

Abstract
Thrombosis is a common complication of end-stage renal disease, particularly in patients on hemodialysis. Although substantial progress has been made in preventing thrombotic complications in various other groups of patients, the mechanisms of thrombosis during hemodialysis require clarification. In this report, we demonstrate that complement activation triggered by hemodialysis biomaterials, and the subsequent generation of the complement anaphylatoxin C5a, results in the expression of functionally active tissue factor (TF) in peripheral blood neutrophils. Because TF is a key initiator of coagulation in vivo, we postulate that the recurring complement activation that occurs during long-term hemodialysis contributes to thrombosis in dialyzed end-stage renal disease patients. Furthermore, we found that complement contributed to the induction of granulocyte colony-stimulating factor, which has been implicated in the pathogenesis of thrombosis in patients treated with the recombinant form of this molecule. Importantly, the inhibition of complement activation attenuated the TF expression and granulocyte colony-stimulating factor induction in blood passing through a hemodialysis circuit, suggesting that the complement system could become a new therapeutic target for preventing thrombosis in patients with chronic renal failure who are maintained on hemodialysis.
Introduction
Several primary kidney and systemic disorders lead to end-stage renal disease (ESRD), which is manifested as renal failure.1 The treatment of choice in ESRD is kidney transplantation. However, the shortage of available kidney donors, combined with contraindications to transplantation in some patients, results in the necessity for sustaining these persons on renal dialysis for various periods of time. Although renal dialysis is a lifesaving procedure, patients undergoing this treatment are at risk for various complications, including thrombosis. Thrombotic complications are thought to be a net outcome of both underlying kidney disease, complicated by renal failure, and its management through dialysis.2
The most common thrombotic complication in hemodialyzed ESRD patients is thrombosis of vascular access, which is a major cause of hemodialysis-associated morbidity.1,3 In addition, recent studies have found that age-adjusted rates of hospitalization resulting from pulmonary embolism are higher in patients on dialysis than in the general population.4 Importantly, thrombotic events fatally complicate the course of various cardiovascular diseases that are the leading cause of death among patients with chronic kidney diseases.5 For example, coronary artery thrombosis increases the risk of myocardial ischemia and subsequent myocardial infarct. Cardiovascular diseases contribute to more than half of deaths in patients with ESRD.1,6 Furthermore, mortality resulting from cardiovascular diseases is notably higher in patients on dialysis than in the general population.7
These epidemiologic data have triggered research efforts to elucidate the pathogenesis of cardiovascular disorders and accompanying thrombotic complications in patients with chronic kidney diseases who are undergoing dialysis, and to provide plausible therapeutic strategies to reduce mortality associated with these complications. However, although remarkable progress has been made in reducing the risk of cardiovascular death and thrombotic complications in the general population, these pathologies remain a major clinical challenge in patients with ESRD, particularly those on dialysis regimens.8
The mechanisms contributing to an increased risk of thrombosis during hemodialysis are complex and still not well understood. However, several studies have pointed to inflammation as an important contributor to thrombotic complications in hemodialyzed patients. Indeed, ESRD is currently perceived as a state of chronic or recurrent inflammation.9 Furthermore, the inflammatory response is exacerbated in hemodialyzed patients as a result of bioincompatibility. Elevated levels of inflammatory mediators and acute-phase reactants, such as tumor necrosis factor (TNF), interleukin-6 (IL-6), C-reactive protein, and fibrinogen, have been repeatedly demonstrated in patients on dialysis.10
Another contributor to dialysis-associated inflammation is complement. Its activation during the initial phase of dialysis was demonstrated more than 3 decades ago.11 Although considerable progress has been made in reducing this activation through modifications of the surfaces of the biomaterials used to manufacture hemodialysis filters and elements of extracorporeal circuits, activation of complement as a result of bioincompatibility still induces adverse reactions in patients maintained on hemodialysis.12
Because we have found that activation of complement and subsequent generation of the complement anaphylatoxin C5a contribute to up-regulation of tissue factor (TF), a major trigger of coagulation in vivo, in patients with antiphospholipid syndrome (APS)13 and acute respiratory distress syndrome (ARDS),14 and because TF levels are elevated in ESRD patients, we hypothesized that complement activation might contribute to the pathogenesis of hemodialysis-associated thrombosis by up-regulating TF. Endothelial cell damage in ESRD patients also contributes to the up-regulation of TF in these cells. However, considering that a low-grade inflammatory response is constantly present in these patients and that hemodialysis exacerbates this inflammation, it is probable that inflammatory mediators, including complement effectors, also activate peripheral blood leukocytes to produce functionally active TF. Because monocytes and neutrophils have been shown to produce TF when activated by inflammatory mediators and complement effectors, we have postulated that these cells are an important source of TF in the hemodialysis setting.
In this report, we provide evidence that C5a, generated through the activation of complement, stimulates the production of TF by peripheral blood neutrophils in patients with chronic kidney diseases who are on hemodialysis; this mechanism may contribute to hemodialysis-associated thrombosis. We have also found that complement regulates the levels of granulocyte colony stimulating factor (G-CSF), which, when administrated as a recombinant protein to patients with neutropenia, has been suggested to increase risk of thrombosis.15 Furthermore, we have demonstrated, in clinically relevant models of hemodialysis, that inhibiting the complement system at the C3 level with compstatin analogs can efficiently reduce the amount of TF produced by neutrophils and attenuate G-CSF induction. These findings suggest that inhibiting the complement system represents a potentially novel treatment strategy for decreasing the risk of thrombosis in hemodialysis patients with chronic kidney disease.
Methods
More detailed methodology is provided in supplemental Methods (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Patients
Blood samples were collected from 3 female (mean age, 65.8 ± 5.18 years) and 3 male (mean age, 62.5 ± 9.16 years) ESRD patients maintained on hemodialysis in the Department of Nephrology, the University Hospital of Alexandroupolis, Greece. ESRD in these patients was a consequence of chronic glomerulonephritis (2 patients), chronic pyelonephritis (2 patients), diabetes mellitus type II (one patient), and essential arterial hypertension (one patient). Three patients have a history of thrombotic cardiovascular complications, and 2 patients have had vascular access thrombosis. Antiplatelet therapy was administered to 3 persons, whereas 3 other patients were treated with low molecular weight heparin. All patients were maintained on low-flux hemodialysis using polysulfone filters. Blood samples from 9 healthy donors were also used. Informed consent was granted by all involved persons. The study was performed in compliance with institutional guidelines approved by the Institutional Review Board of the University Hospital of Alexandroupolis and in accordance with the Declaration of Helsinki.
