

Abstract
Metabolism of β-amyloid peptide (Aβ) is closely associated with the pathology and etiology of Alzheimer’s disease (AD). Our previous studies on aging primates and rodents have revealed that early life lead exposure increases the expression of the β-amyloid precursor protein (AβPP), elevates Aβ levels, and promotes neurodegeneration in old age. These effects were attributed to de novo synthetic pathways; however, the impact on Aβ degradation was not explored. Neprilysin (NEP), a rate-limiting catabolic peptidase is involved in Aβ metabolism in vivo. In the present study we sought to investigate whether accumulation of Aβ induced by Pb exposure is partially due to its ability to subdue NEP expression and consequently NEP activity. SH-SY5Y cells were exposed to Pb concentrations of 0, 5, 10, 20, and 50 μM for 48 h and AβPP, NEP protein and mRNA levels were measured. Additionally, NEP enzymatic activity and Aβ levels were also assessed. Western blot and RT-PCR analysis indicated significant increases in the protein and mRNA expression of AβPP, which appeared to be concentration and time-dependent, while the protein and mRNA expression of NEP as well as NEP activity declined. These actions of Pb were specific and were not observed when substituted by another metal. These results suggest that Pb causes both the overexpression of AβPP and repression of NEP resulting in the buildup of Aβ.
Keywords: Alzheimer’s, Neprilysin, Pb, Aβ, β-amyloid precursor protein, SH-SY5Y cells
1. Introduction
Alzheimer's disease (AD) is the most common form of dementia that affects aging individuals. This age-related disease is characterized by a range of changes in brain anatomy, biology, and function (Goedert and Spillantini, 2006; Lahiri et al., 2003; Sambamurti et al. 2006; Selkoe, 1991; Tanzi and Bertram, 2008). Currently, about 25 million people worldwide are affected by this disorder, of which 4.5 million people live in the United States (Hebert et al., 2003). Similar to many other neurodegenerative diseases, AD is a genetically complex and heterogeneous disorder. The majority of AD cases (~95%) are non-familial, late onset sporadic forms (LOAD, >65 years) that possess no clear genetic association (Migliore and Coppede, 2009). Nutritional, metabolic, environmental and social factors have been linked with the onset and progression of the disease, but, despite the extent of inquiry in this area, the cause of LOAD remains obscure.
Lead (Pb) is widely recognized as a potent central neurotoxin that interferes with neuronal functions and causes a wide variety of long lasting adverse effects, especially in developing brains (Toscano and Guilarte, 2005; White et al., 2007). Epidemiological investigations have repeatedly associated developmental Pb exposure with several adverse effects on the neurobehavioral system (Counter et al., 2005; Shih et al., 2007). Furthermore, past and recent research have established a definite relationship between prenatal and postnatal low level Pb exposure and children’s cognitive functional disabilities (Braun et al., 2006; Lidsky and Schneider, 2003; McGlothan et al., 2008). Although it has been restricted three decades ago in the United States, Pb remains one of the most widespread and insidious environmental burdens (White et al., 2007).
Previous work from our lab has shown that expression of AD-related genes as well as their transcriptional regulator specificity protein 1 (Sp1) are altered in rodents and primates exposed to Pb as infants. These characteristics were manifested by an increase in the expression of the β-amyloid precursor protein (AβPP), β-amyloid cleaving enzyme (BACE) and the β-amyloid peptide (Aβ), followed by increased levels of oxidative damage to DNA, and a reduction in DNA methyltransferase activity (Wu et al., 2008). Studies have been devoted to support a linkage between Pb exposure and the pathogenesis of LOAD; however, the underlying molecular mechanisms behind the subtle neurotoxic effects of lower levels of Pb and the onset of LOAD are not fully defined.
It is well established that the extracellular deposition of Aβ peptide plays a central role in the development of AD (Kayed et al., 2003; Kounnas et al., 2010; Roher et al., 1996). The steady state levels of Aβ are determined by the metabolic balance between its rate of synthesis and its rate of clearance. Synthesis of Aβ is determined by the levels of AβPP from which it is processed and that is also dependent on the de novo synthesis of AβPP as well as its stability. On the other hand, proteases like neprilysin (NEP) and insulin degrading enzyme (IDE), along with others, have been implicated in Aβ degradation (El-Amouri et al., 2008; Higuchi et al., 2005; Iwata et al., 2001; Marr et al., 2004; Miners et al., 2006).
In this study we have focused on AβPP as a generator of Aβ and on NEP as a remover of Aβ. AβPP is an integral membrane protein with a role in neurite outgrowth, post-natal somatic growth and neurobehavioral development (Glass et al., 2010; Heber et al., 2000). Proteolytic processing of AβPP by secretases results in the production of Aβ peptide (Gervais et al., 1999). NEP is a 97 kDa type II membrane-associated protein which is predominantly localized at the presynaptic terminal and is involved in degrading the monomeric and the oligomeric forms of Aβ peptide (El-Amouri et al., 2007; Kanemitsu et al., 2003).
