

Abstract
δ-Opioid receptors are G-protein-coupled receptors that regulate nociceptive and emotional responses. It has been well established that distinct agonists acting at the same G-protein-coupled receptor can engage different signaling or regulatory responses. This concept, known as biased agonism, has important biological and therapeutic implications. Ligand-biased responses are well described in cellular models, however, demonstrating the physiological relevance of biased agonism in vivo remains a major challenge. The aim of this study was to investigate the long-term consequences of ligand-biased trafficking of the δ-opioid receptor, at both the cellular and behavioral level. We used δ agonists with similar binding and analgesic properties, but high [SNC80 ((+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide)]- or low [ARM390 (N,N-diethyl-4-(phenyl-piperidin-4-ylidenemethyl)-benzamide)]-internalization potencies. As we found previously, a single SNC80—but not ARM390—administration triggered acute desensitization of the analgesic response in mice. However, daily injections of either compound over 5 d produced full analgesic tolerance. SNC80-tolerant animals showed widespread receptor downregulation, and tolerance to analgesic, locomotor and anxiolytic effects of the agonist. Hence, internalization-dependent tolerance developed, as a result of generalized receptor degradation. In contrast, ARM390-tolerant mice showed intact receptor expression, but δ-opioid receptor coupling to Ca2+ channels was abolished in dorsal root ganglia. Concomitantly, tolerance developed for agonist-induced analgesia, but not locomotor or anxiolytic responses. Therefore, internalization-independent tolerance was produced by anatomically restricted adaptations leading to pain-specific tolerance. Hence, ligand-directed receptor trafficking of the δ-opioid receptor engages distinct adaptive responses, and this study reveals a novel aspect of biased agonism in vivo.
Introduction
Opioid receptors regulate numerous physiological processes, including reward, pain, and stress. δ-Opioid receptors show a functional profile distinct from that of μ- or κ-opioid receptors (Kieffer and Gavériaux-Ruff, 2002) and play an important role in chronic pain. δ agonists are poor analgesics in acute pain (Gallantine and Meert, 2005), but are highly effective following chronic inflammatory or neuropathic pain (Fraser et al., 2000; Hurley and Hammond, 2000; Cahill et al., 2003). The analysis of δ receptor knock-out mice has further established the existence of an inhibitory δ receptor tone, which reduces nociceptive responses under conditions of persistent pain (Nadal et al., 2006; Gavériaux-Ruff et al., 2008). δ-Opioid receptors also modulate emotional responses. Genetic deletion of either the receptor (Filliol et al., 2000) or its endogenous ligand, enkephalin (König et al., 1996), results in anxiogenic and depressive-like behaviors. Further, exogenous δ agonists produce anxiolytic and antidepressant effects (Broom et al., 2002; Saitoh et al., 2004; Perrine et al., 2006). Altogether, evidence from both pharmacological and genetic approaches indicates that the δ-opioid receptor is an important player in both nociceptive and emotional processing, and represents a promising therapeutic target for pain and mood disorders.
Opioid receptors are members of the G-protein-coupled receptor (GPCR) superfamily. GPCRs exist under multiple active conformations, and extensive in vitro pharmacological evidence indicates that distinct agonists acting at a single receptor may produce different active receptor states, which in turn trigger differential effects at the receptor or cellular level (Kenakin, 2003). These effects include the modulation of specific signaling pathways, or the induction of certain regulatory responses, such as receptor trafficking and desensitization. These ligand-directed responses have been defined as “functional selectivity” or “biased agonism”. The concept of biased agonism has important implications, both in terms of understanding the complexity of GPCR pharmacology and for facilitating drug development (Bosier and Hermans, 2007; Galandrin et al., 2007). However, evidence is mostly based on in vitro experiments using recombinant cell systems. A major challenge in GPCR research, today, is to demonstrate the physiological relevance of agonist-biased signaling and regulation in vivo.
The existence of ligand-biased internalization at opioid receptors has been demonstrated in vitro, and the in vivo relevance of this phenomenon is under intense investigation and debate (Kieffer and Evans, 2002; Kim et al., 2008). In this study, we address biased agonism for the δ opioid receptor in vivo at the level of receptor regulation. The aim of the study was to determine whether agonist-biased receptor trafficking produced differential long-term behavioral adaptations. To correlate ligand-induced receptor internalization with receptor function in vivo, we took advantage of a knock-in mouse line expressing functional δ-opioid receptors with a fluorescent tag (DOR-eGFP) (Scherrer et al., 2006). We examined the pain-relieving effects of two δ-opioid receptor agonists with similar binding and analgesic properties, but high or low internalization potencies. We found that chronic treatments with the two compounds led to entirely distinct adaptive responses at both the receptor, cellular, and behavioral levels.
Materials and Methods
DOR-eGFP and wild-type mice
All experiments were performed in accordance with the European Communities Council Directive of 24 November 1986 and with AALAC. In this study both DOR-eGFP knock-in (male and female) and wild-type C57BL/6J (male) mice were used. Knock-in mice were produced by homologous recombination. In these mice the eGFP cDNA was introduced into exon 3 of the δ-opioid receptor gene, in frame and 5′ from the stop codon. C57BL/6J mice were purchased from Charles River. Mice aged 12 weeks on average, were housed in a temperature- and humidity-controlled animal colony on a 12 h dark-light cycle. Food and water were available ad libitum.
δ agonists
SNC80 [(+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] (Calderon et al., 1994) is a nonpeptidic δ agonist with high selectivity for the δ-opioid receptor. AR-M1000390 (ARM390, N,N-diethyl-4-(phenyl-piperidin-4-ylidenemethyl)-benzamide) is an SNC80 derivative (Wei et al., 2000), synthesized at AstraZeneca Research and Development Montreal. ARM390 was administered per os by gavage, as it is an irritant when injected intraperitoneally (AstraZeneca, personal communication).
Ex vivo tissue analysis of DOR-eGFP mice
Membrane preparations were performed as described previously (Befort et al., 2001). Whole brain and the lumbar segment of the spinal cord were removed, immediately frozen in isopentane or dry ice, and stored at −80°C before use. For the brain, [3H]naltrindole and [35S]GTPγS assays were performed on whole brain or spinal cord membranes. Whole brain membranes were prepared by homogenizing the brain in ice-cold 0.25 m sucrose solution 10 vol (ml/g wet weight of tissue). Samples were then centrifuged at 1100 × g for 10 min. Supernatants were collected and diluted 5 times in buffer containing 50 mm Tris-HCl, pH 7.4, and 1 mm EDTA, following which they were centrifuged at 35,000 × g for 30 min. The pellets were homogenized in 2 ml of ice-cold sucrose solution (0.32 m), aliquoted and kept at −80°C until further use.
For saturation binding studies, 50 μg of membrane proteins were incubated with variable concentrations of [3H]naltrindole (0.1–2 nm) for 1 h at 25°C. Nonspecific binding was determined in the presence of 10 μm naloxone. Incubation was terminated by rapid filtration through 0.1% polyethyleneimine-presoaked microplate filters (Unifilter GF/B), and washing with ice-cold buffer (50 mm Tris-HCl, pH 7.4). Bound radioactivity was quantified using a liquid scintillation counter. Bmax and Kd values were calculated using Microsoft Excel Software.
