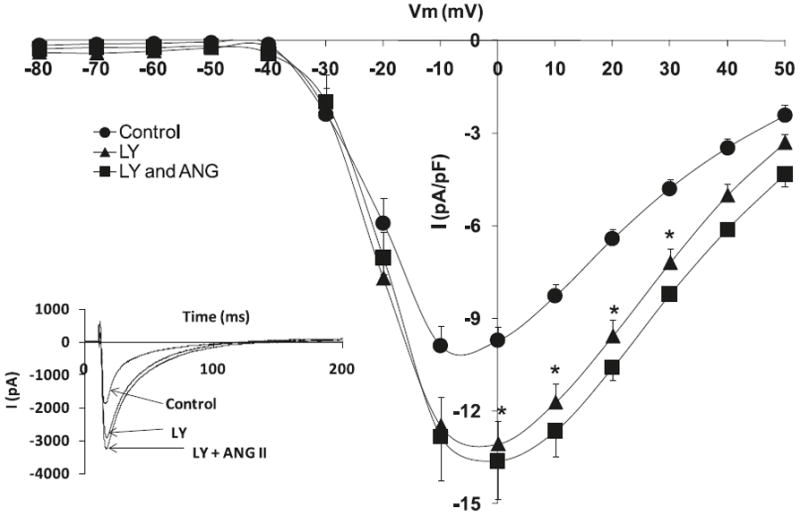
Effects of angiotensin II (Ang II) (10−6 mol/L) and phosphoinositide 3-kinase (PI3K) inhibitor, LY 294002 (1 μmol/L), on L-type Ca+2 current (ICa,L) voltage relationship for volume overload-induced hypertrophied cardiomyocytes, with the respective stimulation protocol shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa,L. Data are presented as average current density ± SEM with n = 10. *, p < 0.05 vs. Control.
Ang II effect on the Akt activation levels in normal sham and hypertrophied shunt hearts
We performed Western blot analysis to assess the level of activation of the PI3-kinase/Akt pathway in sham and shunt hearts that had been treated with Ang II versus untreated. The activation level of Akt was expressed as the ratio of phosphorylated Akt over total Akt protein expression. We found that the basal activation level of Akt in hypertrophied hearts was significantly higher than in the sham ones (Normal 1.00 ± 0.00 vs. Hypertrophied 2.06 ± 0.05; p < 0.01), as shown in Fig. 6. Treatment with Ang II did not have an effect on the activity level of Akt in the normal cardiomyocytes (1.06 ± 0.16 units); however, it downregulated the Akt activation level in the hypertrophied cardiomyocytes (1.84 ± 0.13; p < 0.05).
Fig. 6.
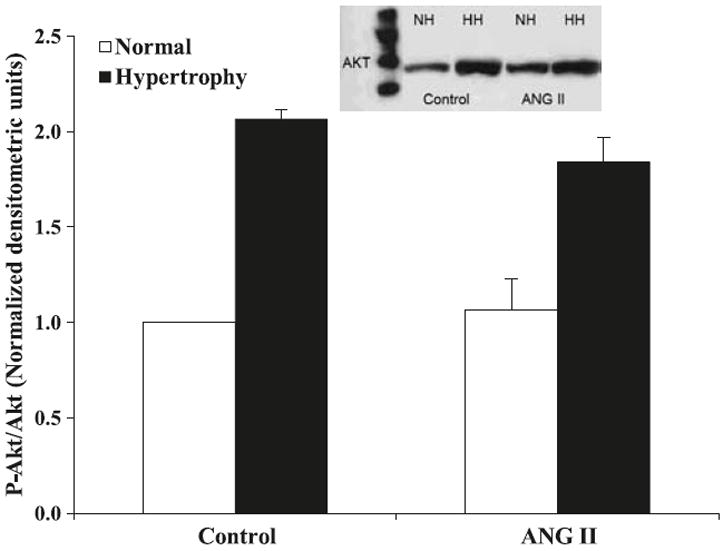
Effects of angiotensin II (Ang II) (10−6 mol/L) on the activation levels of Akt in sham (white bars) and volume overload-induced hypertrophied hearts (black bars). Data are expressed as the ratio of phosphorylated over total protein normalized to control untreated hearts. The inset shows representative Western blot. The data are presented as average ± SEM with n = 3 (from 3 different heart samples repeated 3 times). *, p < 0.05 Sham vs. Shunt; Δ, p < 0.05 Ang II-treated vs. Untreated.