Complement inhibition
A potent compstatin analog (Ac-I[CVW(Me)QDWGAHRC]T-NH2)16 or a newly designed 2.2 times more active analog of this inhibitor (Ac-I[CVW(Me)QDWSarAHRC]I-NH2), termed compstatin-10 (H.Q., unpublished observation, January 2010) were used to block complement activation. A linear analog of compstatin (H-IAVVQDWGHHRAT-NH2) served as a control.17 SB-290157 was used to block C3a receptor (C3aR).18 The C5a receptor (C5aR) was blocked by a cyclic hexapeptide (AcF-[OPdChaWR]).19 These inhibitors and the appropriate controls were synthesized as described.16–19 For the complement inhibition studies, lepirudin-anticoagulated blood (50 μg/mL) or isolated serum was pretreated for 10 minutes with either compstatin analog or its linear analog (25μM), and the shredded filter hollow fibers (15 mg/mL) were then added. The blood was incubated for 60 minutes at 37°C. For the experiments that used serum, 50 μL of the serum, treated as described in this section, was added to the cell suspension and incubated for 90 to 120 minutes at 37°C. C3aR or C5aR inhibitor or the appropriate control (10μM) was added for 10 minutes, and the serum from patients on hemodialysis or filter fiber-activated serum was then added to the cell suspension. Approximately 0.8 to 1 × 106 cells from healthy donors were used for these experiments. Preliminary experiments showed that the effects of complement inhibition were dose-dependent, peaking at the doses described in this section.
Ex vivo hemodialysis model
To mimic the hemodialysis conditions ex vivo, we designed a blood recirculation system consisting of a peristaltic pump (Easyload Masterflex; Millipore), a mini-dialyzer (HPH Junior polysulfone dialyzers, Minntech Corporation) and standard hemodialysis bloodline components that served as a tubing system (Set a/v for Fresenius 2008/4008, HMC Premedical SpA). To prevent any ultrafiltration, the dialysate compartment of the filter was filled with saline solution (0.9% NaCl) and clamped. The circuits were prerinsed with saline for approximately 30 minutes before being filled with 30 mL of blood. The blood sample from one donor, collected with lepirudin, was divided, with half preincubated with either compstatin analog (40μM, 3 experiments) or compstatin-10 (20μM, 1 experiment), and half preincubated with an equal concentration of control peptide for 10 minutes at 37°C. “Predialysis” samples were collected before compstatin analogs or control peptide-treated blood was placed into the circuit system. For each experiment (donor), 2 identical circuit systems were used in parallel: one for blood treated with compstatin and the other for blood with the control peptide. Blood circulated in these systems at 37°C with a flow rate of 150 mL/min for 120 minutes. Samples from these 2 systems were collected at 2, 15, 30, 60, 90, and 120 minutes after the circulation began.
mPT assay
Previous studies have shown that the modified prothrombin time assay (mPT) can be used to evaluate the TF-dependent procoagulant properties of bronchoalveolar lavage fluid and cell culture supernatants.14,20 TF in these fluids is probably an alternatively spliced TF isoform20 or originates from TF microparticles.21 Therefore, we used this method to estimate the procoagulant activity (TF/FVIIa binding activity) of supernatants from blood leukocytes as previously described.13,14 The supernatants of leukocytes incubated with sera from ESRD patients, hemodialysis filter fiber-treated sera, and/or various complement inhibitors were isolated by centrifugation at 1000g for 10 minutes; supernatants were additionally checked to confirm the absence of cells. After performing the classic prothrombin time test (100 μL of platelet poor plasma plus 200 μL of thromboplastin; Instrumentation Laboratory), the mPT assay was conducted. Briefly, 125 μL of leukocyte supernatant and 75 μL of thromboplastin were added to 100 μL of platelet-poor plasma prothrombin time was measured. A total of 125 μL of phosphate-buffered saline (PBS) was used instead of cell supernatant as negative control. To verify that the thromboplastic activity was the result of TF, supernatants were incubated for 30 minutes at room temperature with a specific mouse antihuman TF monoclonal antibody (no. 4509; American Diagnostica) and isotype controls13 at 10 μg/mL before the assay.
Complement activation assays
The magnitude of the complement activation was evaluated by enzyme-linked immunosorbent assays measuring the amounts of the terminal complement complex (TCC) or C3 cleavage products. TCC formation was quantified using monoclonal antibody aE11 recognizing a neoepitope of C9 exposed when it is incorporated into the fluid-phase sC5b-9 complex, as previously described.22 C3 products were quantified using monoclonal antibody C3-9 as previously described.23 This antibody binds to a neoantigen exposed in C3(H2O), C3b, and C3c, but not to nonactivated C3.
Statistical analysis
Data are presented as mean plus or minus SD or mean plus or minus SEM. The statistical significances of observed differences were tested using paired and unpaired t test, Wilcoxon matched-pairs, Mann-Whitney tests, and 2-way analysis of variance. The Bonferroni post test correction was applied to control for the occurrence of false-positives. All statistical analyses were performed with GraphPad Prism 4 (GraphPad Software). P values of less than or equal to .05 were considered significant.
Results
Sera of hemodialyzed ESRD patients stimulate neutrophils and monocytes to produce functionally active TF
The increased thrombogenicity of the blood in patients with ESRD, particularly those on hemodialysis, is probably the result of up-regulation of TF because TF activity is a major trigger of coagulation in vivo and TF levels are increased in these patients.24 However, the sources of TF in hemodialyzed patients remain elusive. Peripheral blood monocytes produce TF in various inflammatory conditions.25 In addition, recent studies have shown that complement-activated neutrophils constitute an important source of functionally active TF in patients with APS13 and ARDS.14 Therefore, we hypothesized that these cells are stimulated in the circulation of hemodialyzed ESRD patients to produce TF. To test this hypothesis, we measured the procoagulant activity of supernatants from polymorphonuclear leukocytes (PMNs) and peripheral blood mononuclear cells (PBMCs) obtained from healthy donors (n = 4) and incubated with ESRD patients' sera (n = 6), which were collected before and at various times after the beginning of hemodialysis.
Supernatants from PMNs incubated with sera of ESRD patients before hemodialysis exhibited a moderate procoagulant activity, as illustrated by the shortening of the mPT compared with supernatants from unstimulated cells or PMNs incubated with serum from healthy persons (Figure 1A). A more pronounced shortening of the mPT was observed when supernatants from PMNs incubated with sera from hemodialyzed ESRD patients were assayed (Figure 1A), indicating that hemodialysis increases the procoagulant properties of the blood in these persons. The increase in these procoagulant properties was observed immediately after hemodialysis was begun, reaching a maximum at 15 minutes and returning to levels observed before hemodialysis after 240 minutes. This transient and rapid increase in procoagulant activity indicates that an increased thrombogenicity of blood during hemodialysis is an acute and transient phenomenon, the opposite of the steady and more pronounced increase in the prothrombotic properties of the blood that was observed in APS patients. Given these properties of APS patients' blood, their sera were chosen for use as positive controls (Figure 1A).
Figure 1.