For each [35S]GTPγS binding assay 5 μg of protein was used per well. Samples were incubated with and without δ-opioid receptor agonists (10−4 to 10−12 m), for 1 h at 25°C in assay buffer containing 50 mm Tris-HCl, pH 7.4, 3 mm MgCl2, 100 mm NaCl, 0.2 mm EGTA, 30 μm GDP and 0.1 nm [35S]GTPγS. Incubation was terminated by rapid filtration and washing in ice-cold buffer (50 mm Tris-HCl, 5 mm MgCl2, 50 mm NaCl, pH 7.4). Bound radioactivity was quantified using a liquid scintillation counter. Nonspecific binding was defined as binding in the presence of 10 μm GTPγS, and basal binding indicates binding in the absence of agonist.
To determine the subcellular distribution of DOR-eGFP after agonist stimulation, mice were anesthetized with ketamine/xylazine (100/10 mg/kg) and intracardially perfused with 10 ml of 9.25% sucrose in ddH2O followed by 30 ml of 4% paraformaldehyde in 0.1 m phosphate buffer (PB; pH 7.4). Brains, spinal cords and dorsal root ganglia were then postfixed for 2 h at 4°C in the fixative solution. The tissue was then cryoprotected at 4°C in a 30% sucrose, 0.1 m PB solution until the tissue sank. Tissue was then frozen in isopentane and stored at −80°C until cut. Freely floating sections were cut at 30 μm in a cryostat. Sections were mounted on Superfrost glass slides in 0.01 m PBS, and DOR-eGFP receptor distribution was immediately examined in different δ receptor-rich regions. All samples were observed under Leica confocal microscopes (SP1 or SP2UV; 63× objective and numerical aperture of 1.32), and the LCS (Leica) software was used for image acquisition. Quantification of cell surface mean fluorescence intensity was determined using ImageJ software. Nuclear fluorescence defined the background level and was subtracted from the cell membrane fluorescence measures (for further details, see Scherrer et al., 2006). In total, 3–4 neurons/region/mouse were analyzed, and there were 3–5 mice/group.
Behavioral testing
Inflammatory pain model.
All experiments were performed between 8:00–16:00 h. In all cases mice were habituated to the testing area for 20 min daily for 2 d before baseline testing. Two different variations of the Complete Freund's Adjuvant (CFA) model of inflammatory pain were used (Pradhan et al., 2009). To assess mechanical pain CFA was injected into the paw. To assess thermal pain CFA was injected into the tail. Separate groups of animals were used for each endpoint.
For mechanical responses, the threshold for responses to punctate mechanical stimuli (mechanical allodynia) was tested according to the up-and-down method (Chaplan et al., 1994). In this case, the plantar surface of the animal hindpaw was stimulated with a series of eight von Frey filaments (bending force ranging from 0.01 to 2 g). Before the injection of CFA baseline mechanical responses (dashed line) were determined. Inflammation was induced by injecting 15 μl of CFA into the plantar surface of the paw, and animals were subsequently tested 48 h later (Abbadie et al., 2003).
For thermal responses, heat hyperalgesia was assessed by immersing the tail (5 cm from the tip) into a 46°C water bath. Tail withdrawal latencies were determined, and a cutoff of 40 s was established. Before the injection of CFA, baseline mechanical responses (dashed line) were determined. Inflammation was induced by injecting 20 μl of CFA 3 cm from the tip of the tail, and all drug tests occurred at least 48 h later.
To ensure that all animals were treated similarly, each mouse received both intraperitoneal and per os injections. Therefore, animals challenged with SNC80 (10 mg/kg, i.p.) also received a per os injection of dH2O (SNC80 group), those challenged with ARM390 (10 mg/kg, p.o.) received an intraperitoneal injection of 0.9% saline (ARM390 group), and control animals were injected with intraperitoneal saline and per os dH2O (control group). Mice were treated chronically with drug or vehicle for a maximum of 6 d. Drug was administered every 24 h, unless otherwise stated, and pain responses were assessed 45 min after drug treatment.
Quantification of locomotor activity.
Before locomotor testing, mice were chronically tested in the CFA tail model to ensure that tolerance to δ agonists was established. CFA was applied to the tail to ensure intact locomotor responses. Locomotor activity was measured using clear Plexiglas boxes (21 cm × 11 cm × 17 cm) placed over an infrared platform. Locomotor activity was assessed using the ViewPoint videotracking software. Counts were integrated in 5 min bins and added to obtain total locomotor activity for a 2 h period. Animals were habituated for 10 min to the locomotor test cages 24 h before the test day. On the test day, basal (in the absence of drug) locomotion was assessed for 1 h, mice were then injected with either vehicle or SNC80 (3 mg/kg, i.p.) following which locomotion was assessed for 2 h.
Elevated plus maze test.
Before testing in the elevated plus maze, mice were chronically tested in the CFA tail model to ensure that tolerance to δ agonists was established. CFA was applied to the tail to ensure intact locomotor responses. The elevated plus maze was made of Makrolon plastic sheeting, and consisted of a cross shape, comprising two open arms, and two closed arms (15 cm high walls, Viewpoint, Lyon, France). Each runway was 5 cm wide and 45 cm long. All testing occurred in low lighting conditions of 25 lux, as determined at the level of the center square. Time spent, and entries into each arm were determined using the ViewPoint videotracking system. Animals were initially placed in the central square of the maze, facing toward a closed arm, and allowed to explore the maze freely for 5 min. Mice were treated with either SNC80 (10 mg/kg, s.c.) or vehicle 60 min before the test. Any animals that fell off the maze were not included in subsequent data analysis.
Electrophysiological recording
The whole-cell patch-clamp technique was used to record voltage-dependent Ca2+ channel (VDCC) activity from DRG neurons (Axopatch 200A amplifier, Molecular Devices). Wild-type C57BL/6J mice were injected with CFA into the paw, and 48 h later were made tolerant to vehicle, SNC80 or ARM390 over 5 d. Mice were killed 6 h after the last drug injection (day 5), and DRGs were dissected, chemically and physically dissociated, and plated. All recordings were performed within 24 h after plating. Culture medium was replaced by an external solution that contained (in mm): 130 TEA-Cl, 10 CaCl2, 5 HEPES, 25 d-glucose and 2.5 × 10−4 tetrodotoxin at pH 7.2. Recording electrodes contained (in mm): 105 CsCl, 40 HEPES, 5 d-glucose, 2.5 MgCl2, 10 EGTA, 2 Mg2+-ATP and 0.5 Na+-GTP, pH 7.2. The potential difference between the open electrode and the bath ground was zeroed before establishing a ≥1-G61527 resistance seal. No compensation was made for the cancellation of liquid junction potential. Ca2+ currents were activated by depolarizing neurons from −80 to 10 mV for 100 ms at 10 s intervals. Currents were low-pass filtered at 2 kHz and digitized (Digidata, Molecular Devices) at 10 kHz. Leak currents were nulled using the P/4 subtraction method. DRG neurons were rapidly and continuously superfused (5 ml/min) with external solution. SNC80 (1 μm) was diluted into external solution on the day of the experiment and applied through the perfusion system. Experiments were performed at room temperature (22–24°C). Mean Ca2+ current amplitudes were measured (pCLAMP 9.0, Molecular Devices) between 5 and 10 ms after initiating the depolarizing step. Mean current amplitudes were then plotted against time. Stable recordings were fitted by a linear function to compare, by extrapolation, control current amplitude to the current amplitude recorded in the presence of opioid receptor agonists.
