

Abstract
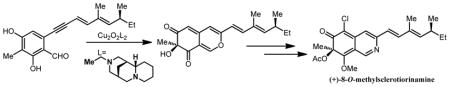
Enantioselective syntheses of the azaphilone natural products (+)-sclerotiorin and (+)-8-O-methylsclerotiorinamine which possess the natural R-configuration at the quaternary center are reported. The syntheses were accomplished using copper-mediated asymmetric dearomatization employing bis-μ-oxo copper complexes prepared from readily available (+)-sparteine surrogates and feature site selective O-methylation of a vinylogous pyridone to access to the isoquinolin-6(7H)-one core of (+)-8-O-methylsclerotiorinamine.
Introduction
The azaphilones1 are a structurally diverse family of natural products containing a highly oxygenated bicyclic core and a quaternary center. Our laboratory has previously reported the enantioselective synthesis of the azaphilone natural product S-15183a 1 (Figure 1) bearing a (R)-quaternary center.2 The synthesis was achieved utilizing enantioselective oxidative dearomatization3 mediated by a [(−)-sparteine]2Cu2O2 complex.4 A number of azaphilone natural products including (+)-sclerotiorin 2 and 8-O-methylsclerotiorinamine 3 possess the opposite stereochemistry at C7 compared to 1 (Figure 1).5 Since (+)-sparteine is not readily available or readily synthesized,6 it was necessary to identify an appropriate (+)-sparteine surrogate for use in asymmetric oxidative dearomatization. In this Article, we report the first asymmetric syntheses of (+)-2 and (+)-3 employing readily available (+)-sparteine surrogates 7 in copper-mediated oxidative dearomatization.
Figure 1.

Azaphilone Natural Products.
Results and Dicussion
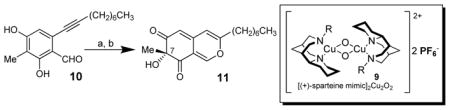
In our initial studies, we investigated the utility of (+)-sparteine surrogates 5–8 developed by O’Brien and co-workers7 for formation of L2Cu2O2 complexes 9 to mediate oxidative dearomatization of alkynylbenzaldehyde 102 in the presence of N,N-diisopropylethylamine (DIEA) as base and 4-dimethylaminopyridine (DMAP) as additive (Table 1). Gratifyingly, (+)-sparteine surrogates 5–7 were found to be suitable for this transformation and provided the desired (S)-enantiomer 11 in good to excellent enantiomeric excess (Table 1).8 N-ethyl-(+)-sparteine mimic 6 was found to be the optimal ligand to afford azaphilone core 11 in 70% yield and 95% e.e, (entry 3, Table 1) and was therefore employed in syntheses of (+)-sclerotiorin 2 and (+)-8-O-methylsclerotiorinamine 3 (vide infra). Use of (+)-sparteine surrogate 7 bearing an N-isopropyl substituent decreased both the yield and ee of azaphilone 11 (entry 4, Table 1). Likewise, ligand 8 bearing an N-neopentyl group was not effective, likely due to steric hindrance in the formation of the putative bis-μ-oxo complexes (entry 5, Table 1). (+)-Sparteine surrogates were also found to be more susceptible to oxidative modification8,9 under the reaction conditions than the alkaloid (−)-sparteine which may explain the slightly lower yields observed relative to (−)-sparteine. Through mass spectrometry analysis, we determined that the (+)-sparteine surrogates are oxidized preferentially on the interior ring system and not on the N-alkyl side chain.8
Table 1.