In order to test the impact of Pb on both the synthetic and degradative pathways of Aβ, we exposed differentiated SH-SY5Y cells to a series of Pb concentrations and monitored the expressions of AβPP and NEP, two primary proteins that regulate Aβ levels.
2. Materials and methods
2.1. Cell culture
SH-SY5Y cells were obtained from American Type Culture Collection (ATCC, VA) and were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium (Invitrogen, MD) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2mM L-glutamine in a CO2 incubator maintained at 5% CO2 and 37°C. In order to differentiate SH-SY5Y cells, they were stimulated with 10 μM all-trans retinoic acid (Sigma-Aldrich, MO) in DMEM/F12 medium containing 1% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine in the dark, and were examined for neurite outgrowth at 48, 72 h, and 6 days (Jamsa et al., 2004). The medium was changed every 3 days. The morphology of cultured cells was examined and photomicrographs were obtained with a 200× objective lens on a Nikon ECLIPSE camera (TE2000-E) adapted to the microscope.
2.2. Pb exposure
Differentiated SH-SY5Y cells were exposed to Pb as follows: A 10 mM Pb stock solution was prepared by dissolving the appropriate amount of Pb-acetate in sterile double-distilled H2O. Experimental Pb concentrations were prepared by dilution of stock solution in DMEM/F12 medium containing 1% FBS, sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. Furthermore, differentiated cells were incubated with 0, 5, 10, 20, 50 μM of Pb for different time periods (24, 48 and 72 h) at 37 C, with 0 μM Pb samples serving as the control group.
2.3. Cell viability assay
3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazoliumbromide (MTT) was obtained from Sigma-Aldrich, MO (Product No. TOX1-1KT). After the third passage, 1×104 cells/well were seeded and differentiated on collagen coated 96-well plates with eight replicates per treatment group. Cells were exposed for 24, 48, or 72 h to a series of Pb concentrations (0, 1, 5, 10, 50, 100, 500, 10000, 1, 5, 10, 50, 100, 500, 2000, and 5000 μM Pb) and were incubated at 37 C with 5% CO2 and 90% humidity. A 5mM Pb working solution was prepared by dilution of 10mM stock solution in the ratio of 1:1 in 2% FBS, hence keeping the concentration of the nutrient comparable to control. After incubation for the appropriate time points, 10 μl of MTT-labeling reagent (included with the kit) was added to each well. Plates were incubated with MTT-labeling reagent for 4 h, followed by addition of 100 μl of solubilizing solution (included with the kit) to each well. Plates were incubated overnight and on the following day absorbance of samples was measured using a microplate reader (Spectra max M2, Molecular Devices, CA). The wavelength for measuring formazan product was 570 nm, and the reference wavelength was 690 nm.
2.4. Western blot analysis
For AβPP protein, cells were lysed with RIPA lysis buffer (150 mM NaCl, 25 mM Tris-HCl at pH 8.0, 1% NP-40, 10 mM NaF, 1mM Na3VO4) containing 1% protease inhibitors. Homogenates were sonicated and vortexed for 5 min before centrifugation at 10,000 × g for 20 min. For NEP protein, the total membrane protein proportion of SH-SY5Y cells was extracted by Eukaryotic Membrane Extraction Reagent Kit (Pierce, IL). Total protein concentration was determined by using BCA kit (Thermo Scientific, IL) and samples were electrophoresed on 8% SDS/PAGE and then transferred to polyvinylidiene diflouride (PVDF) membrane at 20 V for 30 min. Non-specific binding was blocked by incubation with 5% BSA in TBST at room temperature for 1 h. Membranes were incubated with MAB348 N-terminal specific antibody mouse mAb overnight (diluted at 1:1000, Millipore, MA) or neprilysin (56C6) antibody (diluted at 1:1000, Abcam, MA) with gentle agitation on a shaker at 4°C. On the following day, membranes were washed and exposed to HRP conjugated anti-mouse antibody (diluted at 1:5000, Pierce Biotechnology, IL) for 1 h. The blots were developed using Amersham ECL plus system and exposed with Typhoon™ 9410 MultiMode Imager (GE Healthcare Biosciences, NJ). As a control for equal protein loading, membranes were stripped and reprobed with rabbit β-actin antibody (diluted at 1:2500, Sigma-Aldrich, MO) and exposed to HRP conjugated anti-rabbit antibody (diluted at 1:5000, Pierce Biotechnology, IL).