Statistical analysis
Data are expressed as mean ± SEM. All nonlinear regression analysis was performed with GraphPad Prism v4, and all statistical analysis by SigmaStat software. For pain experiments, two-way repeated-measures ANOVAs were performed followed by Tukey's post hoc analysis. For associative tolerance nonparametric multiple comparisons (Mann–Whitney U tests) were made with a Holm-Bonferroni correction. For locomotor stimulation and elevated plus maze analysis, multiple comparisons were made using Bonferroni corrected t tests. For electrophysiological recordings, responses were compared using ANOVA followed by a Tukey's post hoc analysis.
Results
Two selective δ agonists were used throughout the study: SNC80 (Calderon et al., 1994), and ARM390 (Wei et al., 2000). Both compounds show similar receptor binding and G-protein activation properties (Wei et al., 2000; Marie et al., 2003; Pradhan et al., 2009), and produce comparable pain-relieving effects in models of inflammatory pain (Pradhan et al., 2009). The two compounds penetrate the blood brain barrier following systemic injection and produce centrally mediated emotional responses (our unpublished results). However, in both cell systems and in vivo, SNC80 triggers robust δ receptor internalization, whereas ARM390 does not produce significant receptor trafficking (Marie et al., 2003; Pradhan et al., 2009).
Acute SNC80, but not ARM390 treatment produces transient desensitization of the analgesic response
We previously showed that SNC80, but not ARM390, produced acute behavioral desensitization 4 h after acute administration; and that this response was restored 24 h after the initial injection (Pradhan et al., 2009). Here we first examined whether acute behavioral desensitization could be observed at a later time point (12 h) and also confirmed the recovery of receptor function after 24 h. CFA was injected into the tail of DOR-eGFP mice, and strong heat hyperalgesia was observed 48 h later (Fig. 1A, controls vs dashed lines). An initial injection of either SNC80 or ARM390 reversed the CFA-induced hyperalgesia, and the two compounds were equally effective (Fig. 1A,B, Injection 1). Twelve hours after the first injection, a second injection of SNC80 was ineffective, while 24 h after the first injection the antihyperalgesic effect of the compound was restored (Fig. 1A, Injection 2, 12 H and 24 H). ARM390 produced potent antihyperalgesia at the second injection, both 12 and 24 h after the first injection (Fig. 1A, Injection 2, 12 H and 24 H). A parallel group of DOR-eGFP animals were killed at the time that they would have received the second drug injection. We observed robust DOR-eGFP internalization 12 h after SNC80 (Fig. 1B, 12 H), and cell surface fluorescence was restored at the 24 h time point (Fig. 1B, 24 H). In contrast, ARM390 treatment did not induce receptor internalization at any time examined (Fig. 1B, 12 H and 24 H).
Figure 1.

Acute SNC80, but not ARM390, produces transient receptor internalization and behavioral desensitization in DOR-eGFP mice. A, Desensitization (12 H) and recovery (24 H) of the analgesic response in the CFA tail model of inflammatory pain. Injection 1, Thermal responses in DOR-eGFP mice treated with vehicle (control), SNC80 (10 mg/kg), or ARM390 (10 mg/kg). Injection 2, Animals rechallenged 12 (left) or 24 h (right) following Injection 1. Dashed lines represent baseline thermal responses pre-CFA. For drug effects, ***p < 0.001, two-way repeated-measures ANOVA, n = 4–6 mice/group. Acute behavioral desensitization occurs for SNC80 only. B, Internalization and recovery of surface receptors 12 and 24 h after drug treatment. Brain and dorsal root ganglia were analyzed by confocal microscopy, and representative images of these regions are shown. Mean cell surface fluorescence was quantified in 3–4 sections per region per mouse. White bars, control group; gray bars, SNC80 group; black bars, ARM390 group; ***p < 0.001, one-way ANOVA, n = 3–4 mice/group. In vivo internalization is triggered by SNC80 only.
Thus, as we showed previously (Pradhan et al., 2009), receptor internalization (SNC80 but not ARM390) leads to acute desensitization of the analgesic response, observable 4H and 12H after treatment. Importantly, 24 h following treatment with either SNC80 or ARM390, DOR-eGFP was detected on the cell surface and these receptors were fully functional, regardless of whether receptor internalization had occurred previously or not.
Chronic SNC80 and ARM390 treatments produce full analgesic tolerance, independent of receptor internalization
Because SNC80- and ARM390-exposed animals appeared indistinguishable 24 h after the first treatment, we examined the consequences of chronic SNC80 or ARM390 treatment every 24 h. We first tested heat hyperalgesia in DOR-eGFP mice (Fig. 2, left). Animals were injected with CFA into the tail, and 48 h later were treated with vehicle, SNC80 (10 mg/kg) or ARM390 (10 mg/kg) every 24 h for 5 d. Heat hyperalgesia was stably expressed over the 5 d, as observed in control animals. As expected, both SNC80 and ARM390 efficiently reduced heat hyperalgesia on day 1. The drug effect gradually decreased over the 5 d, regardless of the internalization properties of the compounds. The time course for the development of tolerance was comparable for the two drugs, and complete tolerance was observed at day 5. We repeated the same experiment using wild type C57BL/6J mice (Fig. 2, middle). Tolerance developed similarly for the two δ agonists indicating that this observation was not specific to the DOR-eGFP strain. Finally, we determined whether tolerance to SNC80 and ARM390 was endpoint specific. We injected wild type mice with CFA in the paw, and tested mechanical allodynia by von Frey hair stimulation after 48 h (Fig. 2, right). Again, tolerance was observed in both SNC80- and ARM390-treated animals. As with heat hyperalgesia, tolerance developed to the two drugs at the same rate, and was complete by day 5. Together, our results show that analgesic tolerance develops for both SNC80 and ARM390, independent of the high- or low-internalizing properties of the agonist.
Figure 2.

Chronic SNC80 and ARM390 both produce analgesic tolerance. Development of tolerance in two mouse strains and two pain models. All animals were tested every 24 h with vehicle (control), SNC80 (10 mg/kg), or ARM390 (10 mg/kg) for 5 d. Left, CFA tail, thermal responses in DOR-eGFP mice. Middle, CFA tail, thermal responses in wild-type C57BL/6J mice. Right, CFA paw, mechanical responses in wild-type C57BL/6J mice. Dashed lines represent baseline mechanical or thermal responses pre-CFA. n = 5–10 mice/group. Analgesic tolerance developed similarly for SNC80 and ARM390, independent of mouse strain or nociceptive endpoint.