Oxidative dearomatization employing sparteine surrogates.a
 | |||
|---|---|---|---|
| entry | ligand | product | yieldb (ee) |
| 1 |
 4 |
 (R)-11 |
84% (98%)2 |
| 2 |
5 |
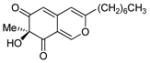 (S)-11 |
63% (92%) |
| 3 |
6 |
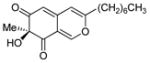 (S)-11 |
70% (95%) |
| 4 |
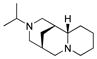 7 |
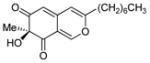 (S)-11 |
54% (74%) |
| 5 |
8 |
 (R)-11 |
33% (11%) |
Conditions: (a) 2.2 equiv. Cu(CH3CN)4PF6, 2.4 equiv. ligand, 1.6 equiv. DIEA, 2.4 equiv. DMAP, O2, −78°C to −10°C; (b) aq. H2PO4/K2HPO4 buffer (pH 7.2), CH3CN, RT [b] Isolated yield after silica gel chromatography. DIEA = N,N-diisopropylethylamine, DMAP = 4-(dimethylamino)pyridine.
Following development of an efficient route to the S-azaphilone core, we initiated studies towards the synthesis of (+)-sclerotiorin 2 and 8-O-methylsclerotiorinamine (3, Figure 1). Azaphilone 2 was the first azaphilone reported in the literature5a and has been shown to be an inhibitor of several important protein targets.10 Azaphilone 3 has been shown to significantly inhibit binding between the Grb2-SH2 domain and the phosphopeptide derived from the Shc protein.5d According to our retrosynthetic analysis (Figure 2), we anticipated that 8-O-methylsclerotiorinamine 3 could be obtained by amination and methylation of sclerotiorin 2 which itself could be derived from acylation and chlorination of 12. Azaphilone core structure 12 may be obtained via diastereoselective oxidative dearomatization3 of alkynylbenzaldehyde 13 followed by cycloisomerization.2
Figure 2.

Retrosynthetic analysis for 8-O-methylsclerotiorinamine 3.
Synthesis of the requisite alkynyl benzaldehyde substrate 13 was initiated with preparation of an iodo-diene side-chain fragment (Scheme 1). Swern oxidation of the commercially available chiral alcohol 14, followed by treatment of the intermediate aldehyde with (carbethoxyethylidene)triphenylphosphorane, afforded allylic ester 15 (85 % yield, >20:1 E:Z selectivity).11 DIBAL-H reduction of 15 provided alcohol 16 (55 %) which was cleanly oxidized with manganese oxide to produce enal 17.12 Chlorous chromium-mediated Takai olefination13 of 17 produced iodo-diene 18 (65 % yield over two steps, 10:1 E:Z selectivity).
Scheme 1.

Synthesis of iodo diene 18.
a) (COCl)2, DMSO, Et3N, CH2Cl2 0°C, 3 h; b) (carboethoxyethylidene)triphenylphosphorane, reflux, 12 h, 85%, 20:1 E:Z; c) DIBAl-H, CH2Cl2,rt, 2 h, 55%; d) MnO2, THF, 24h e) CrCl2, CHI3, dioxane:THF 6:1, 2 h, 0°C, 65% 2 steps, 10:1 E:Z.
For expedient access to the Sonogashira coupling partner for 18, we developed an optimized route to iodobenzaldehyde 19 (Scheme 2).14 We have previously reported a six step route to the corresponding bromobenzaldehyde involving installation of a nitro group to 3-methyl-2,4-dimethoxybenzaldehyde 20 to allow for ortho-halogenation of the aldehyde.15 A more concise route was developed utilizing the directed lithiation conditions originally reported by Comins and coworkers.16 Treatment of 20 with N,N,N′-trimethylethylenediamine, followed by ortho-lithiation of the amino-alkoxide intermediate with n-butyl lithium and subsequent treatment with 1,2-diiodoethane, produced the desired protected iodobenzaldehyde. Deprotection using boron tribromide proceeded smoothly to yield iodobenzaldehyde 19 (33% yield, three steps) using a single purification step. Sonogashira coupling of iodobenzaldehyde 19 with trimethylsilylacetylene, followed by silyl deprotection, afforded alkynylbenzaldehyde 21 (80 % yield, Scheme 3).17 Alkynylbenzaldehyde 21 was subjected to Sonogashira conditions with vinyl iodide 18 providing alkynylbenzaldehyde 13 in 55% yield.
