

Abstract
Atom transfer radical addition of haloalkanes and α-halocarbonyls to olefins is efficiently performed with the photocatalyst Ir[(dF(CF3)ppy)2(dtbbpy)]PF6. This protocol is characterized by excellent yields, mild conditions, low catalyst loading, and broad scope. In addition, the atom transfer protocol can be used to quickly and efficiently introduce vinyl trifluoromethyl groups to olefins and access 1,1-cyclopropane diesters.
Atom transfer radical addition (ATRA) of haloalkanes and α-halocarbonyls to olefins serves as an atom-economical1 method of simultaneously forming C-C and C-X bonds. Following Kharasch's seminal work,2 Curran,3 Oshima,4 and others have developed ATRA into a useful tool in organic chemistry. However, typical ATRA initiators include toxic and hazardous reagents, such as peroxides,2 organotin reagents,3 and triethylboron.4 Other less common initiators have also been used, including p-methoxybenzene-diazonium tetraflouroborate with TiCl3,5 dimanganese decacarbonyl,6 copper,7 iron,8 bimetallic Rh–Ru complexes,9 and chromium(II) acetate,10 but these methods employ harsh conditions and/or lack broad functional group tolerance. In this regard, we sought to develop a protocol capable of effecting ATRA with a broad scope under mild conditions and utilizing safer reagents using photoredox catalysis (Figure 1).11
Figure 1.
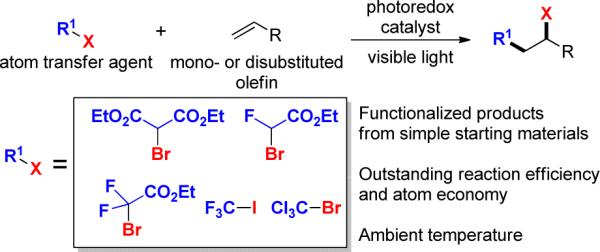
Intermolecular atom transfer radical addition mediated by photoredox catalysis
MacMillan,12 Yoon,13 and our group14 have recently demonstrated the ability of photoredox catalysts, such as Ru(bpy)3Cl2, to initiate organic transformations.15 Specifically, we have utilized the reductive quenching cycle of such photocatalysts to functionalize C-H bonds and reduce activated C-Br bonds to afford intra- and intermolecular radical reactions (Figure 2, Path A).12 However, the intermolecular processes are difficult due to competitive side reactions that result from the generation of reactive intermediates by the reductive quenching process.14e
Figure 2.
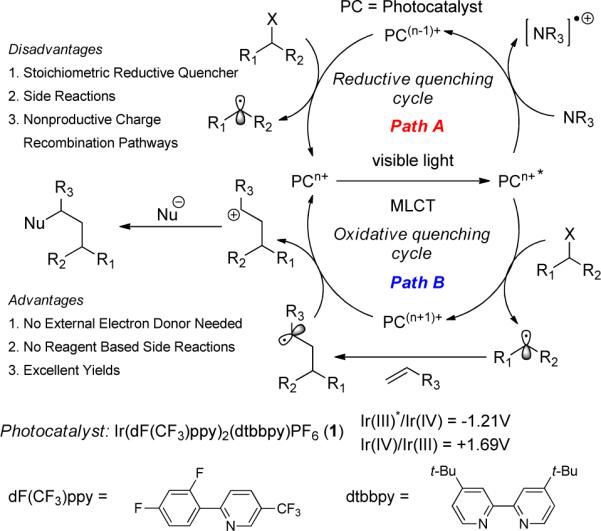
Applications of the reductive and oxidative photocatalytic cycle in initiating organic transformations
We reasoned that these side reactions could be suppressed by utilizing the substrate itself as the excited state quencher via an oxidative quenching pathway (Figure 2, Path B).12c,13c,14f,16 During our survey of photoredox catalysts, we identified Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 (1) as a complex capable of mediating intermolecular ATRA.17 Herein, we disclose the first intermolecular ATRA reaction of haloalkanes and α-halocarbonyls onto olefins under mild conditions to be catalyzed by a visible light active photoredox catalyst.
Our first attempt at intermolecular ATRA involved using tosylated allylamine (2a), diethyl 2-bromomalonate (4, 2.0 equiv) and 1 (1.0 mol %) in DMF and gave 17% yield after 24 h (entry 1, Table 1). The addition of a Lewis acidic additive, LiBF4, resulted in an increased yield, but with incomplete conversion of starting material.11a,b Optimization of the additive and solvent led to increased yield along with greater conversion of the starting material (entries 3–5). However, increasing the nucleophilicity of the olefin, by utilizing Boc-protected allylamine (2b) and 5-hexen-1-ol (2c), afforded complete consumption of the starting materials and nearly quantitative yield of the atom transfer products (entries 6 and 7).