PI3K inhibition and Ang II effects on captopril-treated ICa,L channels in sham and shunted hearts
Figure 7 shows that treatment with the ACE inhibitor, captopril, caused a significant reduction in peak ICa,L density (−7.2 ± 0.8 pA/pF, p < 0.05) as well as a slight negative shift of peak current from 0 mV to −10 mV. Acute administration of Ang II (10−6 mol/L) to normal adult cardiomyocytes routinely treated with captopril (14 days) did not result in any significant changes in the L-type calcium current density nor treatment with PI3K inhibitor, LY294002 (Fig. 7).
Fig. 7.
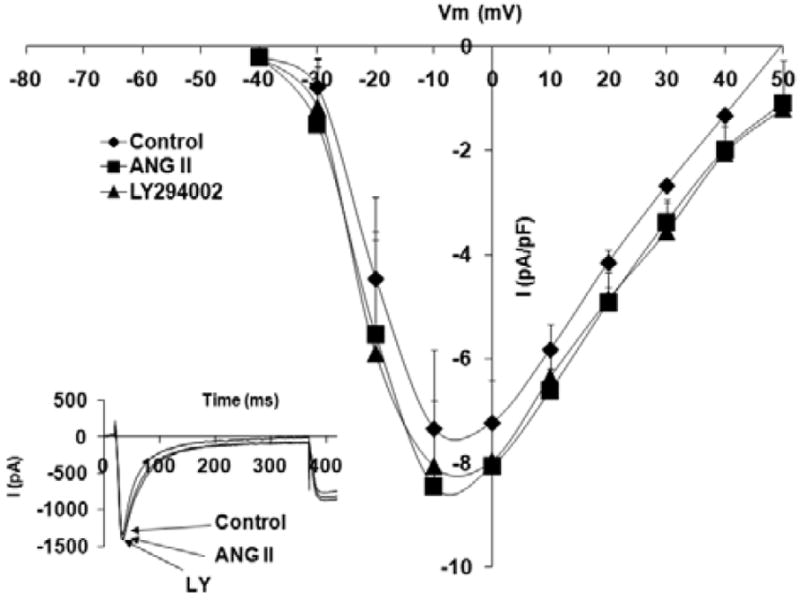
Effects of angiotensin II (Ang II) (10−6 mol/L) and phosphatidylinositol 3-kinase (PI3K) inhibitor, LY 294002 (1 μmol/L), on L-type Ca+2 current (ICa,L) voltage relationship for normal cardiomyocytes, pretreated with captopril for 14 days; the respective stimulation protocol is shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa,L. Data are presented as average current density ± SEM with n = 6.
However, hypertrophied cardiomyocytes from shunted adult rats treated with captopril showed a significant recovery in the ICa,L current density (captopril-treated hypertrophied −10.7 ± 1.5 pA/pF; n = 6, p < 0.05) (Fig. 8). The addition of Ang II induced a reduction in the L-type calcium current (Ang II −7.1 ± 1.0 pA/pF; n = 6, p < 0.05) of hypertrophied cardiomyocytes, which was reverted by PI3K inhibition with LY294002.
Fig. 8.

Effects of angiotensin II (Ang II) (10−6 mol/L) and phosphatidylinositol 3-kinase (PI3K) inhibitor, LY 294002 (1 μmol/L), on L-type Ca+2 current (ICa,L) voltage relationship for volume overload-induced hypertrophied cardiomyocytes pretreated with captopril for 14 days; the respective stimulation protocol is shown as an inset. The inset in the graph shows the respective representative current responses at 0 mV for ICa.L. Data are presented as average current density ± SEM with n = 6. *, p < 0.05 vs. Control; #, p < 0.05 vs. LY294002.