Scheme 2.

Synthesis of alkynylbenzaldehyde 13.
a) N,N,N′-trimethylethylenediamine, n-BuLi, THF, 0°C 30 min.; n-BuLi, THF, −20°C, 16 h, then 1,2-diiodoethane; BBr3, CH2Cl2, 18 h (33% yield three steps b) CuI, trimethylsilylacetylene, (t-Bu)3P-HBF4, PdCl2(CH3CN)4, diisopropylamine, rt, 12 h, then K2CO3, MeOH, rt, 1 h 80%, two steps; c) Pd(PPh3)4, CuI, Et3N, 12 h, rt, 55%.
Scheme 3.

Synthesis of (+)-sclerotioin 2.
a) 2.2 equiv. Cu(CH3CN)4PF6, 2.4 equiv. N-ethyl-(+)-sparteine mimic 6, 1.6 equiv. DIEA, 2.4 equiv. DMAP, O2, −78°C to −10°C; b) aq. H2PO4/K2HPO4 buffer (pH 7.2), CH3CN, rt 76% over two steps c) acetic anhydride, DMAP, DIEA, 0°C, 30 min d) NCS, CH3CN, rt, 24 h 65% over two steps; e.) NH4OAc, MeOH, 30 min, 60%.
(+)-Sclerotiorin 2 was obtained in four steps (49% yield) from alkynylbenzaldehyde substrate 13 (Scheme 4). Copper-mediated, diastereoselective oxidative dearomatization of 13 afforded azaphilone 12 (76% yield, d.r.=12:1) with (S)-stereochemistry at C-7 based on the sign of the optical rotation ([α]D22=+163) after conversion to 2 (lit. reported [α]D22=+133)5a utilizing the N-ethyl (+)-sparteine surrogate 6 to access the desired L2Cu2O2 complex.7b,8 Since the optical rotation of the final product 8-O-methylsclerotiorinamine (+)-3 was consistent with that reported in the literature (vide infra, [α]D22=+104, literature reported [α]D22=+102),5d the apparent erosion in stereoselectivity in the formation of (+)-12 in comparison to the formation of (+)-11 may be an artifact of an inseparable impurity (vida infra). Azaphilone 12 was acylated (Ac2O/DMAP) and chlorinated (N-chlorosuccinimide, NCS) to afford the natural product sclerotiorin 2 (65% yield for two steps). Amination of 2 with ammonium acetate provided the vinylogous pyridone sclerotiorinamine 22 (60% yield).18
Scheme 4.

Isomerization of the C11-C12 olefin.
a) SiO2, ethyl acetate, 1 h.
During the synthesis of (+)-sclerotiorin 2, we found that the azaphilone core 12 was formed as a mixture of two compounds in a ~10:1 ratio after silica gel purification. Based on comparison with related azaphilones such as isochromophilone I and II which contain the same dienyl sidechain, we determined that the minor compound was the azaphilone containing a Z-double bond at C11–C12.19 The minor isomer is generally seen as a minor component during isolation, however, the two olefin isomers have been reported to interconvert.20 Through analyses of the isolation procedures of azaphilones containing the dienyl sidechain, it was determined that the minor E,Z isomer (cf. Scheme 4) is present in compounds which have been purified using silica gel chromatography. Accordingly, we suspected that azaphilones bearing dienyl sidechains were prone to mild acid-catalyzed olefin isomerization. The latter was confirmed by treating a natural sample of (+)-sclerotiorin 2 with silica gel which resulted in a mixture of 2:23 in ~10:1 ratio (Scheme 4). We believe that the facile isomerization results from protonation of 2 to afford the stabilized carbocation 24 which allows for equilibration between 2 an 23.8 The minor isomer 23 may affect the optical rotation of (+)-sclerotiorin, limiting comparison of optical rotations of synthetic ([α]D22=+163) and natural samples ([α]D22=+133).