ESRD patients' sera stimulate the production of functionally active TF in peripheral blood leukocytes from healthy donors. (A-B) The mPT values of supernatants of normal PMNs (A) and PBMCs (B) incubated with PBS (bar 1 in panels A and B), serum from healthy donors (HI, bar 2 in panels A and B), or ESRD sera isolated before (bar 3 in panels A and B; predialysis [Pred]) or during hemodialysis at various time points (bars 4-8 in panels A and B); mPT values of supernatants of PMNs (A) and PBMCs (B) either incubated with APS serum (bar 10 in panels A and B) or incubated with ESRD sera collected between 15 and 30 minutes after the beginning of hemodialysis and treated with TF monoclonal antibodies (bar 11 in panels A and B). Data are representative of 10 independent experiments (mean mPT values ± SD). A paired t test was applied to examine statistical significance: *P < .05; **P < .001. (C) The relative expression of TF mRNA in normal PMNs cultured in the presence of PBS or ESRD patients' sera isolated before or during hemodialysis. Data are representative of 6 independent experiments (mean fold expression [2−DDCt analysis] ± DCt SD). DDCt indicates Delta Delta Cycle threshold; and DCt, Delta Cycle threshold. The Wilcoxon matched-pairs test was used to assess statistical significance; *P < .01. Horizontal lines above data bars indicate statistical significance.
Addition of neutralizing TF antibody to supernatants obtained from PMNs that had been incubated with sera collected from ESRD patients 15 to 30 minutes after the beginning of hemodialysis completely abolished their procoagulant activity, as illustrated by the return of mPT values to the levels seen in healthy persons' sera (Figure 1A). This effect of TF antibody indicates that the procoagulant activity of PMN supernatants is dependent on TF production by these cells. Furthermore, control antibodies added to PMN supernatants at various concentrations did not affect the mPT values in a similar manner, confirming the specificity of these findings. Thus, we concluded that sera from ESRD patients contain factors that stimulate neutrophils to produce functionally active TF. Given that these sera probably contain small amounts of TF, we needed to confirm that the shortening of the mPT was exclusively dependent on TF released from the neutrophils in vitro and was not affected by serum-derived TF. To achieve this goal, we tested the effect of ESRD sera alone on mPT values. The lack of shortening of the mPT that we observed (data not shown) excluded the contribution of serum-derived TF to the procoagulant activities of PMN supernatants. The same experiments described for PMNs were also conducted using PBMCs to validate the results for PMNs because the monocytes included in the PBMC population are a well-known source of TF in various inflammatory conditions. We found that factors present in ESRD sera stimulated these cells to produce functionally active TF in a manner similar to that observed for PMNs (Figure 1B).
These conclusions were further strengthened by an increase in the TF gene expression in PMNs (Figure 1C) and PBMCs (data not shown) obtained and stimulated in the same way as described in this section for measurements of the procoagulant activity of supernatants from leukocytes. Sera from ESRD patients before hemodialysis induced a moderate increase in TF gene expression in PMNs compared with its expression in cells incubated with sera from healthy persons, whereas stimulation of these cells with sera obtained during hemodialysis resulted in more pronounced increase, peaking at 30 minutes after the beginning of this procedure. This increase in TF gene expression corresponded to the increased procoagulant properties of supernatants from PMNs and PBMCs, as demonstrated by a shortening of the mPT values (Figure 1A-B).
TF is expressed in blood leukocytes obtained from ESRD patients, and hemodialysis transiently enhances its expression
Because our experiments have demonstrated that sera from ESRD patients contain mediator(s) that stimulate PMNs and PBMCs from healthy persons to produce TF in vitro, we next asked whether similar mechanisms are responsible for an increased tendency for thrombosis to occur in vivo. For this purpose, we examined the expression of TF isoforms in leukocytes obtained from ESRD patients before and during hemodialysis. TF and alternatively spliced TF mRNA levels were slightly increased in their PMNs and PBMCs even before hemodialysis, compared with leukocytes from healthy persons (Figure 2Ai-ii). Hemodialysis further transiently up-regulated TF isoform expression. The maximum level of expression was observed at 30 minutes, with asTF being the dominant isoform for PMNs.
Figure 2.
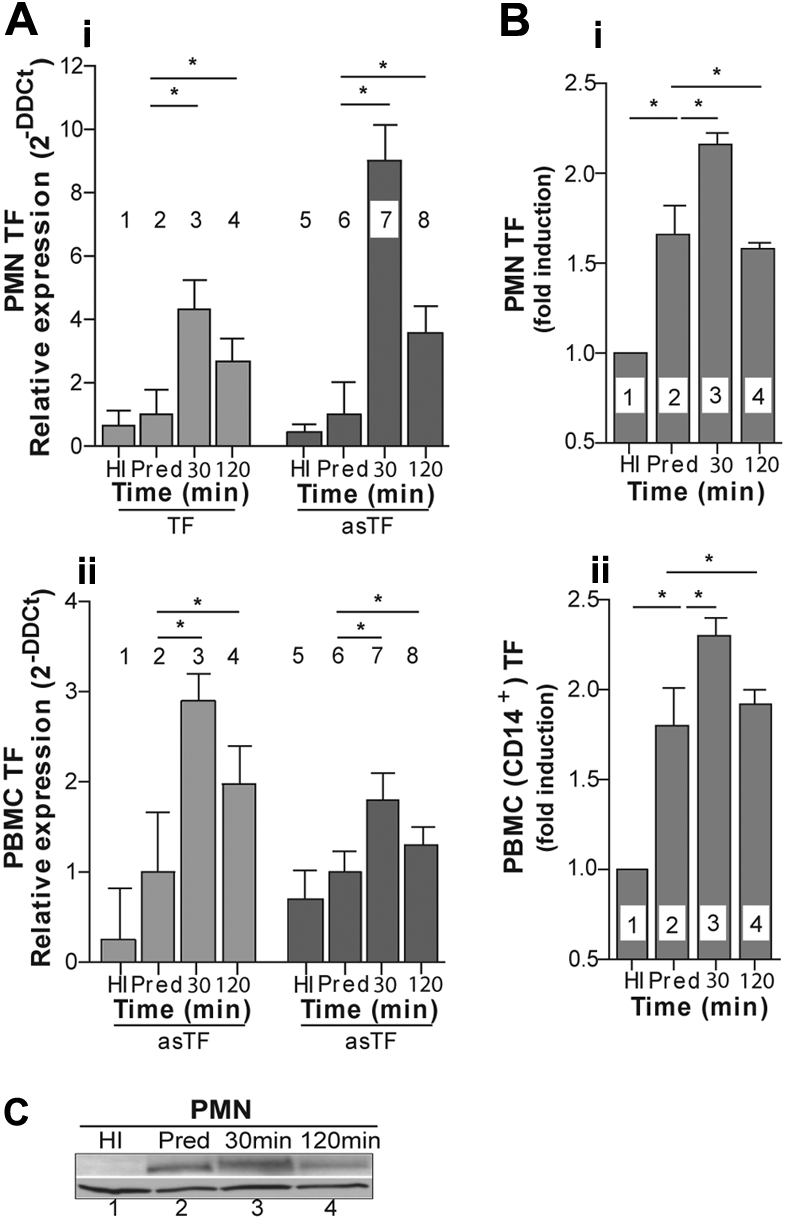
TF is expressed in the peripheral leukocytes of ESRD patients. (A) The relative expression of TF or asTF mRNA in PMNs (i) and PBMCs (ii) obtained from healthy donors (HI, bar 1) or ESRD patients before (predialysis [Pred], bar 2) and during hemodialysis (bars 3, 4). Data are representative of 6 independent experiments (mean fold expression ± DCt SD). The Wilcoxon matched-pairs and Mann-Whitney tests were used to assess statistical significance: *P < .05. (B) The induction of TF expression in PMNs (i) and PBMCs (ii) isolated from ESRD patients before (bar 2) and during hemodialysis (bars 3, 4). The results are presented as ratios of the mean fluorescence intensities of the cells from patients to the cells from healthy donors (bar 1), stained with TF monoclonal antibody, and analyzed by flow cytometry. Data are representative of one experiment (mean ± SD of 4 healthy donors and 6 ESRD patients). The Mann-Whitney and Wilcoxon matched-pairs tests were used to assess statistical significance: *P < .05. (C) TF expression in PMNs isolated from healthy donors (1-HI) or ESRD patients before (2-predialysis [Pred]) and during hemodialysis (lanes 3, 4). One representative Western blot analysis of 4 independent experiments is shown. Horizontal lines above data bars indicate statistical significance.