Table 1.
Optimization of Reaction Conditions/Control Experiments
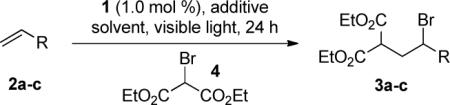
| entry | R | conditionsa | yieldb |
|---|---|---|---|
| 1 | CH2NHTs (2a) | 4 (2.0 equiv), DMF | 17 |
| 2 | 2a | 4 (2.0 equiv), LiBF4 (2.0 equiv), DMF | 25 |
| 3 | 2a | 4 (2.0 equiv), LiBr (2.0 equiv), DMF | 45 |
| 4 | 2a | 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (4:1) | 54 |
| 5 | 2a | 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (1:4) | 67 |
| 6 | CH2NHBoc (2b) | 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (1:4) | 99 |
| 7 | (CH2)4OH (2c) | 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (1:4) | 99 |
| 8 | 2c | 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (1:4), , no light | 0 |
| 9 | 2c | no catalyst, 4 (2.0 equiv), LiBr (2.0 equiv), DMF/H2O (1:4) | 0 |
| 10 | 2c | 4 (1.1 equiv), LiBr (1.1 equiv), DMF/H2O (1:4) | 97 |
| 11 | 2c | 4 (0.95 equiv), LiBr (0.95 equiv), DMF/H2O (1:4) | 95 |
| 12 | 2c | 4 (2.0 equiv), no additive, DMF/H2O (1:4) | 72 |
| 13 | 2c | 4 (2.0 equiv), LiBr (2.0 equiv), DMF | 46 |
Entries 1–6 were degassed (freeze-pump-thaw).
Isolated yield (%) after purification on SiO2.
In order to evaluate the significance of each of the reaction parameters, additional control reactions were run. As expected, no conversion to product was observed in the absence of light or photocatalyst (entries 8 and 9). Furthermore, either component, olefin or 4, can be used as the limiting reagent with no decrease in yield or reaction efficiency (entries 10 and 11). In addition, LiBr and H2O are required to allow the reaction to progress to completion in a 24 h timeframe (entries 12 and 13).18
With the optimized conditions in hand, the atom transfer of 4 with various olefins was examined (entries 1–9, Table 2). Several functional groups are well tolerated under the reaction conditions including free alcohols, silyloxy ethers, benzyl ethers, alkyl bromides, esters, enones, carbamates, and aromatic rings. In addition, monosubstituted and 1,1-disubstituted olefins are competent reaction partners with 4. All reactions are characterized by clean conversion to product within 24 h and can be isolated easily by column chromatography.
Table 2.
ATRA Using Photoredox Catalysisa
| entry | substrate | olefin | product | yieldb |
|---|---|---|---|---|

|
|
|||
| 1 | 4 | R = CH2NHTs | 67c | |
| 2 | 4 | R = CH2NHBoc | 99 | |
| 3 | 4 | R = (CH2)4OH | 95 | |
| 4 | 4 | R = (CH2)3OH | 99 | |
| 5 | 4 | R = (CH2)4OTBS | 90 | |
| 6 | 4 | R = (CH2)4OCH2Ph | 92 | |
| 7 | 4 | R = (CH2)3Br | 92 | |
| 8 | 4 | R = (CH2)4CO2Et | 99 | |
| 9 | 4 |
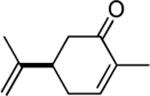
|
|
95d |
| 10 | CF3I |
|
|
90e,f |
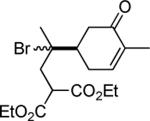
| 11 | CF3I |
|
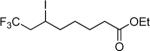
|
81e,f |
| 12 |
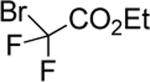
|
|
|
93 |
| 13 |
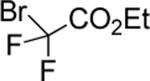
|
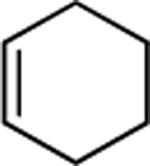
|
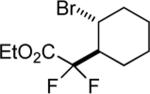
|
75 |
| 14 | CCl3Br |
|
|
87f |
| 15 |
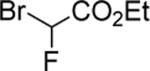
|
|
|
99d |
| 16 |
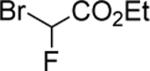
|
|
|
84d |
Reaction conditions: 1 (1.0 mol %), haloalkane (2.0 equiv), and LiBr (2.0 equiv) in DMF/H2O (1:4).
Isolated yield (%) after purification on SiO2.