2.5. RNA Isolation and Real-time PCR
Total RNA from control and exposed cells was extracted with the TRIzol reagent (Invitrogen, CA). First-strand cDNA was synthesized from 1.5 μg of total RNA using the iScript cDNA kit (Bio-Rad, CA). cDNA was then amplified using real-time PCR. The SYBR Green qRT-PCR assay was performed in 25 μl reactions in replicates using 1.5 μl of cDNA template, 1×SYBR Green master mix, 0.4 μM forward and reverse primers, and deionized H2O. The following primer pairs were used: AβPP forward primer 5′-GCC AAA GAG ACA TGC AGT GA -3′, reverse primer 5′-CCA GAC ATC CGA GTC ATC CT -3′; NEP forward primer 5′-CCC AGT GCA TGG TGT ATC AG -3′, reverse primer 5′-TGG CCT ATA GGT TCC ACA CC -3′; GAPDH forward primer 5′-AGC TGA ACG GGA AGC TCA CT -3′, reverse primer 5′-AGG TCC ACC ACT GAC ACG TTG -3′. Amplification was undertaken on the ABI PRISM 7500 machine (Applied Biosystems, CA) with Sequence Detection Software (SDS) version 1.3, and expression was reported relative to GAPDH mRNA with 2-ΔΔCt method.
2.6. Sandwich ELISA measurement of Aβ1–40
Cells were grown to ~80% confluence in 60 mm dishes. After differentiation, the culture medium was replaced with 4 ml fresh culture medium with Pb. The cells were further cultured for 48 h and the culture medium was collected. Human Aβ1–40 was detected in the collected culture medium by ELISA using human Aβ1–40 assay kit JP27713 (IBL, Gunma, Japan). Briefly, 100 μg of total protein from medium samples were placed in each well of a 96-well plate coated with monoclonal antibody specific for the human Aβ35–40 (1A10) and was incubated overnight at 4°C. On the following day, the 96-well plate was given extensive washings with EIA buffer for 7 times followed by addition of 100 μl of labeled antibody to each well containing sample or standard and incubated at 4°C for 1 h. The wells were again washed 9 times with EIA buffer followed by the addition of 100 μl TMB solution and were incubated in the dark for 30 min at room temperature. The reaction was terminated by adding 100 μl of 1NH2SO4 and the colorimetric absorption was taken at 450 nm. The levels of Aβ1–40 in the test samples were calculated relative to the standard curve generated for each plate.
2.7. Fluorometric assay of NEP activity
Proteolytic enzymes have a fundamental role in multiple biological processes and are associated with several pathological conditions. Therefore, targeting these enzymes may be important for a better understanding of their function and development of therapeutic inhibitors. Fluorescence Resonance Energy Transfer (FRET) peptides are convenient tools for the study of peptidases specificity as they allow monitoring of the reaction on a continuous basis, providing a rapid method for the determination of enzymatic activity. FRET is a distance-dependent excited state interaction in which emission of one fluorohore is coupled to the excitation of another. It occurs primarily because the acceptor dipole interacts or resonates with the donor dipole. The use of FRET is to obtain structural maps of complex biological structures, primarily proteins and other macromolecular assemblies such as ribosomes and nucleosomes. Measurements of energy transfer can provide intra- or intermolecular distance data for proteins and their ligands in the range 10–100 Angstrom. Also, FRET can detect change in distance (1–2 Angstrom) between loci in proteins; hence it is a sensitive measure of conformational change.
In order to determine NEP enzymatic activity by FRET method we used the substrate N-dansyl -d-Ala- Gly -p-(nitro)- Phe-Gly (DAGNPG, Sigma-Aldrich, MO), which is principally degraded by NEP, and to a smaller extent by angiotensin-converting enzyme (ACE). Differentiated cells were homogenized in 6 volumes of 50 mM Tris (pH 7.4) and debris was removed by centrifugation at 1000 × g for 10 min. To prevent the fluorogenic substrate cleavage by ACE, 100 μg of cell lysate was pre-incubated with the ACE inhibitor enalapril for 30 min at 37°C in the presence or absence of phosphoramidon, a specific NEP inhibitor. Following this pre-incubation, 50 μM DAGNPG was added and samples were incubated for an additional 1 h in a volume of 200 μl at 37°C. Reactions were terminated by heating samples at 100°C for 5 min, followed by centrifugation at 5000 × g for 5 min to remove the denatured protein. The supernatant was diluted up to 400 μl with 50 mM Tris (pH 7.4) and the fluorescence was determined using a microplate reader (excitation, 342 nm; emission, 562 nm).
2.8. Statistical analysis
All results are represented as mean ± SEM. Statistical analyses were performed with one-way ANOVA followed by significant difference post hoc analysis and t-test with a threshold value of P < 0.05. All statistical analyses were performed using SPSS 12.0 software.
3. Results
3.1. Cytotoxicity of Pb
Recent in vivo studies have demonstrated that low-level Pb exposure during development does not result in neuronal death (Jones et al., 2008); however, we wanted to establish that the Pb concentrations used do not induce cytotoxicity in our in vitro system. Differentiated SH-SY5Y cells were exposed to a series of Pb concentrations (0–5000 μM) and cell viability was assessed using MTT assay following 24, 48 or 72 h of exposure. There was no significant cytotoxicity for 1, 5, 10, 20 and 50 μM of Pb at either 48 or 72 h (Fig. 1A). At the highest concentration of Pb exposure (> 1000 μM) for 72 h, cell viability was significantly reduced and cell damage was enhanced by ~2 fold compared to the control group (P < 0.01). Marked morphological changes in the form of loss of neurites and reduction in the number of cells bearing neurites was also observed at these concentrations at 72 h (Fig. 1B). Thus, Pb exposure at concentrations of 0, 5, 10, 20 and 50 μM for 24 and 48 h were considered low and appropriate for further studies.