The changes in TF protein expression, as measured by flow cytometry (Figure 2B) and Western blotting (Figure 2C), followed the pattern observed for TF mRNA.
Up-regulation of TF in neutrophils of hemodialyzed ESRD patients is complement-mediated
Previous studies have suggested that C5a, which is generated as a result of complement activation, induces production of TF by neutrophils circulating in the blood or accumulating in the pulmonary alveoli of APS13 and ARDS14 patients, respectively. In addition, C5a contributes to fetal loss in an animal model of APS by up-regulating TF in neutrophils.26 Furthermore, hemodialysis activates complement. Therefore, we hypothesized that complement activation and subsequent generation of anaphylatoxins play a role in the up-regulation of TF in the neutrophils of ESRD patients during hemodialysis. To confirm this hypothesis, we initially analyzed complement activation in these patients by monitoring the levels of TCC, which is generated as a result of final steps in the complement cascade activation, in the plasma of ESRD patients before and during hemodialysis. Hemodialysis induced significant complement activation, which reached a maximum 60 minutes after the beginning of this procedure (Figure 3A). In agreement with previous reports,27 we found that this activation was associated with a decrease in C5aR surface expression on PMNs in 3 of 4 analyzed ESRD patients (Figure 3B). The moderate attenuation of C5aR expression, which correlated with low-grade complement activation (Figure 3A), was observed in PMNs from ESRD patients even before hemodialysis; however, a clearer decrease in C5aR expression was observed only after hemodialysis began. In contrast, C5aR expression evaluated in permeabilized leukocytes did not decrease but showed a moderate, although not statistically significant, increase during hemodialysis compared with predialysis status (Figure 3C). These data suggest that C5aR was internalized because of C5a binding and that hemodialysis induced a moderate up-regulation of this receptor compared with its levels in PMNs from patients before hemodialysis. The decreased C5aR expression observed in both experimental settings in ESRD patients compared with healthy donors (Figure 3B-C) also suggests that inflammation associated with chronic kidney disease contributed to moderate down-regulation of this receptor.
Figure 3.

Procoagulant properties of ESRD sera are complement-dependent. (A) The amounts of soluble TCC in plasma from healthy donors (HI, bar 1) or ESRD patients before dialysis (predialysis [Pred], bar 2), and during (bars 3-6) hemodialysis. Data are representative of one experiment (mean ± SD of 4 persons per group). The Mann-Whitney test and paired t test were applied to assess statistical significance: *P < .05. (B-C) The expression of C5a receptor on the surface of PMNs (B) or in permeabilized cells (C) from healthy donors (HI), ESRD patients before dialysis (predialysis [Pred]), and during hemodialysis (5, 30, 60, and 120 minutes). The results are presented as mean fluorescent intensity (MFI) ratios of the cells stained with C5aR monoclonal antibodies to the cells stained with the isotype control. Data are representative of one experiment (mean ± SD of 4 healthy donors and 3 ESRD patients). The unpaired and paired t tests were used to assess statistical significance: *P < .05. (D) The mPT values of supernatants of normal PMNs treated with sera from healthy donors (HI, bar 1), ESRD patients before (predialysis [Pred], bar 2), and during (bar 3) hemodialysis or preincubated with C5a (bar 4) or C3a (bar 5) receptor antagonists and then treated with sera obtained from ESRD patients during hemodialysis. Data are representative of 6 independent experiments (mean ± SD). The Wilcoxon matched-pairs test was used to assess statistical significance: *P < .001. (E) The relative expression of TF mRNA in normal PMNs treated with sera obtained from ESRD patients during hemodialysis or preincubated with C5aR antagonist and then treated with sera from the same patients. Data are representative of 2 independent experiments (mean ± SD of 3 patients). The Wilcoxon matched-pairs test was used to assess statistical significance: P < .01. Horizontal lines above data bars indicate statistical significance.
Next, we examined the effect of the inhibition of anaphylatoxin signaling in PMNs obtained from healthy volunteers and stimulated with sera from hemodialyzed ESRD patients, as described for measurements of the procoagulant activity of supernatants from leukocytes, on the procoagulant properties of supernatants from these cells. We used ESRD sera obtained 15 to 30 minutes after the beginning of hemodialysis because we found that these sera induced the most pronounced shortening of the mPT by PMN supernatants (Figure 1A) and corresponding up-regulation of TF (Figure 1C). Incubation of PMNs with the C5aR inhibitor before stimulation with ERSD sera completely abrogated the procoagulant activity of the supernatants, as demonstrated by a return of the mPT values to those for supernatants from cells incubated with sera from healthy donors (Figure 3D). Furthermore, blocking C5aR inhibited the up-regulation of TF expression in these PMNs (Figure 3E). Conversely, blocking C3aR did not affect the mPT of the supernatants from PMNs stimulated with ESRD serum (Figure 3D). These results indicate that the TF-mediated procoagulant properties of neutrophils are C5a-dependent.