Discussion
The present study underlines the relationship between the L-type calcium channels and the effects of Ang II through the intracellular PI3-kinase pathway in the normal and hypertrophied cardiomyocytes from volume-overloaded adult rat hearts. We have shown that the basal intracellular PI3K signaling pathway seems to have a negative regulatory effect on ICa,L current density in hypertrophied cardiomyocytes. In addition, our data also showed enhanced PI3K/Akt signaling in association with the development of volume-overload cardiac hypertrophy. These findings may partly convey the reduction in the functional ICa,L current density seen in our cardiac hypertrophy model as compared with normal controls. On the other hand, we have also demonstrated that the reduced functional ICa,L current density of hypertrophied cardiomyocytes is improved by Ang II, partly in a PI3K-dependent manner. This corroborates with the fact that Ang II negatively regulates the PI3K pathway in our hypertrophied heart model. Interestingly, treatment of rats with volume overload-induced hypertrophied hearts with the angiotensin-converting-enzyme inhibitor, captopril, regressed the hypertrophy towards normal sham levels and inverted the Ang II effect on ICa,L seemingly through a PI3K upstream effector. In parallel, Ang II did not seem to significantly affect the activity level of the L-type calcium channel in cardiomyocytes of sham rat hearts.
Our data show that eccentric volume overload-induced cardiac hypertrophy is associated with lower L-type calcium channel activity. This is an interesting finding because others have shown similar reduction in ICa,L in the concentric or pressure-overload type of cardiac hypertrophy. Thus, it seems that this reduction in ICa,L current density is a general response to cardiac hypertrophy, which may play a role in setting the stage at the cellular level for the onset of heart failure. The development of cardiac hypertrophy is thought to be associated with a slow progressive build-up of an ischemic condition in the heart muscle (Schaper et al. 1991), which is known to activate the PI3K survival pathway in cardiomyocytes and partly mediates the reduction of cardiomyocyte contractility (Alloatti et al. 2004). The inotropic effect becomes relevant as the major modulator of cardiac contractility, as the L-type calcium channel activity is regulated by phosphorylation (Haddad et al. 1995). There was a clear difference in the response of ICa,L to Ang II between both normal and hypertrophied cardiomyocytes. The L-type calcium current in normal cardiomyocytes was not affected by PI3-kinase or by Ang II. Thus, it seems that ICa,L activity in the normal heart is not regulated by PI3-kinase. In fact, Ang II did not affect the activation level of Akt in normal hearts. This is in agreement with other findings showing that in the normal hearts, there was no significant Ang II-induced inotropic effect (Libonati et al. 1997), and that PI3K inhibition did not modulate the baseline contraction or calcium influx in adult rat cardiomyocytes (Leblais et al. 2004). In another study, it was shown directly that PI3-kinase had no effect on ICa,L of the wild-type normal cardiomyocytes (Lu et al. 2005). Furthermore, there was no alteration in ICa,L of transgenic mice lacking PI3-kinase (Kerfant et al. 2005, 2006). However, our study shows that the development of eccentric cardiac hypertrophy is associated with an enhanced Akt activity, which seems to have a negative modulatory effect on ICa,L channel activity. Hence, inhibition of PI3-kinase (upstream Akt effector) improved the ICa,L current density. It has been shown that Akt directly inhibits cAMP, a positive regulator of ICa,L in the heart (Kerfant et al. 2007; Leblais et al. 2004). As such, strong activation of Akt during eccentric cardiac hypertrophy could convey a significant reduction in ICa,L current density. Others have also shown reductions in ICa,L density during cardiac hypertrophy in other species (Bouron et al. 1992; Nuss and Houser 1991; Shi et al. 2007). In addition, we have shown that PI3-kinase inhibition negated the Ang II-dependent increase of ICa,L current density in the hypertrophied cardiomyocytes. This could suggest that the effect of Ang II on ICa,L in the hypertrophied cardiomyocytes may be PI3-kinase dependent. Angiotensin II effects are known to be mediated through Gqα sarcolemmal protein (Sperelakis et al. 1994), which has been shown to modulate the activity of ICa,L in a PI3K-dependent manner (Lu et al. 2005, 2009).
Surprisingly, treatment of normal cardiomyocytes with an angiotensin-converting-enzyme inhibitor, captopril, quenched the L-type calcium channel activity and induced a leftward shift in its peak current-voltage relationship. This can be translated into a weaker inotropic activity of the cardiomyocyte due to smaller calcium influx and lower action potential plateau, respectively. Nonetheless, neither Ang II nor LY294002 showed any effect on normal cardiomyocytes even after treatment with captopril. Therefore, it may be argued that captopril’s negative effect on ICa,L is not through the Ang II system or PI3-kinase signaling. In support of our data, earlier reports also showed that treatment with captopril decreased the ICa,L level of normal ventricular myocytes (Bryant et al. 1991), as well as reduced calcium influx without affecting the resting baseline Ca2+ concentration of WKY rats (Wang et al. 1996).