With a sample of sclerotiorinamine 22 in hand, we next investigated conditions for O-methylation21 to access the congener 8-O-methylsclerotiorinamine. After screening of various reported methylation procedures, it was found that selective N-methylation could be obtained upon treatment of 22 with trimethyloxonium tetrafluoroborate (Me3OBF4) with diisopropylamine as base (0°C) to afford N-methylsclerotiorinamine 25 (Scheme 5) (85% yield). The selectivity for N-methylation was confirmed by alternative synthesis of 25 by treatment of (+)-sclerotiorin 2 with methylamine (>95%).22 Interestingly, methylation of 22 with Me3OBF4 and 2,6-di-t-butylpyridine (DTBP) as base led to apparent methylation of the C6-oxygen based on the crude NMR spectra. However, the presumed 7,8-dihydroisoquinoline was found to be unstable to purification and isolation. It is possible that utilization of a weaker base may alter the selectivity from N to O-methylation by not significantly deprotonating the N-H of the vinylogous amide until activation by O-alkylation. Other attempts to employ alternant electrophilic methylating agents (e.g. methyl triflate) and various weak bases did not result in isolable products.
Scheme 5.

Synthesis of N-methylsclerotiorinamine 25.
a) Me3OBF4, iPr2NH, THF, 85%; b) methylamine, THF, >95%
A related method for O-methylation of amides involves treatment with diazomethane (CH2N2).23 These reactions generally proceed without the need for base, likely arising from methylation of the enol tautomer of the amide. Gratifyingly, treatment of vinylogous pyridine 22 with trimethylsilyldiazomethane24 in a mixture of dichloromethane and methanol resulted in quantitative formation of 8-O-methylsclerotiorinamine 3 (Scheme 6) which had spectroscopic data consistent with that reported in the literature including the optical rotation ([α]D22=+104, literature reported [α]D22=+102) (Scheme 7).5d In the case at hand, derived enol 26 may be deprotonated by CH2N2 to give ion pair 27 which may expel nitrogen to yield 3 (Figure 3). Presumably, in this case selective methylation at C8 was observed8 due to steric congestion around the C6 ketone.
Scheme 6.

Synthesis of (+)-8-O-methylsclerotiorinamine 3.
a) TMSCHN2, CH2Cl2:MeOH 9:1, quantitative
Figure 3.

Proposed Mechanism for O-methylation
7-Epi-Sclerotiorin 28, a naturally occurring diasteromer of (+)-sclerotiorin varying only at the C7 stereocenter5c and the analogous 7-epi-8-O-methylsclerotiorinamine 29 were synthesized using the same synthetic sequence described above8 by substituting N-ethyl (+)-sparteine surrogate 6 for (−)-sparteine in the copper-mediated, oxidative dearomatization (Figure 4). Comparison of both diastereomers of the natural products with the rotation of the natural samples thus confirms the stereochemistry of 8-O-methylsclerotiorinamine.8
Figure 4.

Epimers of the Azaphilone Natural Products.
Conclusion
We have developed methodology to access azaphilone natural products containing the natural S-configuration at the quaternary center found in various members of this family utilizing (+)-sparteine surrogates. The methodology has been utilized in the asymmetric syntheses of the azaphilone natural products (+)-sclerotiorin 2 and (+)-8-O-methylsclerotiorinamine 3. Further studies towards the synthesis of related natural products and biological evaluation of 2, 3, and related molecules will be reported in due course.