Blocking complement activation abrogates the TF-dependent procoagulant activity of neutrophils
Because hemodialysis filter fibers trigger complement activation, as demonstrated by the presence of C3 cleavage products in plasma obtained from blood after incubation with these fibers (Figure 4A), we used them as a clinically relevant in vitro model to explore therapeutic interventions targeting the complement system as a means of limiting the procoagulant properties of neutrophils. Initial experiments demonstrated that sera from healthy persons incubated with shredded hollow filter fibers gained the ability to induce TF-dependent procoagulant activity in PMNs, as demonstrated by the shortened mPTs seen with supernatants from PMNs after their incubation with these sera, and the lack of this effect when TF antibody was added to the supernatants (Figure 4B). The magnitude of the mPT shortening was correlated with the duration of the incubation with the fibers. Based on these experiments, we concluded that contact between normal donor serum and the fibers induces complement activation, which results in the generation of complement effectors that subsequently up-regulate TF in PMNs incubated with fiber-activated sera. Because a 60-minute incubation had resulted in considerable shortening of mPT, we incubated sera collected from healthy persons and from ESRD patients before hemodialysis with the hemodialysis filter fibers for this same period of time. Supernatants from PMNs stimulated with fiber-activated ESRD sera induced a shortening of the mPT compared with supernatants from untreated cells or cells treated with ESRD patient sera that were not incubated with fibers (Figure 4C). This reduction in mPT values was more pronounced than that observed for normal sera. Adding compstatin analog to normal or ESRD patient sera before their incubation with the fibers prevented this reduction. A similar effect was observed when PMNs were pretreated with the C5aR-blocking peptide before the incubation with fiber-activated sera.
Figure 4.

Hemodialysis filter fibers induce complement activation and enhance TF-dependent procoagulant properties in normal and ESRD sera. (A) The amounts of C3 cleavage products in plasma isolated from normal nontreated blood (bar 1), nontreated blood incubated in 37°C for 60 minutes (bar 2), blood treated with filter fibers (bar 3), or blood treated with filter fibers in the presence of compstatin analog (bar 4) or control peptide (bar 5). Data are representative of 4 independent experiments (mean ± SD). The paired t test was used to assess statistical significance: *P < .05. (B) The mPT values of supernatants of normal PMNs incubated with sera from healthy donors (HI, bar 1), sera activated with filter fibers for various periods of time (bars 2-5), or with filter fiber-activated sera (for 60 minutes) and treated with TF monoclonal antibodies (bar 6). Data are representative of 6 independent experiments (mean ± SD). The Wilcoxon matched-pairs test was used to assess statistical significance: *P < .01. (C) The mPT values of supernatants of normal PMNs incubated with sera from healthy donors (HI, bar 1), ESRD patients before hemodialysis (predialysis [Pred], bar 2), filter fiber-activated sera from healthy donors or patients (bars 3 and 6, respectively), sera from healthy donors or patients incubated with these fibers in the presence of compstatin analog (bars 4 and 7, respectively) or pretreated with C5aR antagonist and then incubated with fiber-activated sera from healthy donors or patients (bars 5 and 8, respectively). Data are representative of 6 independent experiments (mean ± SD). The Wilcoxon matched-pairs test was used to assess statistical significance: *P < .01. (D) The induction of TF expression in normal PMNs incubated with sera from healthy donors (bar 1), fiber-activated sera (bar 2), or sera treated with fibers in the presence of compstatin analog (bar 3). The results are presented as ratios of the mean fluorescence intensities (MFI) of the cells incubated with sera treated with fibers in the presence or absence of compstatin to the cells incubated with sera from healthy donors, stained with TF monoclonal antibodies, and analyzed by flow cytometry. Data are representative of 6 independent experiments (mean ± SD). The Wilcoxon matched-pairs test was used to assess statistical significance: *P < .05. (E) TF expression in normal PMNs incubated with PBS (1), APS sera (2), fiber-treated sera in the presence of compstatin analog (3), or fiber-activated (4) or untreated sera (5). One representative Western blot analysis of 4 independent experiments is shown. Horizontal lines above data bars indicate statistical significance.
Because compstatin analog blocks the complement cascade at the C3 level, these results suggest that inhibiting complement activation could efficiently reduce the TF-dependent procoagulant activity of PMNs during hemodialysis. In addition, therapeutic targeting of C5aR had a similar beneficial effect. These conclusions are supported by the ability of compstatin analog to reduce TF expression in PMNs stimulated with fiber-activated sera (Figure 4D-E).
Hemodialysis-induced complement activation and subsequent TF up-regulation in peripheral blood neutrophils can be efficiently reduced by inhibiting C3 cleavage
To determine whether complement inhibition can be considered as a therapeutic approach to reduce the thrombotic complications during hemodialysis that are associated with TF expression induced by complement activation in neutrophils, we designed an experimental model mimicking hemodialysis. In this setting, a recirculation system was filled with blood from healthy donors. Passing the blood through this circuit induced complement activation, as demonstrated by an increase in C3 cleavage products in the plasma, which was proportional to the duration of the blood contact with the elements of this circuit (Figure 5A). Pretreatment of the blood with compstatin analogs abolished this activation. These inhibitors also reduced TF expression in PMNs (Figure 5B), confirming the functional association between complement activation and the procoagulant properties of these cells during hemodialysis. In addition, inhibition of complement in this experiment decreased the level of CD11b expression in PMNs (Figure 5C), suggesting that complement effectors are potent activators of these cells because CD11b is an established marker of neutrophil activation.
Figure 5.