90% brsm.
dr 1:1.
Excess of haloalkane was used.
No LiBr added.
The success of the intermolecular atom transfer with 4 prompted the examination of other compounds containing activated C-X bonds (entries 10–16, Table 2). The optimized conditions worked well with a number of α-halocarbonyls and haloalkanes. It is noteworthy that cyclohexene, which did not undergo coupling with 4, underwent efficient ATRA with ethyl bromodifluoroacetate (entry 13).19 Several fluorinated compounds are effective reaction partners,12b and demonstrate the applicability of this methodology to the synthesis of molecules that have particular value in medicinal chemistry,20 agrochemicals21 and material science.22
In order to demonstrate the potential synthetic utility of the products accessible via this ATRA, a simple protocol for dehydroiodination of the atom transfer products with CF3I, was realized by subjection of the crude ATRA product to DBU in toluene, providing the trifluoromethyl alkene (E/Z 19:1),236, in excellent yield (Scheme 1). Furthermore, 1,1-cyclopropane diesters could be generated in excellent yields by treating the atom transfer products with Cs2CO3.24 Finally, the coupling reaction remains efficient on a preparative scale and can be conducted with only 0.01 mol % of 1 providing the product of coupling of diethyl bromomalonate with 5-hexen-1-ol on 15 mmol scale in 97% isolated yield.
Scheme 1.
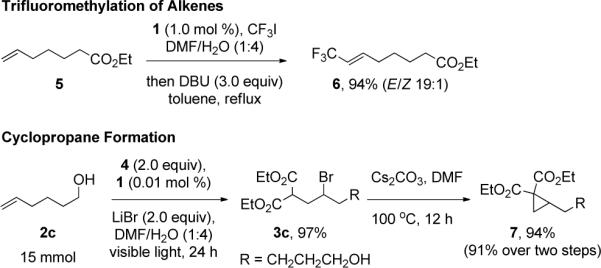
Synthetic Utility of ATRA Products25
Based upon our use of the oxidative quenching cycle, it was anticipated that (1) a C-C bond is formed from the addition of an electron-deficient radical to an olefin and (2) the generation of a carbocation occurs during the turnover of the catalyst (Ir4+→Ir3+).
Competition studies revealed that 1,1-disubstituted olefins react more quickly than the analogous terminal olefins, consistent with an electrophilic radical addition to the olefin.26 In addition, evidence supporting a carbocation intermediate was found during early optimization experiments utilizing 4-penten-1-ol, which resulted in the isolation of 827 in 10% yield. Resubjection of 3d to the reaction conditions or refluxing toluene did not result in the formation of the tetrahydrofuran 8 (eq 1). Therefore, we conclude that 8 is formed by competitive trapping of a carbocation during the course of the ATRA reaction. However, when external nucleophiles, including water and LiCl (in place of LiBr), were added in attempt to quench the carbocation, only the bromohydrin was observed in all cases.26,28
 |
(1) |
Finally, in order to study the possibility of a radical propagation mechanism, diethyl bromomalonate and 5-hexen-1-ol were irradiated under the standard reaction conditions until 50% conversion was achieved (1H NMR), at which point the light source was removed. No further progress was observed after 6 h of stirring in the dark. However, the reintroduction of the external light source led to full conversion to the desired product after 4.5 h. This result indicates the necessity of external light irradiation to allow the reaction to progress.
On the basis of these data, we propose the mechanism outlined in Figure 3 which accounts for the formation of the furan byproduct (eq 1). Oxidative quenching of the visible light induced excited state, Ir3+*, by the haloalkane or α-halocarbonyl generates an electrophilic radical along with the Ir4+ complex, bearing the halide as a counterion. This radical then undergoes an addition to the olefin. The ATRA product can then be formed via two potential pathways. First, oxidation of the alkyl radical (0.47 V vs SCE)29 by Ir4+ generates the carbocation,30,31 pre-associated with the halide, which combine to provide the final product selectively. Alternatively, a radical chain transfer mechanism may also be operative. Each of these potential mechanistic pathways account for the lack of nucleophilic addition of H2O or Cl−.
Figure 3.
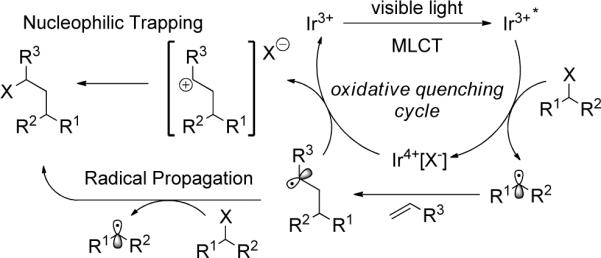
Proposed Mechanism
In conclusion, we have developed a simple protocol for atom transfer radical addition to olefins using the photoredox catalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which is capable of coupling a wide variety of halogenated compounds to terminal olefins and disubstituted olefins under mild conditions to give excellent yields. Conversion of the products to a variety of synthetically and biologically useful moieties has also been demonstrated. Further mechanistic studies are currently underway and will be reported in due course.
