

Abstract

A catalytic stereoselective 1,4-diboration of conjugated dienes with B2(pin)2 was accomplished with Ni(cod)2 and PCy3 as the catalyst. This reaction broadens the substrate scope of current methods for catalytic diene diboration by including internal and sterically hindered dienes, and it proceeds efficiently at low catalyst loadings. The intermediate allylboronate was oxidized to the stereodefined allylic 1,4-diol.
The catalytic diboration of unactivated unsaturated hydrocarbons can be accomplished with many transition metals including Pt, Pd, Rh, Cu, Au, and Ag.1 The 1,4 diboration of 1,3-dienes, in particular, allows for the transformation of simple hydrocarbons to synthetically useful allylic diboronate intermediates.2 Recently, we developed the first catalytic enantioselective version of this type of diboration. This reaction employs a platinum catalyst in conjunction with chiral TADDOL-derived phosphonite ligands and delivers the 1,4-diborylated product in high enantiomeric excess.3 The resultant bis(boronate) esters can be utilized in carbonyl allylation and, through appropriate oxidation strategies, can be converted to butenolides and 2-alkene-1,4-diols. A current limitation of both the enantioselective and the non-enantioselective processes is that they have only been demonstrated with terminal dienes or cyclic dienes and they require the use of moderately expensive catalyst precursors. In this publication, we address these issues and describe a convenient, inexpensive catalytic diene diboration that operates on an expanded substrate scope.
During development of the Ni-catalyzed borylative coupling of dienes and aldehydes,4 we found that Ni(cod)2, in the presence of tricyclohexylphosphine, promotes the 1,4-diboration of a 1,3-diene by bis(pinacolato)diboron (B2(pin)2). The fact that the reaction delivered a single regio- and stereoisomer of product, and that it employed a simple inexpensive catalyst and reagents prompted us to further study this transformation. Our investigation of the reaction began with a brief analysis of ligand effects in the reaction. As depicted in Scheme 1, the diboration of trans-1,3-decadiene was examined in the presence of 2.5 mol % Ni(cod)2 and a near-stoichiometric quantity of B2(pin)2. This survey revealed that strongly donating monodentate phosphine ligands, including both PCy3 and hexamethylphosphorous triamide (HMPT),5 are highly effective in the reaction and after only 15 minutes, catalyzed complete conversion to the 1,4-bis(boronate) product; oxidation delivered the (Z)-2-alkene-1,4-diol in good yield.
Scheme 1.
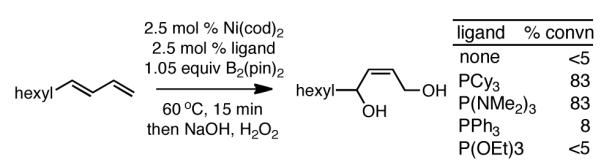
Effect of Ligands on Ni-Catalyzed Diene Diboration
The substrate scope of the Ni/PCy3-catalyzed diboration reaction was investigated with a series of conjugated diene substrates (Table 1). To ensure complete conversion, the reactions were generally allowed to proceed for three hours. As represented in entries 1 and 2, substrates with aromatic and alkyl substitution were converted to the derived 1,4-diol product in high yield and stereoselectivity. Multiply substituted dienes react to provide trisubstituted alkenes with high levels of olefin stereocontrol (entries 3-5). Remarkably, the highly substituted diene in entry 5 was efficiently converted to the derived 1,4-diol in good yield. Notably, silyl ethers and more Lewis basic benzyl ethers are tolerated in the reaction (entries 7 and 8) although an allylic silyl ether was incompatible with the reaction conditions (entry 6). The refractory nature of the allylic ether may arise from the known proclivity for allyl ethers to furnish π-allyl complexes in the presence of Ni(0) complexes.6 Impressively, and in contrast to Pt-catalyzed diboration, there is no decrease in regioselectivity when a quaternary carbon is adjacent to the diene; the 1,4-diol is obtained in high yield and high regioselectivity (entry 8; a 1:1 mixture of 1,4- and 1,2-products are observed in the Pt-catalyzed reaction).3
Table 1.
Ni-Catalyzed 1,4-Diboration of Conjugated Dienes(a)
| entry | substrate | product | yield (%)(b) |
|---|---|---|---|
| 1 |

|
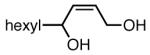
|
84 |
| 2 |

|
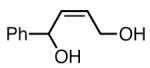
|
84(c) |
| 3 |

|

|
88 |
| 4 |

|
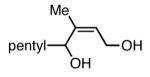
|
94 |
| 5 |
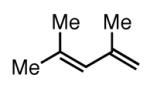
|
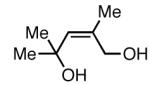
|
71 |
| 6 |
|