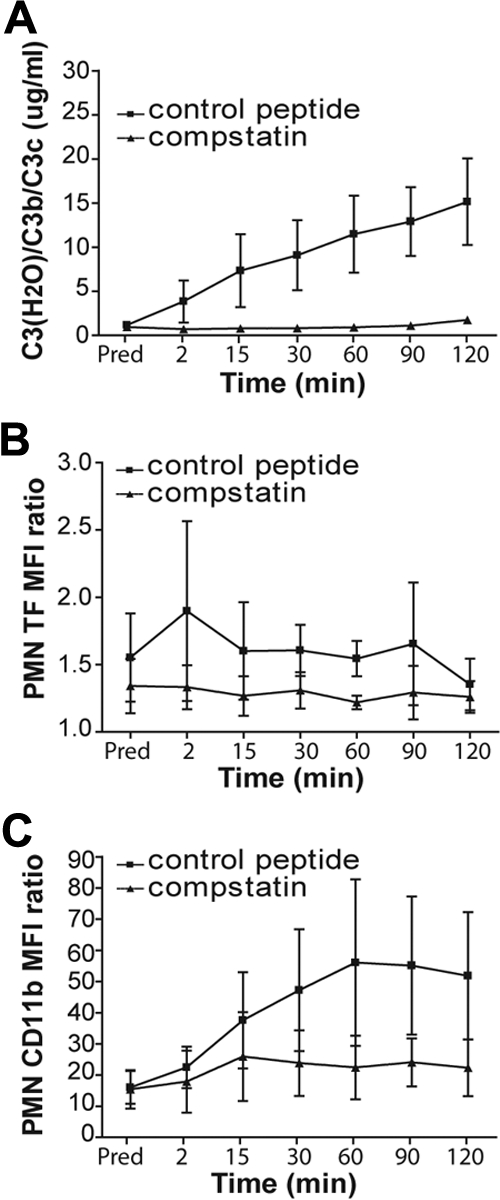
Compstatin analogs attenuate the hemodialysis-associated activation of complement and neutrophils and up-regulation of TF in these cells. Normal blood was recirculated through a hemodialysis simulation system for 2 hours at 37°C. (A) The amounts of C3 cleavage products in plasma isolated from the blood before the beginning of this recirculation (predialysis [Pred]) and at various time points during hemodialysis simulation in the presence of compstatin analogs or control peptide. (B) The induction of TF and (C) CD11b expression in PMNs isolated from the blood before (predialysis [Pred]) and during hemodialysis simulation. The results are presented as ratios of the mean fluorescence intensities (MFI) of the cells stained with TF monoclonal antibodies to the cells stained with the isotype control and analyzed by flow cytometry. Data are representative of 4 independent experiments in panel A and 3 in panels B and C (mean ± SEM). The 2-way analysis of variance test was applied to assess statistical significance of the difference between treatment of blood with compstatin analogs (▴) and the control peptide (■): (A) P < .001, (B) P < .034, and (C) P < .021.
Complement inhibition reduces the amount of IFN-γ, IL-1RA, and G-CSF released during hemodialysis simulation
Several cytokines and chemokines are induced in patients with ESRD as a result of hemodialysis.28 Some of these mediators are implicated in thrombotic complications in several diseases.29 In addition, complement regulates the production and release of several cytokines during the inflammatory response to infections.30 Therefore, we hypothesized that complement, in addition to its direct regulation of TF expression in neutrophils, can indirectly contribute to thrombosis by regulating cytokine production and/or release. To test this hypothesis, we performed a high-throughput screen of cytokines released from blood cells and evaluated the effect of complement inhibition on the levels of these mediators in blood passing through a hemodialysis circuit. We found that hemodialysis simulation triggered the induction of TNF, IL-17, basic fibroblast growth factor, platelet-derived growth factor-BB, granulocyte-macrophage colony-stimulating factor, macrophage inflammatory protein-1α, regulated on activation normal T expressed and secreted (RANTES; supplemental Figure 1A-G), IFN-γ, IL-1RA, and G-CSF (Figure 6). However, inhibition of complement significantly decreased the induction of only IFN-γ, IL-1RA, and G-CSF.
Figure 6.
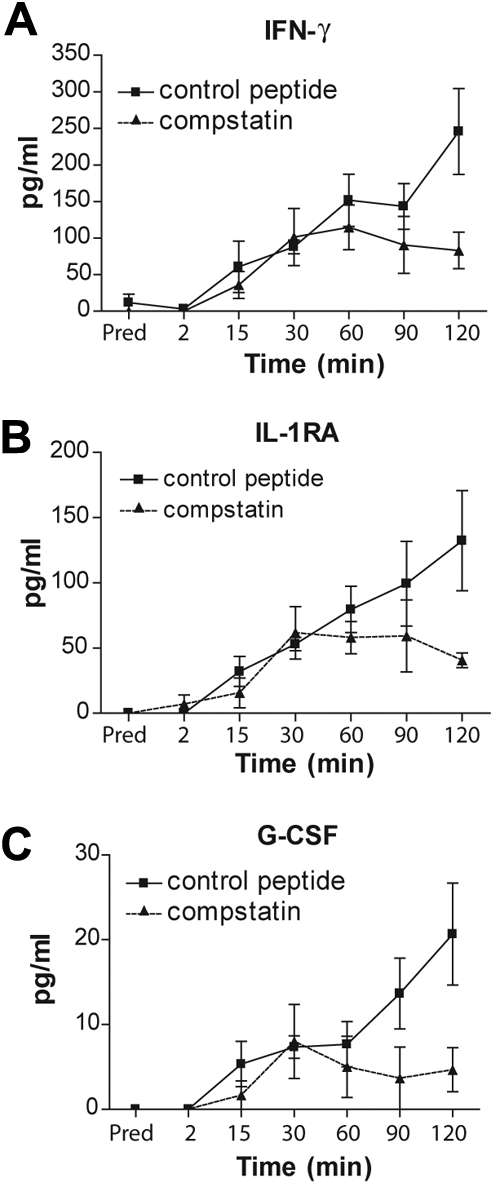
Complement regulates cytokine production and/or release during a hemodialysis simulation. The amounts of (A) IFN-γ, (B) IL-1RA, and (C) G-CSF during hemodialysis simulation. Data are representative of 3 independent experiments (mean ± SEM). The 2-way analysis of variance test was used to assess statistical significance of differences in the amounts of cytokines after treatment of the blood with compstatin analogs (▴) or the control peptide (■): (A) P < .018, (B) P < .030, and (C) P > .008.
Discussion
In the present study, we have demonstrated that serum obtained from ESRD patients during hemodialysis induces the production of functionally active TF by neutrophils and monocytes from healthy donors, indicating that mediators responsible for this effect were generated in the circulation of the patients during hemodialysis. We also found that TF is transiently up-regulated in neutrophils and monocytes from ESRD patients undergoing hemodialysis. These findings led us to conclude that peripheral blood leukocytes are stimulated to produce TF during hemodialysis. Because TF is a major trigger of coagulation in vivo, we postulate that this mechanism contributes to an increased blood thrombogenicity in these patients.
Activation of complement in the blood of ESRD patients and the failure of their sera to induce the expression of functionally active TF in neutrophils pretreated with C5aR antagonist indicate that C5a, generated as a result of complement activation, contributes to thrombosis in hemodialyzed patients with ESRD. This hypothesis was further confirmed by our observation that the activation of complement was triggered by fibers used to manufacture hemodialysis filters. We also found that, in the presence of the compstatin analog, sera incubated with these fibers could no longer induce TF-dependent procoagulant activity by neutrophils. To explore the potential of complement inhibition to attenuate thrombosis in patients on hemodialysis, we designed an ex vivo model of extracorporeal circulation that mimicked this procedure. We found that inhibition of complement reduced the activation of neutrophils and TF expression in these cells that had been triggered by contact with the biomaterial surfaces. Importantly, the compstatin derivative has already been tested in patients with age-related macular degeneration. Therefore, the results of this preclinical research should facilitate further clinical investigation.