SNC80- and ARM390-tolerant animals show distinct receptor modifications
We then investigated the status of δ-opioid receptors after the establishment of tolerance to either SNC80 or ARM390. We examined DOR-eGFP receptor subcellular localization, ligand binding and G-protein coupling. As in Figure 2, DOR-eGFP mice were subjected to CFA injection in the tail, and treated for 5 d with vehicle, SNC80 or ARM390. Full tolerance was observed by day 5 (data not shown), and animals were killed 24 h after the last injection (day 6) for ex vivo analysis. SNC80-tolerant animals showed cell surface expression of DOR-eGFP in all areas examined (Fig. 3A). Importantly however, the fluorescent signal was significantly reduced, suggesting that receptor downregulation had occurred. Further, [3H]naltrindole binding was undetectable in brain membranes (Fig. 3B), and [35S]GTPγS binding was severely attenuated in the brain (Fig. 3C) and spinal cord (Fig. 3D). These results indicate that repeated SNC80 treatment leads to strong receptor downregulation throughout the nervous system. This result is consistent with our previous observation of lysosomal targeting of DOR-eGFP following agonist-induced receptor internalization (Pradhan et al., 2009, see references therein).
Figure 3.

Chronic SNC80 and ARM390 produce distinct adaptive responses at the receptor level. A, Subcellular DOR-eGFP localization. Brain and dorsal root ganglia were analyzed 24 h after the last drug treatment (day 6) by confocal microscopy, and representative images are shown. Mean cell surface fluorescence was quantified in 3–4 sections per region per mouse. White bars, control group; gray bars, SNC80 group; black bars, ARM390 group; ***p < 0.001, one-way ANOVA, n = 3–4 mice/group. Both SNC80 and ARM390 animals show surface fluorescence, but the signal is significantly reduced in the SNC80 group. B, Scatchard plot of [3H]naltrindole saturation binding performed on brain membranes. Experiments were performed in triplicate; n = 3–5 mice/group. Kd (nm) and Bmax (pmol/mg) values were comparable for control (0.13 ± 0.004 nm, 0.20 ± 0.02 pmol/mg) and ARM390 (0.10 ± 0.006 nm, 0.20 ± 0.03 pmol/mg) groups, while specific binding was undetectable in the SNC80 group. C, D, SNC80-induced [35S]GTPγS binding in brain (C) or spinal cord (D) membrane preparations. The y-axis shows mean ± SEM specific [35S]GTPγS binding expressed as percentage basal binding (i.e., absence of agonist). n = 3 mice/group. For brain, EC50 (nm) and Emax (percentage basal) values are comparable for control (466 ± 1.1 nm, 231 ± 3.3%) and ARM390-tolerant (302 ± 1.1 nm, 245 ± 3.5%) groups, while [35S]GTPγS binding was strongly attenuated (239 ± 1.3 nm, 140 ± 1.5%) in the SNC80-tolerant group. Similar results were observed in the spinal cord. EC50 and Emax values were as follows: control, 433 ± 1.2 nm, 167.4 ± 1.9%), ARM390-tolerant group (455 ± 1.2 nm, 176.2 ± 2.8%), and SNC80-tolerant group (220 ± 1.4 nm, 124.6 ± 1.4%). Chronic SNC80 produces DOR-eGFP receptor downregulation, while ARM390 leaves receptors at the cell surface.
In sharp contrast, ARM390-tolerant animals showed intense DOR-eGFP surface fluorescence in both DRG and hippocampus, comparable to the control group (Fig. 3A). Further, ARM390-tolerant animals showed [3H]naltrindole (Fig. 3B) and [35S]GTPγS binding in brain (Fig. 3C), and spinal cord (Fig. 3D) membrane preparations that were comparable to the control group. Hence, δ-opioid receptor surface expression, receptor number and receptor coupling to G-proteins are intact in ARM390-tolerant animals. In contrast to SNC80, tolerance to ARM390 does not seem to involve adaptive mechanisms at the receptor level.
SNC80- and ARM390-tolerant animals show cross-tolerance to analgesic responses
At this stage of the study, the finding that δ-opioid receptors were intact and expressed on the cell surface in ARM390-tolerant animals, was most intriguing. This observation led us to examine analgesic responses of these animals to SNC80, using a cross-over design. A first experiment was performed in DOR-eGFP mice, using the CFA paw model (Fig. 4A). As in the previous experiments, mice were made tolerant to SNC80 or ARM390 by daily treatments over 5 d. On the sixth day, SNC80-tolerant animals were challenged with ARM390, and ARM390-tolerant animals were challenged with SNC80. An antiallodynic effect was not detected for either compound, indicating that cross-tolerance had established in both SNC80- and ARM390-tolerant groups. We performed a second cross-tolerance experiment, using wild-type C57BL/6J mice in the CFA tail model (Fig. 4B). Again, none of the compounds showed antihyperalgesic activity in tolerant animals, indicating that cross-tolerance developed independent of mouse strain or analgesic endpoint (mechanical or thermal).
Figure 4.

Chronic SNC80 and ARM390 both produce analgesic cross-tolerance. SNC80-tolerant mice were challenged with ARM390 (10 mg/kg), and ARM390-tolerant mice were challenged with SNC80 (10 mg/kg). Graphs show tolerance and cross-tolerance data. A, CFA paw, mechanical response in DOR-eGFP mice. B, CFA tail, thermal response in C57BL/6J wild-type mice. Dashed lines represent baseline mechanical responses pre-CFA. n = 5 mice/group. Cross-tolerance occurs in both treatment groups. C, Repeated nociceptive testing produced associative tolerance. Inflammatory pain was induced in the paw of C57BL/6J mice, and 48 h later they were either tested daily for 5 d with vehicle (tested) or left in their home cage (novice). On day 6, mice were challenged with vehicle, SNC80 (10 mg/kg), or ARM390 (10 mg/kg). Dashed lines represent baseline mechanical responses pre-CFA. n = 5–6 mice/group. Antiallodynic effects of SNC80 or ARM390 were reduced in the habituated (tested) groups compared with corresponding novice drug groups (*p < 0.05, **p < 0.01), indicating that associative tolerance had developed. Nevertheless, in the habituated/tested animals, the two agonists continued to produce significant antiallodynic effects (##p < 0.01 compared with habituated vehicle controls), demonstrating that associative tolerance only partially accounts for the full tolerance and cross-tolerance observed in chronically SNC80- or ARM390-treated animals.