Experimental Section
Azaphilone core 11
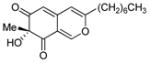
Alkynylbenzaldehyde 10 (27 mg, 0.10 mmol) and N,N-diisopropylethylamine (DIEA) (28 μL, 0.16 mmol) in 400 μL CH2Cl2 solution were added to the Cu2L2O2 complex prepared from Cu(CH3CN)4PF6 (82 mg, 0.22 mmol) and (−)-sparteine (56 μL, 0.24 mmol) in 1.6 mL anhydrous CH2Cl2 under an oxygen atmosphere at −78°C. After 5 min, 4-dimethylaminopyridine (29.2 mg, 0.24 mmol) in 400 μL CH2Cl2 was added and the mixture was warmed and stirred at −10°C for 30 h under an oxygen atmosphere. The reaction was quenched at −10°C with 2 mL of 10% sulfuric acid and 2 mL sat. brine. The mixture was extracted three times with EtOAc, and the combined extracts were concentrated in vacuo. The crude residue was dissolved in 2.0 mL of CH3CN, and 1.0 mL of 1:1.6 aqueous KH2PO4/K2HPO4 buffer (pH 7.2, prepared by dissolving 1.7 g KH2PO4 and 3.5 g K2HPO4 in 50 mL of distilled water) was added to the solution. The resulting heterogeneous mixture was stirred at room temperature for 1 h. 10% H2SO4 was added dropwise to the reaction mixture to adjust the pH to 2–3. The resulting mixture was extracted three times with EtOAc. The organic extracts were dried over anhydrous Na2SO4, concentrated, and purified on silica gel (hexane: EtOAc = 10:1 to 2:1) to afford azaphilone core 11. The 1H, 13C NMR, IR, and CIHRMS were found to be identical to those previously reported.2 The enantiomeric excess for 11 was determined using a HPLC (Chiralcel OD, 15% iPrOH in hexane, 1.0 mL/min) using UV detection at 320 nm and are listed in Table 1 corresponding to the ligand used.
2-Iodo-4,6-dihydroxy-5-methylbenzaldehyde 19
N,N,N′-trimethylethylenediamine (4.0 mL, 30.5 mmol) was dissolved in 200 mL of THF. A solution of 1.9M nBuLi (15 mL, 36.7 mmol) was added at 0°C. After stirring for 15 minutes, the reaction was cooled to −20°C and a solution of benzaldehyde 20 (5.0 g, 27.7 mmol) in 50 mL THF was added. The mixture was stirred for 30 minutes and then a solution of 1.9M nBuLi (45 mL, 73.3 mmol) was added dropwise. The reaction was stirred overnight. The solution was cooled to −40°C and a solution of diiodoethane (23.5 g, 83.2 mmol) in 50 mL THF was added dropwise. After 5 minutes, the reaction was warmed to room temperature and the reaction was quenched by addition of saturated ammonium chloride (100 mL), followed by saturated sodium thiosulfate (40 mL), and finally extracted three times with diethyl ether (3 X 120 mL). The organic layer was concentrated in vacuo and purified on silica (ethyl acetate:hexanes 1:10) to yield 19 (2.8 g, 33% yield, three steps) as a faint yellow powder. mp 75–77°C; 1H NMR (400 MHz, CDCl3) δ 10.25 (1H, s), 6.91 (1H, s), 3.88 (3H, s), 3.79 (3H, s), 2.08 (3H, s); 13C NMR (75.0 MHz, CDCl3) δ 189.6, 162.7, 161.8, 124.0, 120.68, 120.65, 112.2, 62.3, 55.9, 8.1; IR (thin film) 2944, 1685, 1584, 1559, 1456, 1376, 1283, 1234, 1133, 1019, 816 cm−1; CIHRMS M+ calculated for C8H7IO3 278.9518, found 278.9520.
Vinyl iodide 18
To CrCl2 (5.4 g, 43.7 mmol) was added anhydrous THF (75 mL) at 0°C. In a separate flask, unsaturated aldehyde 17 (1.0 g, 7.9 mmol), and iodoform (4.3 g, 11.0 mmol) were added to anhydrous THF (45 mL). The aldehyde mixture was added to the reaction mixture via cannula dropwise and the resulting mixture was stirred for 1 h. The reaction was filtered through Celite® and then washed with ether. The filtrate was finally washed with water and brine, the organic layer dried with sodium sulfate, filtered, and concentrated in vacuo. The concentrated material was pass through a plug of neutral alumina (hexanes) to afford vinyl iodide 18 as a light yellow oil 1.28 g (65% yield, 10:1 E:Z by 1H NMR analysis). Due to stability issues, 18 was used immediately and was not fully characterized. 1H NMR (400 MHz, CDCl3) δ 7.05 (1H, d, J=15.3 Hz), 6.12 (1H, d, J=15.3 Hz), 5.24 (1H, d, J=10.2 Hz), 2.35 (1H, m), 1.70 (3H, s), 1.40 (2H, m), 0.95 (3H d), 0.83 (3H,t, J=7.5 Hz).