Although TF is a key initiator of coagulation in vivo and plays a central role in pathologic clotting thrombosis,25 data concerning its role in hemodialysis-related thrombosis are surprisingly limited.31 Recently, increased plasma TF levels have been demonstrated in patients on dialysis, but potential mechanisms linking inflammation with coagulation were not explored.24 In addition, the sources of the increased TF levels in the blood of hemodialyzed patients and the stimuli responsible for this increase have not yet been determined. Thus, our findings point to novel mechanisms involving complement activation, the generation of C5a, and the subsequent induction of functional TF in blood neutrophils that can contribute to thrombosis in the hemodialysis setting.
Given that ESRD is associated with persistent low-grade inflammation even without hemodialysis, it is probable that other inflammatory mediators also contribute to up-regulation of TF expression in PMNs in ESRD patients. However, C5a, generated as a result of complement activation triggered by the recurrent contact of blood with biomaterials, appears to be a primary agent causing TF induction in these leukocytes during hemodialysis. Therefore, we postulate that C5a is an important inflammatory mediator that bridges inflammation and thrombosis in this clinical situation. This conclusion is further strengthened by our finding that complement inhibition decreases the amount of the potent chemokine G-CSF in blood passing through a hemodialysis circuit. The importance of G-CSF for hemodialyzed patients was initially suggested by data showing that this mediator is strongly induced in these persons.32 Although G-CSF has not yet been linked to hemodialysis-associated thrombosis, patients treated with recombinant G-CSF for various conditions associated with neutropenia have been suggested to have an increased risk of thrombosis.15 Therefore, we propose that complement, in addition to enhancing TF expression in neutrophils, can enhance the risk of thrombosis indirectly by regulating G-CSF. This chemokine is a potent activator of neutrophils and mobilizes these cells from the bone marrow. Because during hemodialysis, an initial transient neutropenia is quickly reversed to leukocytosis,33 it is probable that G-GSF induced by hemodialysis contributes to this process. In consequence, a high number of hyperactivated neutrophils, which are a source of TF, further heightens the risk of thrombosis. In addition, G-CSF enhances platelet aggregation,34 increasing the tendency of the blood to clot; an increase in platelet-derived growth factor-BB, which we observed, confirms this process. Although we found that complement inhibition also attenuated the induction of IFN-γ in blood passed through the hemodialysis circuit, the significance of this finding for hemodialyzed patients needs to be established because increased amounts of this cytokine have not yet been reported in such persons. Similarly, the role of IL-17 induction remains unclear, although recent studies have implicated this cytokine in the pathogenesis of atherosclerosis,35 which is greatly enhanced in ESRD patients. We also found that complement inhibition attenuated the release/production of IL-1RA during hemodialysis stimulation. This mediator has anti-inflammatory activity, and its levels are increased in hemodialyzed patients.36 However, the associations between IL-1RA and coagulation are unknown. High amounts of TNF and chemokines, such as granulocyte-macrophage colony-stimulating factor, macrophage inflammatory protein-1α, and RANTES, can be associated with thrombosis because TNF is a well-established enhancer of coagulation,37 and chemokines can contribute to trafficking and activation of peripheral blood leukocytes.
A thorough understanding of the interconnections between complement and coagulation that are triggered by blood-biomaterial interactions during hemodialysis should provide new insights into the adverse effects of bioincompatibility in other clinical settings that use artificial surfaces. Intense efforts are being directed toward eliminating these adverse effects.38 Complement activation has been described as a primary event inducing and enhancing these undesirable reactions.39 Thus, we anticipate that mechanisms similar to those reported here for hemodialysis are applicable to other conditions with bioincompatibility-associated complications. Therefore, the development of biomaterials with only limited capacity to induce complement activation or coated with complement inhibitors, the blocking of complement activation, or the disabling of C5aR signaling may prove beneficial in preventing thrombotic complications in various groups of patients, in addition to those on hemodialysis.
The induction of TF expression by C5aR signaling during hemodialysis places ESRD in a group of acquired thrombotic disorders mediated by C5a, including APS,13 sepsis,40 and ARDS,14 and indicating that the close relationship between complement and thrombosis may be a rather common phenomenon that is present in various as-yet unexplored conditions.41 Thus, the cross-talk between complement and hemostasis may have important therapeutic implications for reducing the frequency of thrombotic complications in a variety of diseases. The development of several complement inhibitors and the safe use of these drugs in humans42,43 make the inhibition of complement activation a promising strategy for preventing thrombosis in the clinic.
Supplementary Material
Acknowledgments
The authors thank Dr Deborah McClellan for her help in editing the manuscript.
This work was supported by the Governing Board of the University Hospital of Alexandroupolis (K.R.) and National Institutes of Health grants EB003968, AI068730, and GM062134 (J.D.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: I.K. performed the stimulation studies, real-time PCR, flow cytometry, and Western blot experiments, analyzed the data, and wrote the manuscript; M.M.M. analyzed the data and wrote the manuscript; M.D. performed the mPT assays; S.R. performed flow cytometry experiments; K.K. and I.M. carried out flow cytometry and real-time PCR experiments; S.P., P.P., and V.V. selected patients for the study and provided patient samples; P.M. synthesized and characterized the complement receptor inhibiting peptides; H.Q. synthesized and characterized compstatin analogs; T.E.M. performed the cytokine assays and analyzed data; and K.R. and J.D.L. conceived and supervised the project; and all authors critically revised the manuscript and approved the final version.
Conflict-of-interest disclosure: J.D.L. and K.R. are coinventors on the following provisional patent application: Inhibiting pro-coagulant effect of biomaterials. The remaining authors declare no competing financial interests.
Correspondence: John D. Lambris, Department of Pathology and Laboratory Medicine, School of Medicine, University of Pennsylvania, 422 Curie Blvd, Philadelphia, PA 19104; e-mail: lambris@upenn.edu.