Fig. 1.
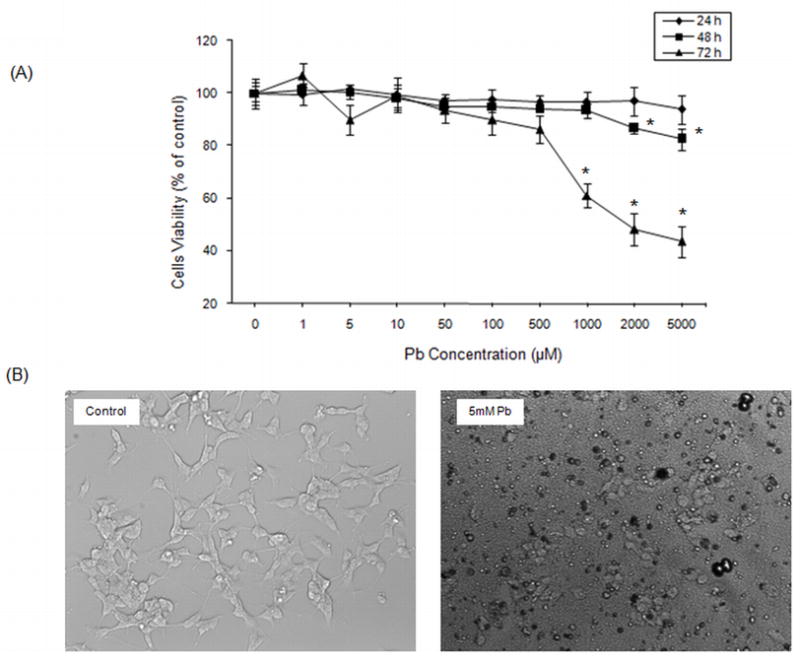
Pb exposure and cytotoxicity in differentiated SH-SY5Y cells. Cells were exposed for 24, 48, or 72 h to a series of Pb concentrations and cell viability was monitored using the MTT assay. After cells were exposed to Pb (0–5000 μM), images were collected with a Nikon ECLIPSE phase-contrast microscope (200×). Viability data shown in (A) are for the mean ± SEM. *P<0.05. (B) Photomicrographs showing the morphological effects of 5 mM Pb on SH-SY5Y cells after 72 h.
3.2. Pb altered protein expression and mRNA levels of AβPP and NEP
Western blot analysis using an antibody directed against AβPP or NEP was used to study the protein expression of AβPP and NEP in control and Pb exposed cells. Our results indicated an increase in AβPP protein expression within 48 h of Pb exposure, with a significant elevation observed at 20 μM and 50 μM Pb concentrations (P < 0.05) (Fig. 2A). We also observed a decline in NEP protein expression following Pb exposure in these cells with the most significant decrease at 48 h for the 20 μM and 50 μM Pb (P < 0.05) (Fig. 2B).
Fig. 2.
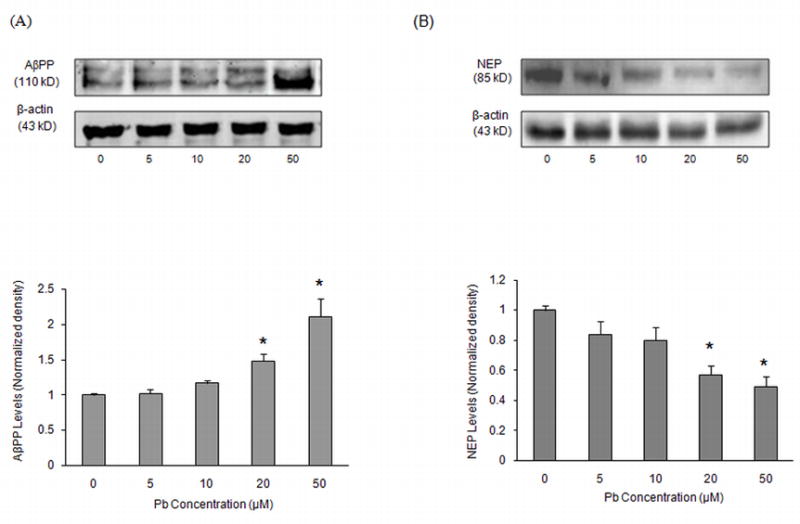
AβPP and NEP levels in SH-SY5Y cells after exposure to different concentrations of Pb. Cells were exposed to Pb (0–50 μM) for 48 h. Above are representative Western blots of AβPP (A) and NEP (B) and below is their quantification after normalization to β-actin levels. Data shown are for the mean ± SEM for each protein. Three independent experiments were performed in triplicate; *P < 0.05 versus control at same time points.