As ARM390 produced tolerance and cross-tolerance in the absence of changes at the receptor level, we explored the possibility that this tolerance was due to associative tolerance resulting from our repeated testing paradigm. Consistent with previous reports (Gamble and Milne, 1989; Williams et al., 2001, see references therein), repeatedly testing mice for 5 d with vehicle resulted in an attenuation of analgesic effects, and this was observed for both SNC80 and ARM390 (Fig. 4C). However, this associative tolerance only partially contributed to the full tolerance produced by either δ agonist as both still produced a significant antiallodynic effect in the habituated group. These results indicate that the observed tolerance to either SNC80 or ARM390 resulted from both associative and nonassociative (drug) factors, and confirmed the development of drug-induced tolerance in response to chronic ARM390.
Altogether, these data show that neither SNC80-tolerant animals, whose receptors are downregulated, nor ARM390-tolerant animals, whose receptors are intact at the cellular level, respond to any δ agonist in a model of pain. We conclude that repeated agonist treatment abolishes δ-opioid analgesia, either via internalization-dependent or internalization-independent mechanisms.
ARM390-tolerant mice respond to SNC80 at receptor trafficking and behavioral levels
We further investigated δ-opioid receptor function in ARM390-tolerant animals. We first examined SNC80-induced receptor internalization in both SNC80- and ARM390-tolerant animals. As in previous experiments, mice were made tolerant to SNC80 or ARM390 over 5 d. On the sixth day SNC80-tolerant animals were challenged with ARM390, ARM390-tolerant animals were challenged with SNC80, and tissue from central and peripheral nervous systems were analyzed by confocal microscopy. In accordance with our previous observation (Fig. 3A), SNC80-tolerant animals showed DOR-eGFP downregulation, as evidenced by significantly reduced fluorescence in the hippocampus, striatum and dorsal root ganglia (Fig. 5A). Interestingly, robust DOR-eGFP internalization was observed in ARM390-tolerant animals upon challenge with SNC80, in both the central and peripheral nervous system (Fig. 5A). In combination with receptor binding and G-protein coupling data from tolerant animals (Fig. 2D,E), this result confirms that chronic treatment with ARM390 leaves δ receptors functionally intact at the cell surface and readily responsive to agonist-induced internalization.
Figure 5.
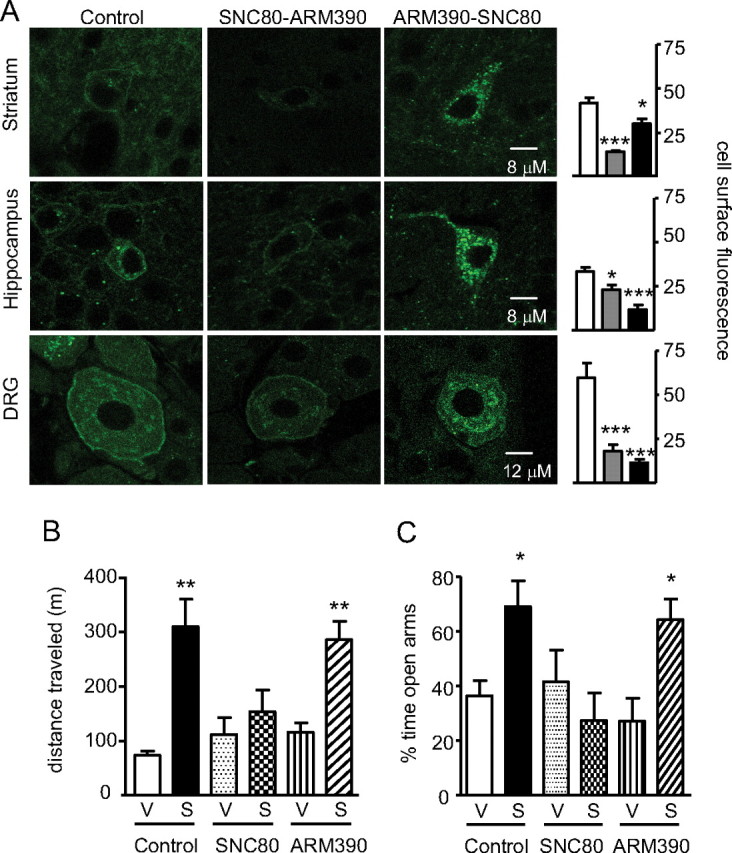
Chronic ARM390 induces analgesic tolerance only, leaving other δ-opioid receptor responses intact. A, SNC80-induced internalization in SNC80- and ARM390-tolerant animals. Striatum, hippocampus, and dorsal root ganglia were analyzed by confocal microscopy, and representative images are shown. Mean cell surface fluorescence was quantified in 3–4 sections per region per mouse. White bars, control group; gray bars, SNC80 group; black bars, ARM390 group; *p < 0.05, ***p < 0.001, one-way ANOVA, n = 3–4 mice/group. After chronic ARM390, surface δ receptors can be internalized by SNC80. B, SNC80-induced locomotor activation in SNC80- and ARM390-tolerant animals. Wild-type C57BL/6J mice were chronically treated with vehicle, SNC80, or ARM390, following which they were challenged with either vehicle or SNC80 (3 mg/kg, i.p.). For drug effects, **p < 0.01, as determined by multiple t tests with Bonferroni corrections, n = 5–6 mice/group. Control and ARM390-tolerant animals, but not SNC80-tolerant animals, showed SNC80-induced locomotor activation. C, SNC80-induced anxiolysis in SNC80- and ARM390-tolerant animals. Wild-type C57BL/6J mice were made tolerant to SNC80 or ARM390, and all groups were challenged with either vehicle or SNC80 (10 mg/kg, s.c.), and tested in the elevated plus maze. Data represent the percentage time spent in the open arms compared with total time spent in open and closed arms. For drug effects, **p < 0.01, *p < 0.05 as determined by multiple t tests with Bonferroni corrections, n = 5–6 mice/group. Control and ARM390-tolerant animals, but not SNC80-tolerant animals, showed an SNC80-induced anxiolysis. Altogether, chronic SNC80 produces generalized behavioral tolerance, while chronic ARM390 induces analgesic tolerance only.
Because SNC80 was able to internalize δ-opioid receptors in the ARM390-tolerant animals, but was unable to produce antiallodynic or antihyperalgesic effects (Fig. 4), we tested whether SNC80 could elicit other behavioral responses that do not involve pain processing mechanisms. SNC80 is known to produce locomotor activation (Spina et al., 1998) and anxiolytic responses (Saitoh et al., 2004) and we therefore tested both behaviors. We treated three groups of wild-type C57BL/6J mice with either vehicle (control), SNC80 (SNC80-tolerant) or ARM390 (ARM390-tolerant) and established tolerance over 5 d, as described previously. On day 6, mice in each group were challenged in a locomotor test with either vehicle or SNC80 (Fig. 5B). SNC80-tolerant animals were unresponsive to the locomotor stimulant effect of SNC80, consistent with a previous report (Jutkiewicz et al., 2005). In contrast, robust SNC80-induced locomotor stimulation was observed in both control and ARM390-tolerant animals. The anxiolytic effect of SNC80 was examined in another experimental group, and similar results were obtained (Fig. 5C). Acute SNC80 challenge did not modify the behavior of SNC80-tolerant animals in the elevated plus maze. However, control and ARM390-tolerant animals did respond to acute SNC80 and spent significantly more time in the open arms, indicating a reduction in anxiety levels. Therefore, after a 5 d chronic regimen, ARM390 treatment produced tolerance and cross-tolerance at the level of pain responses, while agonist-induced locomotor activation and anxiolysis remained intact.