|
- |
| 7 |

|
|
95 |
| 8 |

|

|
88 |
| 9 |

|
no reaction | - |
| 10 |
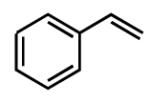
|
no reaction | - |
Reactions conducted at [substrate] = 0.25 M and oxidized with 30% H2O2 and 3 M NaOH.
Isolated yield of purified material. Values are an average of two experiments.
1 mol % catalyst Ni(cod)2 and 1 mol% PCy3 employed for this experiment.
Similar to Pt-catalyzed diboration, only dienes able to adopt the S-cis conformation participate in 1,4-diboration.3 As seen in entry 9 (Table 1), an A(1,3) interaction destabilizes the Sc-is conformation of the substrate, and this is likely the reason the reaction does not proceed. However, the more hindered diene in entry 5 contains off-setting A(1,3) interactions in the S-cis and S-trans conformers, and this reaction proceeds efficiently. Unlike Pt-catalyzed diboration,7 however, simple alkenes do not react at all under these conditions (entry 10).
To further test the hypothesis that the S-cis conformation is the reactive one, diborations of cis-and trans-1,3-pentadiene were compared by 1H NMR as the reaction progressed (Scheme 2). After one hour of reaction, trans-pentadiene was completely converted to the derived 1,4-bis(boronate) ester. In contrast, the reaction of cis-pentadiene was incomplete (82% conversion). Further, the remaining starting material was found to consist of a roughly equal mixture of the trans and cis isomer. Thus while cis-dienes can participate in the 1,4-diboraiton, they appear to do so at a slower rate and the reactive pathway may involve prior isomerization to the trans diene isomer.
Scheme 2.

Comparison of the Diboration of Cis and Trans-1,3-Pentadiene
Recent examples of catalytic diene diboration have surrounded terminal dienes and cyclic substrates; internal dienes have not been studied. To learn whether Ni catalysts might effectively expand the scope of diene diboration, a series of internal dienes was examined under the above described reaction conditions. Preliminary experiments suggested that longer reaction times are required, but with this modification, internal acyclic dienes were found to be willing participants in the diboration (Table 2). Generally, the internal dienes react efficiently and provide the internal Z-syn-1,4-diol after oxidation. As depicted in Table 2, both aryl- and alkyl-substituents are tolerated (entries 1-4). Cyclic dienes also participate in the Ni-catalyzed diboration, but with somewhat inferior yields (entries 5 and 6).
Table 2.
Ni-Catalyzed 1,4-Diboration of Internal Dienes(a)