Supplementary Material
Acknowledgement
Financial support from start-up funds from Boston University and partial support from the NIGMS (P50-GM067041) is gratefully acknowledged. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the NSF. The authors thank Dr. Jagan Narayanam for helpful suggestions. J.D.N. and M.D.K. thank Boston University for Graduate Fellowships. J.W.T. thanks Vertex for a Graduate Fellowship.
Footnotes
Supporting Information Available: Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- (2).(a) Kharasch MS, Skell PS, Fischer P. J. Am. Chem. Soc. 1948;70:1055. [Google Scholar]; (b) Kharasch MS, Jensen EV, Urry WH. Science. 1945;102:128. doi: 10.1126/science.102.2640.128. [DOI] [PubMed] [Google Scholar]
- (3).(a) Curran DP, Bosch E, Kaplan J, Newcomb M. J. Org. Chem. 1989;54:1826. [Google Scholar]; (b) Curran DP, Chang C-T. J. Org. Chem. 1989;54:3140. [Google Scholar]; (c) Curran DP, Chen M-H, Spleterz E, Seong CM, Chang C-T. J. Am. Chem. Soc. 1989;111:8872. [Google Scholar]; (d) Curran DP, Seong CM. J. Am. Chem. Soc. 1990;112:9401. [Google Scholar]; (e) Curran DP, Tamine J. J. Org. Chem. 1991;56:2746. [Google Scholar]; (f) Curran DP, Kim D. Tetrahedron. 1991;47:6171. [Google Scholar]; (g) Curran DP, Kim D, Ziegler C. Tetrahedron. 1991;47:6189. [Google Scholar]
- (4).(a) Yorimitsu H, Nakamura T, Shinokubo H, Oshima K. J. Org. Chem. 1998;63:8604. [Google Scholar]; (b) Yorimitsu H, Nakamura T, Shinokubo H, Oshima K, Omoto K, Fujimoto H. J. Am. Chem. Soc. 2000;122:11041. [Google Scholar]; (c) Yorimitsu H, Shinokubo H, Matsubara S, Oshima K, Omoto K, Fujimoto H. J. Org. Chem. 2001;66:7776. doi: 10.1021/jo010652l. [DOI] [PubMed] [Google Scholar]
- (5).Cao L, Li C. Tetrahedron Lett. 2008;49:7380. [Google Scholar]
- (6).Gilbert BC, Kalz W, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. J. Chem. Soc., Perkin Trans. 2000;1:1187. [Google Scholar]
- (7).(a) Yang Z-Y, Burton DJ. J. Org. Chem. 1991;56:5125. [Google Scholar]; (b) Metzger JO, Mahler R. Angew. Chem., Int. Ed. 1995;34:902. [Google Scholar]
- (8).Forti L, Ghelfi F, Libertini E, Pagnoni UM, Soragni E. Tetrahedron. 1991;53:17761. [Google Scholar]
- (9).Quebatte L, Scopelliti R, Severin K. Angew. Chem., Int. Ed. 2004;43:1520. doi: 10.1002/anie.200353084. [DOI] [PubMed] [Google Scholar]
- (10).Lübbers T, Schäfer HJ. Synlett. 1991:861. [Google Scholar]
- (11).For recent reviews on photoredox catalysis, see: Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102. doi: 10.1039/b913880n.. Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527. doi: 10.1038/nchem.687.. Zietler K. Angew. Chem., Int. Ed. 2009;48:9785. doi: 10.1002/anie.200904056.