The cellular effects of Pb are widespread and are elicited through a variety of different mechanisms, including altered gene transcription (Garza et al., 2006). Pb is thought to interfere with gene expression by competing for the metal binding sites of transcription factors, such as zinc finger proteins (Basha et al., 2003; Zawia, 2003). We performed quantitative real-time RT-PCR to evaluate the effects of Pb on intracellular AβPP and NEP mRNA levels, relative to GAPDH. Fig. 3 depicts an increase in AβPP mRNA levels following exposure to 5, 10, 20, and 50 μM Pb for 48 h, which was observed to be significant in the range of 10–50 μM (P < 0.05). However for NEP, the mRNA expression was down-regulated following Pb exposure. Our data further revealed that relative intensities of NEP mRNA were more sensitive to Pb than AβPP, which was depicted by a significant decrease in NEP mRNA levels at 5–10 μM of Pb for 48 h (P < 0.05). No further decline in NEP was observed with higher Pb concentrations (20–50 μM).
Fig. 3.
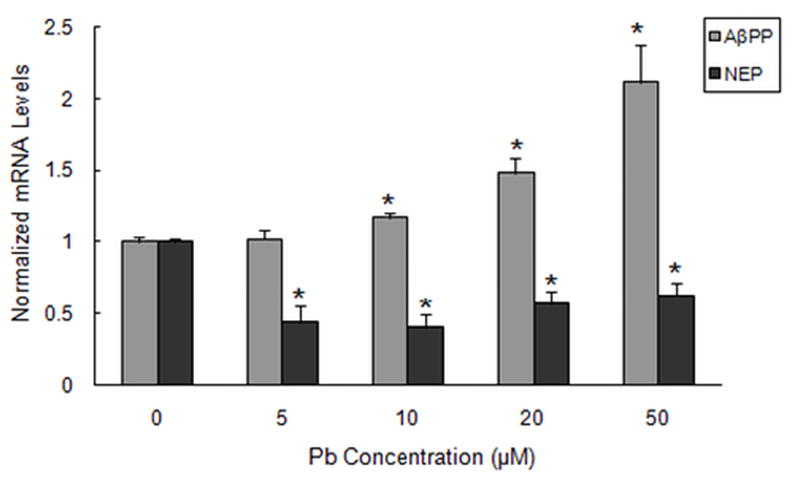
Effect of Pb exposure on the mRNA levels of AβPP and NEP in SH-SY5Y cells. Cells were exposed to Pb (0–50 μM) for 48 h. Total mRNA levels were analyzed by real-time RT-PCR and normalized to GAPDH. Results shown are for the mean ± SEM of relative mRNA level to control values. Three independent experiments were performed in triplicate; *P < 0.05 versus control at same time points.
3.3. Effect of Pb on secreted Aβ1–40
The above data displayed that increased AβPP expression is associated with decreased NEP expression at 48 h following Pb exposure. We thus predicted that a corresponding increase in Aβ1–40 levels would occur since both AβPP and NEP play a central role in regulating Aβ metabolism. We compared secreted Aβ1–40 levels in the control and Pb-exposed cells. SH-SY5Y cells were exposed to 5–50 μM Pb for 48 h and the Aβ1–40 secreted into the medium was measured by ELISA (Fig. 4A). Our results indicate increased Aβ1–40 levels in response to Pb exposure in a concentration-dependent manner. Thus, the observed increase in Aβ1–40 levels can be correlated with the increased AβPP and decreased NEP expression after 48 h of exposure to Pb.
Fig. 4.
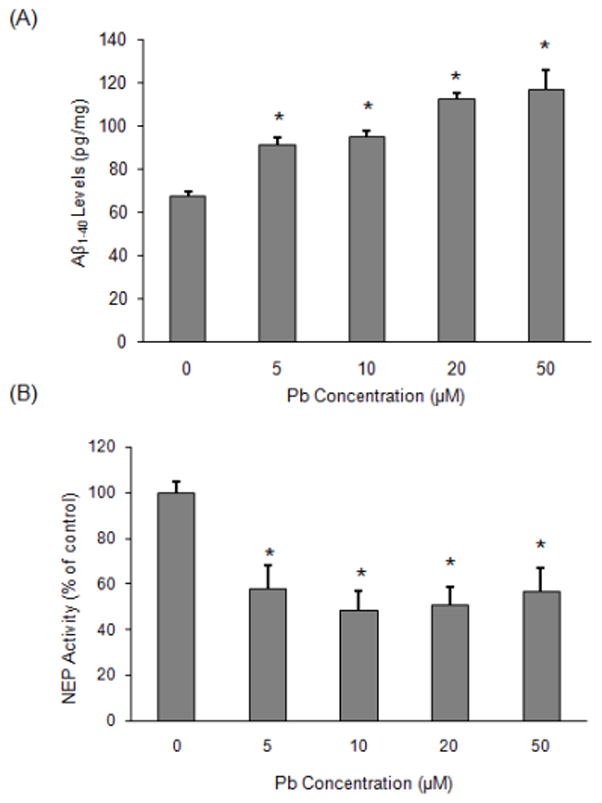
Quantification of Aβ levels and NEP activity in SH-SY5Y cells in response to Pb. Cells were exposed for 48h to Pb (0–50 μM). Extracellular Aβ was measured by sandwich ELISA. Whole cells were harvested and NEP activity was analyzed for each group by fluorescence resonance energy transfer. Specific NEP activity was calculated by subtracting residual fluorescent intensity after incubation with the NEP inhibitor phosphoramidon. (A) Aβ levels, (B) NEP enzyme activity. Values expressed as a percent of control for the mean ± SEM. Three independent experiments were performed in triplicate; *P < 0.05 versus control.