References
- 1.United States Renal Data System. Minneapolis, MN: United States Renal Data System; 2009. 2009 Annual Data Report, Atlas of ESRD. [Google Scholar]
- 2.Yao Q, Axelsson J, Stenvinkel P, Lindholm B. Chronic systemic inflammation in dialysis patients: an update on causes and consequences. ASAIO J. 2004;50(6):lii–lvii. doi: 10.1097/01.mat.0000147958.87989.eb. [DOI] [PubMed] [Google Scholar]
- 3.Vazquez MA. Vascular access for dialysis: recent lessons and new insights. Curr Opin Nephrol Hypertens. 2009;18(2):116–121. doi: 10.1097/MNH.0b013e328325d635. [DOI] [PubMed] [Google Scholar]
- 4.Tveit DP, Hypolite IO, Hshieh P, et al. Chronic dialysis patients have high risk for pulmonary embolism. Am J Kidney Dis. 2002;39(5):1011–1017. doi: 10.1053/ajkd.2002.32774. [DOI] [PubMed] [Google Scholar]
- 5.Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension. 2003;42(5):1050–1065. doi: 10.1161/01.HYP.0000102971.85504.7c. [DOI] [PubMed] [Google Scholar]
- 6.VI. Causes of death in ESRD. Am J Kidney Dis. 1999;34(2) suppl 1:S87–S94. doi: 10.1053/AJKD034s00087. [No authors listed] [DOI] [PubMed] [Google Scholar]
- 7.Collins AJ. Cardiovascular mortality in end-stage renal disease. Am J Med Sci. 2003;325(4):163–167. doi: 10.1097/00000441-200304000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Appel LJ. Beyond (or back to) traditional risk factors: preventing cardiovascular disease in patients with chronic kidney disease. Ann Intern Med. 2004;140(1):60–61. doi: 10.7326/0003-4819-140-1-200401060-00013. [DOI] [PubMed] [Google Scholar]
- 9.Silverstein DM. Inflammation in chronic kidney disease: role in the progression of renal and cardiovascular disease. Pediatr Nephrol. 2009;24(8):1445–1452. doi: 10.1007/s00467-008-1046-0. [DOI] [PubMed] [Google Scholar]
- 10.Kaysen GA. The microinflammatory state in uremia: causes and potential consequences. J Am Soc Nephrol. 2001;12(7):1549–1557. doi: 10.1681/ASN.V1271549. [DOI] [PubMed] [Google Scholar]
- 11.Craddock PR, Fehr J, Brigham KL, Kronenberg RS, Jacob HS. Complement and leukocyte-mediated pulmonary dysfunction in hemodialysis. N Engl J Med. 1977;296(14):769–774. doi: 10.1056/NEJM197704072961401. [DOI] [PubMed] [Google Scholar]
- 12.Hoenich NA. Membranes for dialysis: can we do without them? Int J Artif Organs. 2007;30(11):964–970. doi: 10.1177/039139880703001104. [DOI] [PubMed] [Google Scholar]
- 13.Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794–4802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 14.Kambas K, Markiewski MM, Pneumatikos IA, et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol. 2008;180(11):7368–7375. doi: 10.4049/jimmunol.180.11.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zakarija A, Kwaan HC. Adverse effects on hemostatic function of drugs used in hematologic malignancies. Semin Thromb Hemost. 2007;33(4):355–364. doi: 10.1055/s-2007-976171. [DOI] [PubMed] [Google Scholar]
- 16.Katragadda M, Magotti P, Sfyroera G, Lambris JD. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J Med Chem. 2006;49(15):4616–4622. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- 17.Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage displayed random peptide library. J Immunol. 1996;157(2):884–891. [PubMed] [Google Scholar]
- 18.Ames RS, Lee D, Foley JJ, et al. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol. 2001;166(10):6341–6348. doi: 10.4049/jimmunol.166.10.6341. [DOI] [PubMed] [Google Scholar]
- 19.Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD. A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol. 2001;166(4):2479–2486. doi: 10.4049/jimmunol.166.4.2479. [DOI] [PubMed] [Google Scholar]
- 20.Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res. 2005;96(12):1233–1239. doi: 10.1161/01.RES.0000171805.24799.fa. [DOI] [PubMed] [Google Scholar]
- 21.Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N. Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost. 2009;7(7):1092–1098. doi: 10.1111/j.1538-7836.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mollnes TE, Lea T, Harboe M, Tschopp J. Monoclonal antibodies recognizing a neoantigen of poly(C9) detect the human terminal complement complex in tissue and plasma. Scand J Immunol. 1985;22(2):183–195. doi: 10.1111/j.1365-3083.1985.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 23.Hammel M, Sfyroera G, Ricklin D, et al. A structural basis for complement inhibition by Staphylococcus aureus. Nat Immunol. 2007;8(4):430–437. doi: 10.1038/ni1450. [DOI] [PubMed] [Google Scholar]
- 24.Adams MJ, Irish AB, Watts GF, Oostryck R, Dogra GK. Hypercoagulability in chronic kidney disease is associated with coagulation activation but not endothelial function. Thromb Res. 2008;123(2):374–380. doi: 10.1016/j.thromres.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(8):1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 26.Redecha P, Tilley R, Tencati M, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110(7):2423–2431. doi: 10.1182/blood-2007-01-070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Himmerfarb J, Gerard NP, Hakim RM. Intradialytic modulation of granulocyte C5a receptors. J Am Nephrol. 1991;2(4):920–926. doi: 10.1681/ASN.V24920. [DOI] [PubMed] [Google Scholar]
- 28.Friedrich B, Alexander D, Janessa A, et al. Acute effects of hemodialysis on cytokine transcription profiles: evidence for C-reactive protein-dependency of mediator induction. Kidney Int. 2006;70(12):2124–2130. doi: 10.1038/sj.ki.5001865. [DOI] [PubMed] [Google Scholar]
- 29.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95(9):858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 30.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171(3):715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemanova P, Opatrny K, Vit L, Sefrna F. Tissue factor, its inhibitor, and the thrombogenicity of two new synthetic membranes. Artif Organs. 2005;29(8):651–657. doi: 10.1111/j.1525-1594.2005.29103.x. [DOI] [PubMed] [Google Scholar]
- 32.Sato H, Ohkubo M, Nagaoka T. Levels of serum colony-stimulating factors (CSFs) in patients on long-term haemodialysis. Cytokine. 1994;6(2):187–194. doi: 10.1016/1043-4666(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 33.Kaplow LS, Goffinet JA. Profound neutropenia during the early phase of hemodialysis: 1968. J Am Soc Nephrol. 1999;10(2):425–428. [PubMed] [Google Scholar]
- 34.Kaptan K, Ifran A, Beyan C, Sertkaya D. Recombinant human granulocyte colony-stimulating factor (rhG-CSF) promotes in vitro platelet aggregation. Hematology. 2007;12(5):441–444. doi: 10.1080/10245330701384286. [DOI] [PubMed] [Google Scholar]
- 35.Eid RE, Rao DA, Zhou J, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119(10):1424–1432. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira BJ. Cytokine production in patients on dialysis. Blood Purif. 1995;13(3):135–146. doi: 10.1159/000170196. [DOI] [PubMed] [Google Scholar]
- 37.Esmon CT. The impact of the inflammatory response on coagulation. Thromb Res. 2004;114(5):321–327. doi: 10.1016/j.thromres.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson B, Korsgren O, Lambris JD, Ekdahl KN. Can cells and biomaterials in therapeutic medicine be shielded from innate immune recognition? Trends Immunol. 2009;31(1):32–38. doi: 10.1016/j.it.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chenoweth DE. Complement activation during hemodialysis: clinical observations, proposed mechanisms, and theoretical implications. Artif Organs. 1984;8(3):281–290. doi: 10.1111/j.1525-1594.1984.tb04291.x. [DOI] [PubMed] [Google Scholar]
- 40.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 41.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28(4):184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Mol Immunol. 2009;47(2):185–195. doi: 10.1016/j.molimm.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25(11):1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.