Dienyne-benzaldehyde 13
An oven-dried round-bottom flask containing a mixture of vinyl iodide 18 (639 mg, 2.556 mmol), PdCl2(PPh3)2 (60 mg, 0.084 mmol), and CuI (6.6 mg, 0.033 mmol) was evacuated and refilled with argon. Anhydrous triethylamine (6.0 mL, x mmol) was added dropwise and the reaction was stirred at rt for 10 minutes. Alkynylbenzaldehyde 21 (300 mg, 1.70 mmol) in 3 mL THF was added. The resulting mixture was stirred at room temperature until complete conversion of 21 was observed (TLC analysis). The reaction mixture was diluted with water, carefully neutralized with 0.1 N aqueous HCl, and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification on silica gel (hexane: EtOAc = 10: 1) provided 279 mg (55%) of enyne benzaldehyde 13 as a yellow solid. mp 149–154°C; 1H NMR (400 MHz, CDCl3) δ 12.34 (1H, s), 10.21 (1H, s), 6.72 (1H, d, J=16 Hz), 6.49 (1H, s), 5.69 (1H, d, J=16 Hz), 5.44 (2H, ovrlp), 2.40 (1H, m), 2.10 (3H, s) 1.76 (3H, s), 1.36 (2H, m), 0.97 (3H, d, J=6.8 Hz), 0.84 (3H, t, J=7.6 Hz); 13C NMR (75.0 MHz, acetone-d6) δ 195.3, 163.3, 160.7, 148.8, 144.4, 132.6, 131.9, 128.7, 127.3, 114.3, 112.2, 104.0, 96.3, 85.5, 34.9, 30.4, 20.6, 12.3, 7.4; IR (thin film) 3161 (br), 2922, 2360, 1595, 1457, 1288, 1259cm−1; ESIHRMS [M+H]+ calculated for C19H23O3 299.1667, found C19H23O3 299.1647. [α]D22 = +14.0 (c = 0.3, CHCl3).
Azaphilone 12
Alkynylbenzaldehyde 13 (27 mg, 0.10 mmol) and N,N-diisopropylethylamine (DIEA) (28 μL, 0.16 mmol) in 400 μL CH2Cl2 solution were added to the Cu2L2O2 complex prepared from Cu(CH3CN)4PF6 (82 mg, 0.22 mmol) and N-ethyl-(+)-sparteine mimic 6 (56 μL, 0.24 mmol) in 1.6 mL anhydrous CH2Cl2 under an oxygen atmosphere at −78°C. After 5 min, 4-dimethylaminopyridine (29 mg, 0.24 mmol) in 400 μL was added and the mixture was warmed and stirred at −10°C for 30 h under an oxygen atmosphere. The reaction was quenched at −10°C with 2 mL of 10% sulfuric acid and 2 mL sat. brine. The mixture was extracted three times with EtOAc, and the combined extracts were concentrated in vacuo. The crude residue was dissolved in 2.0 mL of CH3CN, and 1.0 mL of 1:1.6 aqueous KH2PO4/K2HPO4 buffer (pH 7.2, prepared by dissolving 1.7 g KH2PO4 and 3.5 g K2HPO4 in 50 mL of distilled water) was added to the solution. The resulting heterogeneous mixture was stirred at room temperature for 1 h. 10% H2SO4 was added dropwise to the reaction mixture to adjust the pH to 2–3. The resulting mixture was extracted three times with EtOAc. The organic extracts were dried over anhydrous Na2SO4, concentrated, and purified on silica gel (hexane: EtOAc = 10:1 to 2:1) to afford azaphilone core 12 as a red oil (76% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.85 (1H, s), 6.95 (1H, d, J=15.6 Hz), 6.13 (1H, s), 5.90 (1H, d, J=15.6 Hz), 5.60 (1H, d, J=9.6 Hz), 5.51 (1H, s), 3.88 (1H, s), 2.40 (2H, m) 1.75 (3H, s), 1.50 (3H, s), 1.33 (2H, m), 0.93 (3H, d, J=6.4 Hz), 0.79 (3H, s); 13C NMR (75.0 MHz, CDCl3) δ 196.0, 156.9, 152.5, 148.3, 144.2, 142.1, 131.8, 115.9, 108.8, 105.5, 83.4, 35.1, 30.1, 28.7, 12.3, 12.0; IR (thin film) 3431 (br), 2961, 2360, 1616, 1540, 1172, 838cm−1 [α]D22 = +82.0 (c = 0.1, CHCl3).