ARM390-tolerant animals show desensitization at the level of the dorsal root ganglia
We next investigated the mechanism of ARM390-induced analgesic tolerance. We focused on δ receptor activity at the level of the DRG, as these contain first order neurons in pain transmission, but are not primary mediators of locomotor and emotional processing. Due to technical limitations we could not analyze δ receptor radioligand or [35S]GTPγS binding in the DRGs, as performed in brain and spinal cord membranes. A main downstream signaling effector of δ receptors are VDCCs, which are inhibited by δ agonists in DRGs (Walwyn et al., 2005). As in previous experiments, wild-type C57BL/6 mice were treated with CFA in the paw, and made tolerant to SNC80 or ARM390 over 5 d. On day 6, we measured SNC80-induced Ca2+ channel inhibition in DRGs collected from these animals (Fig. 6). The SNC80 challenge was ineffective in DRGs from naive animals, as previously described (Walwyn et al., 2005). In contrast, SNC80 efficiently inhibited Ca2+ channels following CFA-induced inflammation, consistent with enhanced δ receptor function following chronic pain (Cahill et al., 2003). In DRGs from SNC80-tolerant animals, δ-mediated regulation of Ca2+ channel activity was significantly reduced, in agreement with δ receptor downregulation following chronic treatment with this drug (Fig. 3A–D). Remarkably, δ-regulated Ca2+ channel inhibition was also reduced, and even abolished in DRGs from ARM390-tolerant mice. Therefore, chronic treatment with ARM390 does not change surface localization (Fig. 3A), receptor number (Fig. 3B), coupling to G-proteins in the CNS (Fig. 3C,D), or ability to internalize (Fig. 5A), but does desensitize δ receptor-VDCC coupling. This disruption of δ receptor-Ca2+channel interaction in primary nociceptors could explain the analgesic tolerance in ARM390-tolerant animals.
Figure 6.
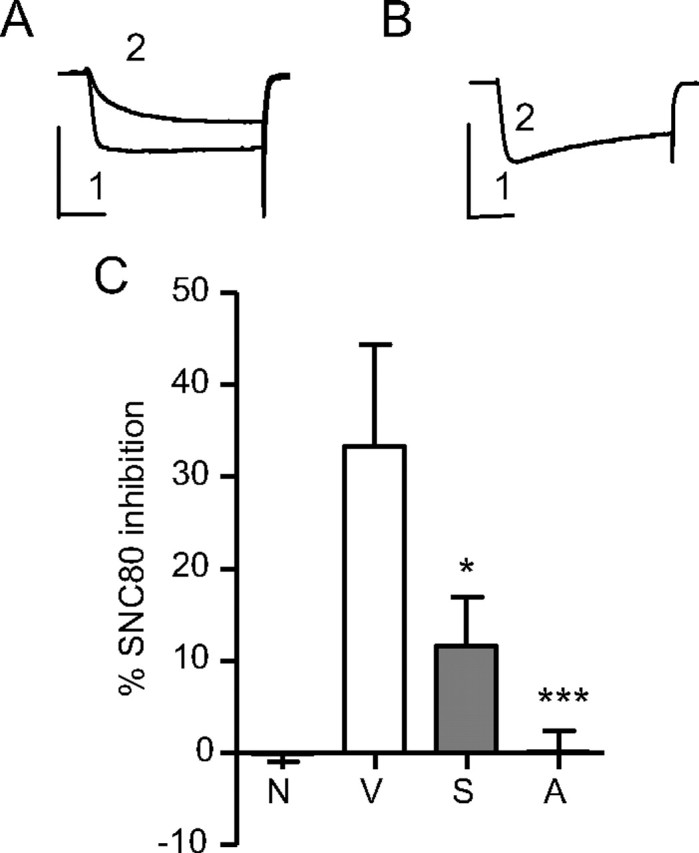
Chronic SNC80 and ARM390 both reduce δ receptor-mediated inhibition of Ca2+ channels in DRGs. The inhibitory effect of SNC80 (1 μm) on voltage-dependent Ca2+ channels was measured in medium-large DRG neurons. A, B, Exemplar currents from SNC80-responsive (A) or -nonresponsive (B) DRGs. (1, baseline depolarization; 2, SNC80-induced inhibition). Calibration: 10 ms, 20 mA on the x- and y-axes, respectively. C, SNC80 inhibited Ca2+ currents in ipsilateral DRGs from CFA (V, 33 ± 11%), but not naive (N, −0.13 ± 0.87%) animals. Both chronic SNC80 (S) and ARM390 (A) treatments reduced this effect to levels seen in naive DRGs (11.7 ± 5.3 and 0.12 ± 2.4%, respectively). Cumulative data from 6–14 cells are presented showing percentage SNC80 inhibition of Ca2+ currents; *p < 0.05, ***p < 0.001.
Discussion
Ligand-biased tolerance and δ-opioid receptors
A major challenge in the field of GPCR research is to understand the implication of ligand-biased signaling and regulation on physiological processes in vivo. To date, only a few examples have demonstrated the existence of in vivo agonist-biased mechanisms leading to specific behavioral responses. Hallucinations induced by specific 5HT2A agonists were shown to involve activation of a specific G-protein signaling pathway at cortical 5HT2A receptors, highlighting the relevance of ligand-directed signaling in psychoactive processes (González-Maeso et al., 2007). In the opioid research field, ligand-directed trafficking at the μ-opioid receptor was recently shown as a predictor of μ-opioid tolerance and dependence (Kim et al., 2008). Here, we show that ligand-biased trafficking at another opioid receptor, the δ-opioid receptor, leads to differential long-term adaptations in vivo and produces two highly distinct forms of behavioral tolerance.
We investigated the in vivo consequences of chronic treatment with either a high (SNC80)- or a low (ARM390)-internalizing agonist, both at the neuronal level and in several δ-opioid receptor-regulated behaviors. Our results indicate that chronic SNC80 treatment results in a robust and widespread downregulation of δ-opioid receptors throughout the nervous system. This adaptive cellular response is accompanied by a complete loss of all δ receptor-mediated behavioral responses, including analgesia, locomotor activation and anxiolysis. In contrast, chronic ARM390 treatment does not modify receptor expression at the cell surface. Nevertheless, tolerance develops specifically to the pain-relieving effects of δ agonists, while other δ receptor-regulated behaviors are unchanged. The intriguing analgesic-specific tolerance elicited by ARM390 is associated with an adaptive response at the level of voltage-dependent Ca2+ channels in DRGs. Together, our results indicate that analgesic tolerance can be established by at least two distinct pathways (Fig. 7).
Figure 7.