- (12).(a) Nicewicz D, MacMillan DWC. Science. 2008;322:70. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shih H, Vander Wal MN, Grange RL, MacMillan DWC. J. Am. Chem. Soc. 2010;132:13600. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Du J, Yoon TP. J. Am. Chem. Soc. 2009;131:14604. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ischay MA, Lu Z, Yoon TP. J. Am. Chem. Soc. 2010;132:8572. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu Z, Shen M, Yoon TP. J. Am. Chem. Soc. 2011;133:1162. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]; (b) Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org. Lett. 2010;12:368. doi: 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]; (c) Condie AG, González-Gómez JC, Stephenson CRJ. J. Am. Chem. Soc. 2010;132:1464. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]; (d) Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem. Commun. 2010;46:4985. doi: 10.1039/c0cc00981d. [DOI] [PubMed] [Google Scholar]; (e) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org. Lett. 2010;12:3104. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]; (f) Dai C, Narayanam JMR, Stephenson CRJ. Nature Chem. 2011;3:140. doi: 10.1038/nchem.949. [DOI] [PubMed] [Google Scholar]
- (15).For additional recent examples, see: Koike T, Akita M. Chem. Lett. 2009;38:166.. Andrews SR, Becker JJ, Gagné MR. Angew. Chem., Int. Ed. 2010;49:7274. doi: 10.1002/anie.201004311.. Neumann M, Fuldner S, Konig B, Zeitler K. Angew. Chem., Int. Ed. 2011;50:951. doi: 10.1002/anie.201002992.. Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem. Commun. doi: 10.1039/c0cc04539j. DOI: 10.1039/C0CC04539J.
- (16).For examples of oxidative quenching in visible light photoredox catalysis, see: Cano-Yelo H, Deronzier A. J. Chem. Soc., Faraday Trans. 1. 1984;80:3011.. Cano-Yelo H, Deronzier A. Tetrahedron Lett. 1984;25:5517.. Zeh JM, Liou SL, Kumar AS, Hsia MS. Angew. Chem., Int. Ed. 2003;42:577. doi: 10.1002/anie.200390166.
- (17).Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr., Malliaras GG, Bernhard S. Chem. Mater. 2005;17:5712. [Google Scholar]
- (18).LiBr is required only for the reactions of α-bromo esters and presumably serves to assist in activating the bromo ester towards reduction. See also refs 13a,b,d for the use of Lewis acid additives in photoredox reactions
- (19).Curran has previously reported that iodomalonates only add efficiently to terminal and 1,1-disubstituted olefins. For details, see reference 3b
- (20).Kirk K. J. Fluorine Chem. 1999;100:127. [Google Scholar]
- (21).Jeschke P. ChemBioChem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- (22).Extrand CW. J. Fluorine Chem. 2003;122:121. [Google Scholar]
- (23).For examples of the preparation of vinyl trifluoromethane using (a) Cross coupling, see: Xiao J, Weisblum B, Wipf P. J. Am. Chem. Soc. 2005;127:5742. doi: 10.1021/ja051002s.; (b) Cross metathesis, see: Imhof S, Randl S, Blechert S. Chem. Commun. 2001:1692. doi: 10.1039/b105031c.; (c) Elimination, see: Bazhin DN, Gorbunova TI, Zapevalov AY, Salouti VI. J. Fluorine Chem. 2009;130:438.
- (24).For synthetic uses of 1,1-cyclopropane diesters, see: Pohlhaus PD, Johnson JS. J. Org. Chem. 2005;70:1057. doi: 10.1021/jo048230+.. Perreault C, Goudreau SR, Zimmer LE, Charette AB. Org. Lett. 2008;10:689. doi: 10.1021/ol702414e.. Lebold TP, Kerr MA. Pure Appl. Chem. 2010;82:1797.
- (25).For additional examples demonstrating the synthetic utility of atom transfer products derived from halo esters, see reference 3b
- (26).See the Supporting Information for more details
- (27).Pastine SJ, McQuaid KM, Sames D. J. Am. Chem. Soc. 2005;127:12180. doi: 10.1021/ja053337f. [DOI] [PubMed] [Google Scholar]
- (28).Nucleophilicity parameters (13.80 for bromide, 11.30 for chloride, 5.20 for water) predict that trapping with bromide is favored. For details on nucleophilicity parameters, see: Minegishi S, Kobayashi S, Mayr H. J. Am. Chem. Soc. 2004;126:5174. doi: 10.1021/ja031828z.. Minegishi S, Loos R, Kobayashi S, Mayr H. J. Am. Chem. Soc. 2005;127:2641. doi: 10.1021/ja045562n.
- (29).Wayner DDM, Houmam A. Acta Chem. Scand. 1998;52:377. [Google Scholar]
- (30).Oxidation of bromide to bromine atom (0.86 V vs SCE) and radical combination cannot be ruled out. Given the facile oxidation of alkyl radicals (see ref. 28), this oxidation appears more likely. For the oxidation potential of bromide, see: Kubiak CP, Schneemeyer LF, Wrighton MS. J. Am. Chem. Soc. 1980;102:6900.
- (31).For a review of radical-polar crossover reactions, see: Murphy JA. The Radical-Polar Crossover Reaction. In: Renaud P, Sibi M, editors. Radicals in Organic Synthesis. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2001. pp. 298–335.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.