| entry | substrate | product | yield (%)(b) |
|---|---|---|---|
| 1 |
|

|
77 |
| 2 |
|

|
66 |
| 3 |
|

|
74 |
| 4 |
|

|
72 |
| 5 |

|

|
44 |
| 6 |

|

|
47 |
Reactions conducted at [substrate] = 0.25 M and oxidized with 30% H2O2 and 3 M NaOH.
Isolated yield of purified material. Values are an average of two experiments.
To examine the synthetic utility of the subsequent diboration, a large scale Ni-catalyzed diboration of 1,3-decadiene was examined (Scheme 3). Noteworthy is that this substrate and other terminal dienes are readily prepared on multigram scale by the Wittig methylenation of enals. In the case of 1,3-decadiene, it was found that on 4.5 gram scale, the Ni-catalyzed reaction proceeded effectively and delivered the derived 1,4-diol in 87% yield. It should be noted that purification of the product, on large scale, is greatly aided by subjection of the crude reaction mixture to three equivalents of NaIO4; this effectively removes pinacol which is the only by-product of this reaction.
Scheme 3.
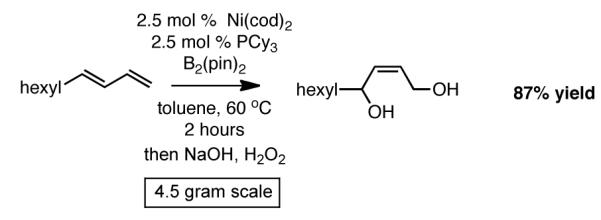
Large Scale Diboration of 1,3-Decadiene
The commonly accepted mechanism for diboration of dienes with group 10 metals involves oxidative addition of the boron species to the metal followed by coordination of the alkene. Subsequent insertion into the M-B bond with concomitant π-allyl formation, followed by reductive elimination provides the product.2 However, the observation that styrene is unreactive suggests that this mechanism may not be operative.8 An alternate possibility involves initial association of Ni(0) with the diene to form complex 1 or 2 (Scheme 4).9 Subsequent reaction with B2(pin)2 would provide the least hindered Ni-C bond which provides the product after reductive elimination. Current studies are aimed at elucidating these features of this reaction.
Scheme 4.
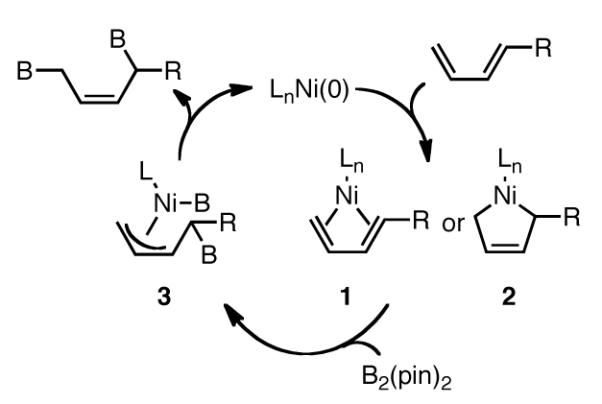
Proposed Catalytic Cycle for Diene Diboration
In summary, we have developed a novel Ni-catalyzed 1,4-diboration of conjugated dienes. This system expands the scope of dienes to provide internal 1,4-bis(boronate)esters, provides more reliable 1,4-selectivity with sterically hindered dienes, and requires short reaction times. Further studies on the mechanism and synthetic utility are underway in our laboratory.
Supplementary Material
Acknowledgment
This work was supported by the NIH (GM 64451).
Footnotes
Supporting Information. Complete experimental procedures and characterization data (1H and 13C NMR, IR, and mass spectrometry). This material is free of charge via the internet at http://pubs.acs.org.
References
- (1)(a).For a review of Pt- and Rh-catalyzed diboration: Norman NC, Marder TB. Top. Catal. 1998;5:63. For Pd: Yang FY, Cheng H. J. Am. Chem. Soc. 2001;123:761. doi: 10.1021/ja005589g. Woodward AR, Burks HE, Sieber JD, Morken JP. J. Am. Chem. Soc. 2004;126:16328. doi: 10.1021/ja044167u. Lillo V, Mas-Marzá E, Segarra A, Carbó JJ, Bo C, Peris E, Fernandez E. Chem. Commun. 2007:3380. doi: 10.1039/b705197b. For Cu:Lillo V, Fructos MR, Braga AAC, Maseras F, Reguejo MM, Pérez PJ, Fernandez E. Chem.-Eur. J. 2007;13:2614. doi: 10.1002/chem.200601146. Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:16630. doi: 10.1021/ja9089928. For Au: Baker RT, Nguyen P, Marder TB, Westcott SA. Angew. Chem. Int. Ed. 1995;34:1336. Corberan R, Ramirez J, Sanau M, Peris E, Fernandez E. Tetrahedron: Asymmetry. 2006;17:1759. Ramirez J, Sanau M, Fernandez E. Angew. Chem. Int. Ed. 2008;47:5194. doi: 10.1002/anie.200800541. For Ag: Corberan R, Ramirez J, Poyatos M, Peris E, Fernandez E. Chem. Commun. 2005:3056. doi: 10.1039/b503239c.
- (2)(a).Ishiyama T, Yamamoto M, Miyaura N. Chem. Commun. 1996:2073. [Google Scholar]; (b) Clegg W, Thorsten J, Marder TB, Norman NC, Orpen AG, Peakman TM, Quayle MJ, Rice CR, Scott AJ. J. Chem. Soc., Dalton Trans. 1998:1431. [Google Scholar]
- (3).Burks HE, Kliman LT, Morken JP. J. Am. Chem. Soc. 2009;131:9134. doi: 10.1021/ja809610h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cho HY, Morken JP. J Am. Chem. Soc. 2008;130:16140. doi: 10.1021/ja806113v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For an excellent description of the electronic properties of HMPT, see: Moloy KG, Petersen JL. J. Am. Chem. Soc. 1995;117:7696.
- (6)(a).Eisch JJ, Im KR. J. Organomet. Chem. 1977;139:C45. [Google Scholar]; (b) Hayashi T, Konishi M, Yokota K, Humada M. Chem. Commun. 1981:313. [Google Scholar]; (c) Yamamoto T, Ishizu J, Yamamoto A. J. Am. Chem. Soc. 1981;103:6863. [Google Scholar]
- (7)(a).Pt-catalyzed alkene diboration: Ishiyama T, Yamamoto M, Miyaura N. Chem. Commun. 1997:689. Iverson CN, Smith MR., III Organometallics. 1997;16:2757. Marder TB, Norman NC, Rice CR. Tetrahedron Lett. 1998;39:155. Mann G, John KD, Baker RT. Organic Letters. 2000;2:2105. doi: 10.1021/ol006008v. Lillo V, Mata J, Ramirez J, Peris E, Fernandez E. Organometallics. 2006;25:5829.
- (8).Similarly, reaction of 2-vinylnaphthalene occurs with <25% conversion to a mixture of compounds, none of which appear to be a diboration product.
- (9)(a).Benn R, Betz P, Goddard R, Jolly PW, Kokel N, Kruger C, Topalovic IZ. Naturforsch. 1991;46:1395. [Google Scholar]; (b) Karsch HH, Leithe AW, Reisky M, Witt E. Organometallics. 1999;18:90. [Google Scholar]; (c) Shirakawa E, Takahashi G, Tsuchimoto T, Kawakami Y. Chem .Commun. 2002:2210. doi: 10.1039/b207185a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.