Two distinct paths toward analgesic tolerance. The scheme summarizes receptor analysis at the cellular level (subcellular localization, receptor number, G-protein coupling, and Ca2+ channel activity) and behavioral data obtained throughout this study. Data highlight the distinct adaptive processes elicited by chronic treatment using either the high-internalizing agonist (SNC80, left) or the low-internalizing agonist (ARM390, right). Top, A single 24 h treatment cycle leads to restored or intact receptors, with seemingly identical functional properties both at cellular and behavioral levels. Bottom, Five 24 h treatment cycles produce highly distinct forms of tolerance. Chronic SNC80 downregulates receptors throughout the nervous system, as classically described in cellular models, leading to generalized tolerance at the behavioral level. Chronic ARM390 does not modify receptor expression at the cell surface, but decreases δ receptor-mediated Ca2+ channel responses in DRGs. Further, tolerance develops only for analgesic but not locomotor or emotional responses, indicating that in vivo adaptations occur specifically at the level of pain processing pathways.
Internalization-dependent tolerance
Chronic treatment by the internalizing agonist (SNC80) resulted in DOR-eGFP downregulation at the cellular level. δ-Opioid receptor downregulation has been described earlier in transfected cell systems (Malatynska et al., 1996; Trapaidze et al., 1996; Ko et al., 1999; Whistler et al., 2002). This process results from receptor targeting to lysosomal compartments following internalization (Pradhan et al., 2009), and is considered a classical GPCR response to chronic agonist exposure (Ferguson, 2001; Law et al., 2004; Christie, 2008). In vivo, we did not observe significant downregulation following acute SNC80, and cell surface receptors were restored within 24 h after internalization had occurred, either by de novo synthesis or recruitment from intracellular stores (Pradhan et al., 2009; this study). However repeated SNC80 injections every 24 h, resulted in profound receptor downregulation, suggesting that degradation ultimately overcame receptor recovery. This phenomenon occurred throughout the nervous system, and all agonist-induced responses at the behavioral level were lost. A previous study also reported tolerance to the convulsant and locomotor stimulant effects of SNC80 following chronic treatment (Jutkiewicz et al., 2005). In that study, however, antidepressant effects of the compound remained and receptor downregulation was not observed throughout the brain. This discrepancy with our analysis of long-term effects of SNC80 may arise from different routes of drug administration (Jutkiewicz et al., 2005) and behavioral measures. Under our experimental conditions, robust receptor downregulation in SNC80-tolerant mice was revealed by four different ex vivo parameters (imaging, radioligand and [35S]GTPγS binding, and Ca2+ channel conductance) and in three behavioral responses (analgesia, locomotion and anxiolysis). Together, our comparison of high- and low-internalizing agonist effects strongly suggests that δ-opioid receptor internalization in vivo is a major regulatory event that leads to generalized tolerance.
Internalization-independent tolerance
Chronic treatment with the low-internalizing agonist (ARM390) produced behavioral tolerance only at the level of pain processing, while leaving locomotor and emotional responses intact. We investigated the underlying cause for this particular type of internalization-independent tolerance focusing on adaptations occurring in DRGs, the first order neurons of the pain processing pathway. Chronic ARM390 treatment affected neither the overall receptor number and cellular localization, nor ability of the receptor to internalize in these cells. Because the receptor seemed unaffected, we hypothesized that internalization-independent tolerance resulted from cellular adaptations occurring downstream of the receptor. Many signaling effector systems have been described, including Gβγ-mediated regulation of voltage-dependent Ca2+ channels (Williams et al., 2001; Christie, 2008) in DRGs (Walwyn et al., 2005). Our data demonstrate that δ receptor inhibition of Ca2+ channel activity, indeed, has desensitized in these cells, providing a mechanism for the ARM390 tolerance to analgesic responses. Importantly, this adaptive response seems specific to DRGs, as we did not observe any alteration of G-protein activation in the brain and spinal cord in ARM390-tolerant animals, and centrally mediated behaviors remained intact. This anatomically restricted adaptation may have resulted from altered δ receptor function in DRGs following CFA administration (Gendron et al., 2006) or altered coupling between δ receptors and Ca2+ channels. Overall our findings demonstrate that chronic receptor activation does not necessarily induce universal adaptive responses in vivo, but can recruit specific processes in restricted neuronal pathways. We also cannot exclude the possibility that, in addition to DRG-specific mechanisms, other adaptations may occur at the systems level through recruitment of opponent processes within the pain neurocircuitry (Ossipov et al., 2005; Simonin et al., 2006).
Cellular or systems level adaptive mechanisms have been proposed in the context of the μ-opioid receptor. The low-internalizing μ agonist morphine produces analgesic tolerance. A proposed hypothesis is that continuous signaling at the receptor is responsible for the development of counter-adaptations that lead to reduced analgesic effects of the drug (Whistler et al., 1999; He et al., 2002; Kieffer and Evans, 2002). However, the parallel between the two opioid receptors remains limited to low-internalizing agonists, because μ-opioid receptors rapidly recycle following internalization while δ-opioid receptors are degraded (Ko et al., 1999; Tanowitz and von Zastrow, 2003; Hanyaloglu and von Zastrow, 2008).
Therapeutic implications
Conclusions from this study have therapeutic implications as δ agonists are being tested clinically for the treatment of pain, anxiety and severe depression (Dondio, 2000; Dondio et al., 2001). From our data, it appears that internalizing and noninternalizing agonists would both produce tolerance in pain management. However, the development of noninternalizing δ agonists may be of particular interest for the treatment of anxiety and depression, as tolerance is less likely to occur in this context (Jutkiewicz et al., 2005).
In conclusion, this study shows that ligand-directed trafficking leads to the development of differential tolerance at δ-opioid receptors. The ligand-specific tolerance observed in this study strengthens the notion that ligand-biased responses have important implications in GPCR biology in vivo. Our findings uncover a novel aspect of biased agonism, which extends the concept of ligand-specific signaling to ligand-biased tolerance.