In contrast, regression of volume overload-induced cardiac hypertrophy by captopril was associated with enhanced L-type calcium channel activity. A recent study (Fernández-Campo et al. 2009) showed a reduction in the activity of Akt after captopril treatment of hypertensive subjects. This may alleviate the negative effect of PI3-kinase/Akt on ICa,L to a certain extent, leading to an enhancement of the activation level of ICa,L after captopril treatment. In another study using the pressure overload-induced cardiac hypertrophy model, reduced ICa,L was restored to control level by an angiotensin-converting-enzyme inhibitor, imidapril (Li et al. 2003). On the other hand, we have shown that inhibition of PI3-kinase seems to avert the Ang II effect on ICa,L of cardiomyocytes from captopril-treated rats. Thus, this may suggest that in the regressed hearts, the effects of Ang II are PI3K dependent to a large extent. It should be noted that captopril treatment of rats with volume overload-induced cardiac hypertrophy inverted the effects of Ang II on the L-type calcium channels. This is not due to a captopril-induced alteration in PI3-kinase signaling to the L-type calcium channels, since LY294002 enhanced ICa,L in the untreated hypertrophied cardiomyocytes and the captopril-treated ones. Thus, the switch in the effect of Ang II seems to reside upstream from PI3-kinase in the Ang II signaling pathway.
Therefore, this study introduces a novel role of the PI3-kinase pathway in the modulation of the functional calcium channel density during the development of volume overload-induced cardiac hypertrophy. In addition, our data indicate that effects of Ang II on the functional calcium channel density may be partly mediated through a PI3-kinase-dependent mechanism. Furthermore, we have shown that regression of volume overload-induced cardiac hypertrophy by captopril improved ICa,L, which seems to be partly regulated through PI3-kinase. Thus, the PI3-kinase pathway may represent a target of opportunity for the modulation of the inotropic activity of volume overload-induced hypertrophied hearts to delay or prevent its progression into overt heart failure.
Acknowledgments
This work was supported in part by grants GM08016–38 NIGMS/NIH, 2G12 RR003048 RCMI, Division of Research Infrastructure, National Center for Research Resources (NCRR) / National Institutes of Health (NIH) and Specialized Neuroscience Research Programs (SNRP) to GEH.
Contributor Information
Zikiar Alvin, Department of Physiology and Biophysics, College of Medicine, Howard University, 520 W Street, WA 20059, USA.
Graham G. Laurence, Department of Physiology and Biophysics, College of Medicine, Howard University, 520 W Street, WA 20059, USA
Bernell R Coleman, Department of Physiology and Biophysics, College of Medicine, Howard University, 520 W Street, WA 20059, USA.
Aiqiu Zhao, Department of Physiology and Biophysics, College of Medicine, Howard University, 520 W Street, WA 20059, USA.
Majd Hajj-Moussa, Université St-Esprit de Kaslik, Kaslik St, Jounieh, Lebanon.
Georges E. Haddad, Department of Physiology and Biophysics, College of Medicine, Howard University, 520 W Street, WA 20059, USA.