3.4. NEP activity is altered following Pb exposure
Next, we investigated NEP activity after exposure of differentiated SH-SY5Y cells to various Pb concentrations. A significant decrease of ~42% and ~51% was observed in NEP activity after the addition of 5 or 10 μM Pb for 48 h in comparison to the control group (P < 0.05) (Fig. 4B). No further decline in NEP activity was observed at higher concentrations of Pb (20 μM or 50 μM). In order to further determine the time course for Pb action, we exposed cells with 10 μM Pb for different time periods. As shown in Fig. 5A, NEP activity was not changed after exposure for up to 24 h; however, at the 48 h time point, NEP activity was down-regulated significantly (P < 0.05). To further scrutinize the specificity of Pb-induced decrease in NEP activity, we exposed cells to 10 μM magnesium (Mg) as a negative control. Compared to the controls, NEP activity was lowered by ~50% in cells exposed to Pb, while no decrease was observed in those exposed to Mg (Fig. 5B).
Fig. 5.
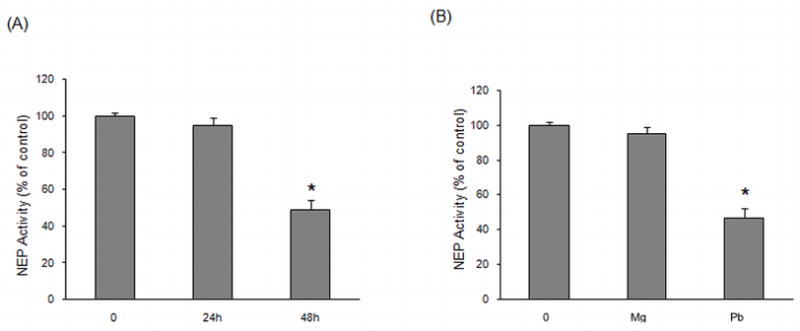
Time course and specificity of the action of Pb on NEP activity in SH-SY5Y cells. Whole cells were harvested and NEP activity was analyzed for each group by fluorescence resonance energy transfer. (A) NEP activity at various times following exposure to 10 μM Pb. (B) NEP activity after exposure to 10 μM Pb or Mg for 48 h. Values are for the mean ± SEM expressed as the percentage of controls. Three independent experiments were performed in triplicate; *P < 0.05 versus control.
4. Discussion
Cerebral deposition of Aβ, an invariant feature of AD, reflects an imbalance between the rates of Aβ production and clearance (Farris et al., 2007). The causes of Aβ elevation in LOAD are largely unknown, although many endogenous metals such as zinc (Zn), copper (Cu), iron (Fe), and Mg are suspected to contribute to the pathogenesis of AD (Lovell et al., 1998; Maynard et al., 2002; Rivera-Mancia et al., 2010). Few studies have focused on environmental heavy metals that are not part of cellular factors. Among such heavy metals, Pb poses widespread public concern. Exposure to Pb occurs through paint, glazed earthenware, lead piping, solder in food containers, moonshine whiskey, and automobile battery casings. Previous reports from our lab have demonstrated that AβPP mRNA and protein expression in the cerebral cortex increases with age and are abnormally elevated in aged rats or primates exposed to Pb as infants (Basha et al., 2005; Wu et al., 2008).
Consistent with our earlier in vivo studies, the present in vitro experiments also displayed a significant increase in AβPP protein expression and mRNA levels following exposure to Pb at 48 h, which was gradually followed by an increase in Aβ levels. Since the steady state levels of the cleavage products of AβPP are dependent on the de novo synthesis of AβPP, proteolytic processing, as well as the clearance of these products (Murphy and Levine, 2009), we also measured NEP levels and found them to be decreased in cells exposed to different concentrations of Pb. Thus our data suggest that Pb interferes in multiple ways to alter the turnover of AβPP and its Aβ products.