Footnotes
This research was supported by the Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale (INSERM), the Université de Strasbourg, the French Agence Nationale de la Recherche Grant IMOP, U.S. National Institutes of Health-National Institute on Drug Abuse Grant DA05010, and the Stefan and Shirley Hatos Research Foundation. A.A.A.P. was supported by INSERM-Fonds de la Recherche en Santé Québec. C.N. was supported by the Fondation pour la Recherche Médicale. We thank AstraZeneca R&D Montreal for the gift of ARM390. We thank J. L. Vonesch and the Institut de Génétique et de Biologie Moléculaire et Céllulaire Imaging Platform. We thank G. Scherrer for development of DOR-eGFP mice. We thank J. Becker, D. Massotte, and C. Gavériaux-Ruff for advice and helpful discussion.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befort K, Filliol D, Decaillot FM, Gaveriaux-Ruff C, Hoehe MR, Kieffer BL. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J Biol Chem. 2001;276:3130–3137. doi: 10.1074/jbc.M006352200. [DOI] [PubMed] [Google Scholar]
- Bosier B, Hermans E. Versatility of GPCR recognition by drugs: from biological implications to therapeutic relevance. Trends Pharmacol Sci. 2007;28:438–446. doi: 10.1016/j.tips.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH. Convulsant activity of a non-peptidic delta-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology (Berl) 2002;164:42–48. doi: 10.1007/s00213-002-1179-y. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain. 2003;101:199–208. doi: 10.1016/s0304-3959(02)00333-0. [DOI] [PubMed] [Google Scholar]
- Calderon SN, Rothman RB, Porreca F, Flippen-Anderson JL, McNutt RW, Xu H, Smith LE, Bilsky EJ, Davis P, Rice KC. Probes for narcotic receptor mediated phenomena. 19. Synthesis of (+)-4-[(alpha R)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC 80): a highly selective, non-peptide delta opioid receptor agonist. J Med Chem. 1994;37:2125–2128. doi: 10.1021/jm00040a002. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondio G. Development of novel pain relief agents acting through the selective activation of the delta-opioid receptor. Farmaco. 2000;55:178–180. doi: 10.1016/s0014-827x(00)00015-x. [DOI] [PubMed] [Google Scholar]
- Dondio G, Ronzoni S, Farina C, Graziani D, Parini C, Petrillo P, Giardina GA. Selective delta opioid receptor agonists for inflammatory and neuropathic pain. Farmaco. 2001;56:117–119. doi: 10.1016/s0014-827x(01)01020-5. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gavériaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Fraser GL, Gaudreau GA, Clarke PB, Ménard DP, Perkins MN. Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br J Pharmacol. 2000;129:1668–1672. doi: 10.1038/sj.bjp.0703248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Gallantine EL, Meert TF. A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin Pharmacol Toxicol. 2005;97:39–51. doi: 10.1111/j.1742-7843.2005.pto_97107.x. [DOI] [PubMed] [Google Scholar]
- Gamble GD, Milne RJ. Repeated exposure to sham testing procedures reduces reflex withdrawal and hot-plate latencies: attenuation of tonic descending inhibition? Neurosci Lett. 1989;96:312–317. doi: 10.1016/0304-3940(89)90397-2. [DOI] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci. 2008;27:2558–2567. doi: 10.1111/j.1460-9568.2008.06223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Lucido AL, Mennicken F, O'Donnell D, Vincent JP, Stroh T, Beaudet A. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. J Neurosci. 2006;26:953–962. doi: 10.1523/JNEUROSCI.3598-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 2000;20:1249–1259. doi: 10.1523/JNEUROSCI.20-03-01249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, Woods JH. Differential behavioral tolerance to the delta-opioid agonist SNC80 ([(+)-4-[(alphaR)-alpha-[(2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl]-(3-methoxyphenyl)methyl]-N,N-diethylbenzamide) in Sprague-Dawley rats. J Pharmacol Exp Ther. 2005;315:414–422. doi: 10.1124/jpet.105.088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. Opioid tolerance—in search of the holy grail. Cell. 2002;108:587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, Waldhoer M, Ou CJ, Taylor S, Ferwerda M, Cado D, Whistler JL. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JL, Arvidsson U, Williams FG, Law PY, Elde R, Loh HH. Visualization of time-dependent redistribution of delta-opioid receptors in neuronal cells during prolonged agonist exposure. Brain Res Mol Brain Res. 1999;69:171–185. doi: 10.1016/s0169-328x(99)00094-7. [DOI] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Law PY, Loh HH, Wei LN. Insights into the receptor transcription and signaling: implications in opioid tolerance and dependence. Neuropharmacology. 2004;47(Suppl 1):300–311. doi: 10.1016/j.neuropharm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Malatynska E, Wang Y, Knapp RJ, Waite S, Calderon S, Rice K, Hruby VJ, Yamamura HI, Roeske WR. Human delta opioid receptor: functional studies on stably transfected Chinese hamster ovary cells after acute and chronic treatment with the selective nonpeptidic agonist SNC-80. J Pharmacol Exp Ther. 1996;278:1083–1089. [PubMed] [Google Scholar]
- Marie N, Landemore G, Debout C, Jauzac P, Allouche S. Pharmacological characterization of AR-M1000390 at human delta opioid receptors. Life Sci. 2003;73:1691–1704. doi: 10.1016/s0024-3205(03)00489-2. [DOI] [PubMed] [Google Scholar]
- Nadal X, Baños JE, Kieffer BL, Maldonado R. Neuropathic pain is enhanced in delta-opioid receptor knockout mice. Eur J Neurosci. 2006;23:830–834. doi: 10.1111/j.1460-9568.2006.04569.x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Massotte D, Gavériaux-Ruff C, Kieffer BL. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS One. 2009;4:e5425. doi: 10.1371/journal.pone.0005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. J Pharmacol Sci. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Tryoen-Tóth P, Filliol D, Matifas A, Laustriat D, Cao YQ, Basbaum AI, Dierich A, Vonesh JL, Gavériaux-Ruff C, Kieffer BL. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci U S A. 2006;103:9691–9696. doi: 10.1073/pnas.0603359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F, Schmitt M, Laulin JP, Laboureyras E, Jhamandas JH, MacTavish D, Matifas A, Mollereau C, Laurent P, Parmentier M, Kieffer BL, Bourguignon JJ, Simonnet G. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A. 2006;103:466–471. doi: 10.1073/pnas.0502090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina L, Longoni R, Mulas A, Chang KJ, Di Chiara G. Dopamine-dependent behavioural stimulation by non-peptide delta opioids BW373U86 and SNC 80: 1. Locomotion, rearing and stereotypies in intact rats. Behav Pharmacol. 1998;9:1–8. [PubMed] [Google Scholar]
- Tanowitz M, von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem. 2003;278:45978–45986. doi: 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- Trapaidze N, Keith DE, Cvejic S, Evans CJ, Devi LA. Sequestration of the delta opioid receptor. Role of the C terminus in agonist-mediated internalization. J Biol Chem. 1996;271:29279–29285. doi: 10.1074/jbc.271.46.29279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Maidment NT, Sanders M, Evans CJ, Kieffer BL, Hales TG. Induction of delta opioid receptor function by up-regulation of membrane receptors in mouse primary afferent neurons. Mol Pharmacol. 2005;68:1688–1698. doi: 10.1124/mol.105.014829. [DOI] [PubMed] [Google Scholar]
- Wei ZY, Brown W, Takasaki B, Plobeck N, Delorme D, Zhou F, Yang H, Jones P, Gawell L, Gagnon H, Schmidt R, Yue SY, Walpole C, Payza K, St-Onge S, Labarre M, Godbout C, Jakob A, Butterworth J, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. N,N-Diethyl-4-(phenylpiperidin-4-ylidenemethyl)benzamide: a novel, exceptionally selective, potent delta opioid receptor agonist with oral bioavailability and its analogues. J Med Chem. 2000;43:3895–3905. doi: 10.1021/jm000229p. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P, Murray SR, Von Zastrow M. Modulation of postendocytic sorting of G protein-coupled receptors. Science. 2002;297:615–620. doi: 10.1126/science.1073308. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]