References
- Ai T, Horie M, Obayashi K, Sasayama S. Accentuated antagonism by angiotensin II on guinea-pig cardiac L-type Ca-currents enhanced by β-adrenergic stimulation. Pflugers Arch. 1998;436(2):168–174. doi: 10.1007/s004240050619. [DOI] [PubMed] [Google Scholar]
- Allen IS, Cohen NM, Dhallan RS, Gaa ST, Lederer WJ, Rogers TB. Angiotensin II increases spontaneous contractile frequency and stimulates calcium current in cultured neonatal rat heart myocytes: insights into the underlying biochemical mechanisms. Circ Res. 1988;62(3):524–534. doi: 10.1161/01.res.62.3.524. [DOI] [PubMed] [Google Scholar]
- Alloatti G, Montrucchio G, Lembo G, Hirsch E. Phosphoinositide 3-kinase γ: kinase-dependent and -independent activities in cardiovascular function and disease. Biochem Soc Trans. 2004;32(2):383–386. doi: 10.1042/BST0320383. [DOI] [PubMed] [Google Scholar]
- Bouron A, Potreau D, Raymond G. The L type calcium current in single hypertrophied cardiomyocytes isolated from the right ventricle of ferret heart. Cardiovasc Res. 1992;26(7):662–670. doi: 10.1093/cvr/26.7.662. [DOI] [PubMed] [Google Scholar]
- Bryant SM, Ryder KO, Hart G. Effects of captopril on membrane current and contraction in single ventricular myocytes from guinea-pig. Br J Pharmacol. 1991;102(2):462–466. doi: 10.1111/j.1476-5381.1991.tb12195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BM, Bassani RA, Franchini KG, Bassani JW. Enhanced calcium mobilization in rat ventricular myocytes during the onset of pressure overload-induced hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291(4):H1803–H1813. doi: 10.1152/ajpheart.01345.2005. [DOI] [PubMed] [Google Scholar]
- Chen X, Piacentino V, III, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91(6):517–524. doi: 10.1161/01.RES.0000033988.13062.7C. [DOI] [PubMed] [Google Scholar]
- De Mello WC. Intracellular angiotensin II regulates the inward calcium current in cardiac myocytes. Hypertension. 1998;32(6):976–982. doi: 10.1161/01.hyp.32.6.976. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Monterrubio J. Intracellular and extracellular angiotensin II enhance the L-type calcium current in the failing heart. Hypertension. 2004;44(3):360–364. doi: 10.1161/01.HYP.0000139914.52686.74. [DOI] [PubMed] [Google Scholar]
- Ding YF, Brower GL, Zhong Q, Murray D, Holland M, Janicki JS, Zhong J. Defective intracellular Ca2+ homeostasis contributes to myocyte dysfunction during ventricular remodelling induced by chronic volume overload in rats. Clin Exp Pharmacol Physiol. 2008;35(7):827–835. doi: 10.1111/j.1440-1681.2008.04923.x. [DOI] [PubMed] [Google Scholar]
- Do KH, Kim MS, Kim JH, Rhim BY, Lee WS, Kim CD, Bae SS. Angiotensin II-induced aortic ring constriction is mediated by phosphatidylinositol 3-kinase/L-type calcium channel signaling pathway. Exp Mol Med. 2009;41(8):569–576. doi: 10.3858/emm.2009.41.8.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19(1):35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2(3):173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- Fernández-Campo L, Grande MT, Diego J, Fuentes-Calvo I, Macías-Núñez JF, Sánchez-Rodríguez A, et al. Effect of different antihypertensive treatments on Ras, MAPK and Akt activation in hypertension and diabetes. Clin Sci (Lond) 2009;116(2):165–173. doi: 10.1042/CS20080119. [DOI] [PubMed] [Google Scholar]
- Freer RJ, Pappano AJ, Peach MJ, Bing KT, McLean MJ, Vogel S, Sperelakis N. Mechanism for the postive inotropic effect of angiotensin II on isolated cardiac muscle. Circ Res. 1976;39(2):178–183. doi: 10.1161/01.res.39.2.178. [DOI] [PubMed] [Google Scholar]
- Habuchi Y, Lu LL, Morikawa J, Yoshimura M. Angiotensin II inhibition of L-type Ca2+ current in sinoatrial node cells of rabbits. Am J Physiol. 1995;268(3 Pt 2):H1053–H1060. doi: 10.1152/ajpheart.1995.268.3.H1053. [DOI] [PubMed] [Google Scholar]