NEP is a plasma membrane glycoprotein of the neutral Zn metalloendopeptidase and is also known as physiologically important Aβ-degrading enzyme in mammalian central nervous system (El-Amouri, 2007; Roques et al., 1980; Zou et al., 2006). NEP expression and activity is modulated by various factors that are associated with AD. An inverse relationship between NEP immunoreactivity and amyloid plaque formation in various brain regions of control and AD patients has been reported (Akiyama et al., 2001). NEP is susceptible to oxidative damage by metal-mediated oxidation and that can increase its susceptibility to proteolysis (Adlard and Bush, 2006).
It has been reported that the expression or activity of NEP was reduced in the rat brain by aluminum (Al) (Luo et al., 2009) and also down-regulated by Cu in WT-7 cells (Li et al., 2010). However, little is known about the impact of Pb on NEP expression. Furthermore, NEP activity was also decreased at 48 h following Pb exposure suggesting that the decrease in activity is a direct result of lowered NEP protein levels and not a direct effect of the metal on the enzyme. Along with the lowered NEP protein levels and activity, there was also a significant down-regulation of NEP mRNA expression. The decrease in mRNA and protein, and activity all occurred after 48 hours suggesting that reduction of NEP gene expression was responsible for the lowering of NEP protein and activity. This decreased Aβ-catabolic activity of NEP induced by Pb would thus result in reduced Aβ degradation, which ultimately leads to enhanced Aβ accumulation. It was particularly interesting to note that we still can detect a gradual rise in Aβ at low levels of Pb exposure (5–10 μM). In recent years, some attention about Aβ buildup has been shifted from AβPP cleavage to the processes responsible for peptide degradation.
Emerging evidence also suggest a regulatory link between Aβ production and regulation of the NEP gene. A direct link to Aβ production and NEP gene regulation was proposed by Pardossi-Piquard and coworkers (2005), as they postulated that the NEP gene was transcriptionally regulated by one of the products of AβPP metabolism, the amyloid intracellular domain (AICD). In addition, recent studies by Belyaev et al. (2009) have verified a direct interaction of AICD with the NEP promoters by binding histone acetylation markers (H4K8, H4K16) in NB7 cells. Furthermore, it was shown that the chromatin of NEP promoter was associated with histone deacetylase binding in SH-SY5Y cells by chromatin immunoprecipitation analysis. Hence, they proposed unique insights into the mechanisms underlying the NEP expression regulation by the chromatin acetylation status of its promoter.
A question posed by these findings relates to how Pb exposure can result in alternate effects on gene expression. While it is still not understood how Pb can induce the expression of some genes and repress that of others, gene array studies on primates in our lab have shown that 80% of the genes altered late in life due to infantile Pb exposure were down-regulated, whilst 20% were up-regulated (data submitted for publication). Genome-wide expression profiling in mice brains also displayed similar results (data submitted for publication).
In conclusion, the present results indicate that the accumulation of Aβ is a product of disturbances in two different pathways: one associated with Aβ production and the other with its elimination. Exposure to Pb up-regulates the expression of the AβPP gene, which is then translated into more AβPP protein products and subsequent Aβ processing. On the other hand, Pb down-regulates the NEP gene, causing a reduction in NEP protein levels and thus reduces available active enzyme acting to degrade Aβ. Both pathways converge and result in the overproduction of Aβ. Future studies will decipher how Pb can have such alternate effects on gene expression.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS) and by grant NIH-5RO1ES015867-03. The research core facility was funded (P20RR016457) by the National Center for Research Resources (NCRR), a component of NIH. The authors would like to thank Lina Adwan and Gehad Subaiea for reviewing and editing the manuscript.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adlard PA, Bush Al. Metal and Alzheimer’s disease. J Alzheimers Dis. 2006;10:145–63. doi: 10.3233/jad-2006-102-303. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kondo H, Ikeda K, Kato M, McGeer PL. Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid beta-protein (Abeta) deposition. Brain Res. 2001;902:277–81. doi: 10.1016/s0006-8993(01)02390-3. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Brydie M, Razmiafshari M, Zawia NH. Lead-induced developmental perturbations in hippocampal Sp1 DNA-binding are prevented by zinc supplementation: in vivo evidence for Pb and Zn competition. Int J Dev Neurosci. 2003;21:1–12. doi: 10.1016/s0736-5748(02)00137-5. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, et al. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaev ND, Nalivaeva NN, Makova NZ, Turner AJ. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 2009;10:94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JM, Kahn RS, Froehlich T, Auinger P, Lanphear BP. Exposures to environmental toxicants and attention deficit hyperactivity disorder in U.S. children. Environ Health Perspect. 2006;114:1904–9. doi: 10.1289/ehp.9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter SA, Buchanan LH, Ortega F. Neurocognitive impairment in lead-exposed children of Andean lead-glazing workers. J Occup Environ Med. 2005;47:306–12. doi: 10.1097/01.jom.0000155717.45594.65. [DOI] [PubMed] [Google Scholar]
- El-Amouri SS, Zhu H, Yu J, Gage FH, Verma IM, Kindy MS. Neprilysin protects neurons against Abeta peptide toxicity. Brain Res. 2007;1152:191–200. doi: 10.1016/j.brainres.2007.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am J Pathol. 2008;172:1342–54. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Schutz SG, Cirrito JR, Shankar GM, Sun X, George A, et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–51. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza A, Vega R, Soto E. Cellular mechanisms of lead neurotoxicity. Med Sci Monit. 2006;12:RA57–65. [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–81. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, et al. Mice with combined gene knockouts reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–63. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Iwata N, Saido TC. Understanding molecular mechanisms of proteolysis in Alzheimer's disease: progress toward therapeutic interventions. Biochim Biophys Acta. 2005;1751:60–7. doi: 10.1016/j.bbapap.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Jamsa A, Hasslund K, Cowburn RF, Backstrom A, Vasange M. The retinoic acid and brain-derived neurotrophic factor differentiated SH-SY5Y cell line as a model for Alzheimer's disease-like tau phosphorylation. Biochem Biophys Res Commun. 2004;319:993–1000. doi: 10.1016/j.bbrc.2004.05.075. [DOI] [PubMed] [Google Scholar]
- Jones LG, Prins J, Park S, Walton JP, Luebke AE, Lurie DI. Lead exposure during development results in increased neurofilament phosphorylation, neuritic beading, and temporal processing deficits within the murine auditory brainstem. J Comp Neurol. 2008;506:1003–17. doi: 10.1002/cne.21563. [DOI] [PubMed] [Google Scholar]
- Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113–6. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, et al. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer's disease. Neuron. 2010;67:769–80. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer's disease. Curr Drug Targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- Li M, Sun M, Liu Y, Yu J, Yang H, Fan D, et al. Copper downregulates neprilysin activity through modulation of neprilysin degradation. J Alzheimers Dis. 2010;19:161–9. doi: 10.3233/JAD-2010-1218. [DOI] [PubMed] [Google Scholar]
- Lidsky TI, Schneider JS. Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain. 2003;126:5–19. doi: 10.1093/brain/awg014. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Luo Y, Niu F, Sun Z, Cao W, Zhang X, Guan D, et al. Altered expression of Abeta metabolism-associated molecules from D-galactose/AlCl(3) induced mouse brain. Mech Ageing Dev. 2009;130:248–52. doi: 10.1016/j.mad.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, et al. Neprilysin regulates amyloid Beta peptide levels. J Mol Neurosci. 2004;22:5–11. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, et al. Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–6. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- McGlothan JL, Karcz-Kubicha M, Guilarte TR. Developmental lead exposure impairs extinction of conditioned fear in young adult rats. Neurotoxicology. 2008;29:1127–30. doi: 10.1016/j.neuro.2008.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Coppede F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667:82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2006;65:1012–21. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Levine Iii H. Alzheimer's Disease and the Amyloid-beta Peptide. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005;46:541–54. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Rivera-Mancia S, Perez-Neri I, Rios C, Tristan-Lopez L, Rivera-Espinosa L, Montes S. The transition metals copper and iron in neurodegenerative diseases. Chem Biol Interact. 2010;186:184–99. doi: 10.1016/j.cbi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, et al. Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996;271:20631–5. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- Roques BP, Fournie-Zaluski MC, Soroca E, Lecomte JM, Malfroy B, Llorens C, et al. The enkephalinase inhibitor thiorphan shows antinociceptive activity in mice. Nature. 1980;288:286–8. doi: 10.1038/288286a0. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Suram A, Venugopal C, Prakasam A, Zhou Y, Lahiri DK, et al. A partial failure of membrane protein turnover may cause Alzheimer's disease: a new hypothesis. Curr Alzheimer Res. 2006;3:81–90. doi: 10.2174/156720506775697142. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid protein and Alzheimer's disease. Sci Am. 1991;265:68–71. 4–6, 8. doi: 10.1038/scientificamerican1191-68. [DOI] [PubMed] [Google Scholar]
- Shih RA, Hu H, Weisskopf MG, Schwartz BS. Cumulative lead dose and cognitive function in adults: a review of studies that measured both blood lead and bone lead. Environ Health Perspect. 2007;115:483–92. doi: 10.1289/ehp.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Alzheimer's disease: The latest suspect. Nature. 2008;454:706–8. doi: 10.1038/454706a. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Brain Res Rev. 2005;49:529–54. doi: 10.1016/j.brainresrev.2005.02.004. [DOI] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, et al. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, et al. Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawia NH. Transcriptional involvement in neurotoxicity. Toxicol Appl Pharmacol. 2003;190:177–88. doi: 10.1016/s0041-008x(03)00161-3. [DOI] [PubMed] [Google Scholar]
- Zou LB, Mouri A, Iwata N, Saido TC, Wang D, Wang MW, et al. Inhibition of neprilysin by infusion of thiorphan into the hippocampus causes an accumulation of amyloid Beta and impairment of learning and memory. J Pharmacol Exp Ther. 2006;317:334–40. doi: 10.1124/jpet.105.095687. [DOI] [PubMed] [Google Scholar]