- Haddad GE, Sperelakis N, Bkaily G. Regulation of the calcium slow channel by cyclic GMP dependent protein kinase in chick heart cells. Mol Cell Biochem. 1995;148(1):89–94. doi: 10.1007/BF00929507. [DOI] [PubMed] [Google Scholar]
- Ikenouchi H, Barry WH, Bridge JH, Weinberg EO, Apstein CS, Lorell BH. Effects of angiotensin II on intracellular Ca2+ and pH in isolated beating rabbit hearts and myocytes loaded with the indicator indo-1. J Physiol. 1994;480(2):203–215. doi: 10.1113/jphysiol.1994.sp020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric D, Yao X, Thandapilly SJ, Louis XL, Cantor E, Chaze B, et al. Defects in ryanodine receptor function are associated with systolic dysfunction in rats subjected to volume overload. Exp Physiol. 2010;95(8):869–879. doi: 10.1113/expphysiol.2009.052100. [DOI] [PubMed] [Google Scholar]
- Kaibara M, Mitarai S, Yano K, Kameyama M. Involvement of Na+-H+ antiporter in regulation of L-type Ca2+ channel current by angiotensin II in rabbit ventricular myocytes. Circ Res. 1994;75(6):1121–1125. doi: 10.1161/01.res.75.6.1121. [DOI] [PubMed] [Google Scholar]
- Kass RS, Blair ML. Effects of angiotensin II on membrane current in cardiac Purkinje fibers. J Mol Cell Cardiol. 1981;13(9):797–809. doi: 10.1016/0022-2828(81)90237-6. [DOI] [PubMed] [Google Scholar]
- Kerfant BG, Gidrewicz D, Sun H, Oudit GY, Penninger JM, Backx PH. Cardiac sarcoplasmic reticulum calcium release and load are enhanced by subcellular cAMP elevations in PI3Kγ-deficient mice. Circ Res. 2005;96(10):1079–1086. doi: 10.1161/01.RES.0000168066.06333.df. [DOI] [PubMed] [Google Scholar]
- Kerfant BG, Rose RA, Sun H, Backx PH. Phosphoinositide 3-kinase γ regulates cardiac contractility by locally controlling cyclic adenosine monophosphate levels. Trends Cardiovasc Med. 2006;16(7):250–256. doi: 10.1016/j.tcm.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Kerfant BG, Zhao D, Lorenzen-Schmidt I, Wilson LS, Cai S, Chen SR, et al. PI3Kγ is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res. 2007;101(4):400–408. doi: 10.1161/CIRCRESAHA.107.156422. [DOI] [PubMed] [Google Scholar]
- Kimura M, Osanai T, Okumura K, Suga S, Kanno T, Kamimura N, et al. Involvement of phosphorylation of β-subunit in cAMP-dependent activation of L-type Ca2+ channel in aortic smooth muscle-derived A7r5 cells. Cell Signal. 2000;12(1):63–70. doi: 10.1016/S0898-6568(99)00068-6. [DOI] [PubMed] [Google Scholar]
- Leblais V, Jo SH, Chakir K, Maltsev V, Zheng M, Crow MT, et al. Phosphatidylinositol 3-kinase offsets cAMP-mediated positive inotropic effect via inhibiting Ca2+ influx in cardiomyocytes. Circ Res. 2004;95(12):1183–1190. doi: 10.1161/01.RES.0000150049.74539.8a. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu Z, Xiao J, Zhang C, Ma J, Liu N. Long-term effects of imidapril on calcium and potassium currents in rabbit left ventricular hypertrophic myocytes. Chin Med J (Engl) 2003;116(12):1795–1798. [PubMed] [Google Scholar]
- Libonati JR, Eberli FR, Sesselberg HW, Apstein CS. Effects of low-flow ischemia on the positive inotropic action of angiotensin II in isolated rabbit and rat hearts. Cardiovasc Res. 1997;33(1):71–81. doi: 10.1016/S0008-6363(96)00185-X. [DOI] [PubMed] [Google Scholar]
- Lu Z, Jiang YP, Ballou LM, Cohen IS, Lin RZ. Gαq inhibits cardiac L-type Ca2+ channels through phosphatidylinositol 3-kinase. J Biol Chem. 2005;280(48):40347–40354. doi: 10.1074/jbc.M508441200. [DOI] [PubMed] [Google Scholar]
- Lu Z, Jiang YP, Wang W, Xu XH, Mathias RT, Entcheva E, et al. Loss of cardiac phosphoinositide 3-kinase p110 α results in contractile dysfunction. Circulation. 2009;120(4):318–325. doi: 10.1161/CIRCULATIONAHA.109.873380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez N, Morel JL, Kalkbrenner F, Viard P, Schultz G, Mironneau J. A βγ dimer derived from G13 transduces the angiotensin AT1 receptor signal to stimulation of Ca2+ channels in rat portal vein myocytes. J Biol Chem. 1997;272(37):23180–23185. doi: 10.1074/jbc.272.37.23180. [DOI] [PubMed] [Google Scholar]
- Macrez N, Mironneau C, Carricaburu V, Quignard JF, Babich A, Czupalla C, et al. Phosphoinositide 3-kinase isoforms selectively couple receptors to vascular L-type Ca2+ channels. Circ Res. 2001;89(8):692–699. doi: 10.1161/hh2001.097864. [DOI] [PubMed] [Google Scholar]
- Malhotra R, Sadoshima J, Brosius FC, III, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin–angiotensin system in cardiac myocytes in vitro. Circ Res. 1999;85(2):137–146. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- Mazzolai L, Nussberger J, Aubert JF, Brunner DB, Gabbiani G, Brunner HR, Pedrazzini T. Blood pressure-independent cardiac hypertrophy induced by locally activated renin-angiotensin system. Hypertension. 1998;31(6):1324–1330. doi: 10.1161/01.hyp.31.6.1324. [DOI] [PubMed] [Google Scholar]
- Mazzolai L, Pedrazzini T, Nicoud F, Gabbiani G, Brunner HR, Nussberger J. Increased cardiac angiotensin II levels induce right and left ventricular hypertrophy in normotensive mice. Hypertension. 2000;35(4):985–991. doi: 10.1161/01.hyp.35.4.985. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Houser SR. Voltage dependence of contraction and calcium current in severely hypertrophied feline ventricular myocytes. J Mol Cell Cardiol. 1991;23(6):717–726. doi: 10.1016/0022-2828(91)90981-Q. [DOI] [PubMed] [Google Scholar]
- Prestle J, Quinn FR, Smith GL. Ca2+-handling proteins and heart failure: novel molecular targets? Curr Med Chem. 2003;10(11):967–981. doi: 10.2174/0929867033457656. [DOI] [PubMed] [Google Scholar]
- Quignard JF, Mironneau J, Carricaburu V, Fournier B, Babich A, Nurnberg B, et al. Phosphoinositide 3-kinase γ mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J Biol Chem. 2001;276(35):32545–32551. doi: 10.1074/jbc.M102582200. [DOI] [PubMed] [Google Scholar]
- Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, et al. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83(2):504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- Shi CX, Wang YH, Dong F, Zhang YJ, Xu YF. Transmural L-type calcium current in a pressure-overloaded mouse model with heart failure. Sheng Li Xue Bao. 2007;59(1):19–26. [PubMed] [Google Scholar]
- Sperelakis N, Xiong Z, Haddad G, Masuda H. Regulation of slow calcium channels of myocardial cells and vascular smooth muscle cells by cyclic nucleotides and phosphorylation. Mol Cell Biochem. 1994;140(2):103–117. doi: 10.1007/BF00926749. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Green KN, Carpenter E, Peers C. Protein kinase C evokes quantal catecholamine release from PC12 cells via activation of L-type Ca2+ channels. J Biol Chem. 2000;275(35):26786–26791. doi: 10.1074/jbc.M003881200. [DOI] [PubMed] [Google Scholar]
- Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, Kodama I. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res. 2000;48(2):300–309. doi: 10.1016/S0008-6363(00)00180-2. [DOI] [PubMed] [Google Scholar]
- Viard P, Exner T, Maier U, Mironneau J, Nurnberg B, Macrez N. Gβγ dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB J. 1999;13(6):685–694. doi: 10.1096/fasebj.13.6.685. [DOI] [PubMed] [Google Scholar]
- Volk T, Nguyen TH, Schultz JH, Ehmke H. Relationship between transient outward K+ current and Ca2+ influx in rat cardiac myocytes of endo- and epicardial origin. J Physiol. 1999;519(3):841–850. doi: 10.1111/j.1469-7793.1999.0841n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang L, Qi JH, Zhang PJ, Wei PJ, Gu PK, Jin ZJ. Effects of captopril and enalaprilat on intracellular Ca2+ content in isolated cardiomyocytes from rats. Zhongguo Yao Li Xue Bao. 1996;17(3):233–235. [PubMed] [Google Scholar]
- Wang X, Ren B, Liu S, Sentex E, Tappia PS, Dhalla NS. Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J Appl Physiol. 2003;94(2):752–763. doi: 10.1152/japplphysiol.00248.2002. [DOI] [PubMed] [Google Scholar]