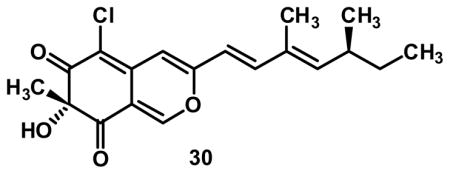
7-epi-Azaphilone 30
Azaphilone 30 was synthesized following the same procedure as above for (+)-12, except for the replacement of ligand 6 with (−)-sparteine 4. All spectroscopic data was indistinguishable from (+)-12. [α]D22 = −62.0 (c = 0.1, CHCl3).
(+)-Sclerotiorin 2
To azaphilone (+)-12 (120 mg, 0.41 mmol) in 3 mL methylene chloride at 0°C was added acetic anhydride (77 μL, 0.82 mmol) and N, N-diisopropylethylamine (80 μL, 0.45 mmol). The resulting mixture was stirred at 0°C for 30 m. The reaction mixture was quenched with water, extracted three times with ethyl acetate, washed with 1 N HCl, washed three times with water, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was dissolved in acetonitrile (3 mL) and N-chlorosuccinimide (129 mg, 0.96 mmol) was added. The reaction vessel was covered with aluminum foil and stirred at room temperature until 1H NMR analysis indicated completion of the reaction. The reaction mixture was diluted with ethyl acetate, washed with water, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to yield sclerotiorin 2 as a red oil (65%, two steps). The synthetic sample matched the 1H, 13C, and mass spectrum of the natural product. However due to the presence of a small amount of olefin isomer the optical rotations were not comparable. 1H NMR (400 MHz, CDCl3) δ 7.91 (1H, s), 7.05 (1H, d, J=15.6 Hz), 6.62 (1H, s), 6.07 (1H, d, J=15.6 Hz), 5.69 (1H, d, J=9.6 Hz), 2.45 (1H, m), 2.15 (3H, s), 1.82 (3H, s) 1.54 (3H, s), 1.39 (2H, m), 1.33 (2H, m), 0.99 (3H, s), 0.85 (3H, t, J=7.2 Hz); 13C NMR (75.0 MHz, CDCl3) δ 196.0, 186.2, 170.3, 158.3, 152.9, 149.1, 143.1, 138.9, 132.2, 115.9, 114.7, 111.0, 106.6, 84.8, 35.3, 30.3, 22.7, 20.4, 20.3, 12.6, 12.2; IR (thin film) 3438 (br), 2924, 2853, 1631cm−1 CIHRMS M+ calculated for C21H24O5Cl 391.1312, found 391.1333. [α]D22 = +163.0 (c = 0.1, EtOH) lit. reported [α]D22 = +133.0° (c = 0.3, EtOH).
7-epi-Sclerotiorin 28
Starting from 30, the same procedure employed to prepare (+)-2 was followed. All spectroscopic data was undistinguishable from (+)-2. [α]D22 = −103.0 (c = 0.1, EtOH).
Sclerotiorinamine (+)-22
To sclerotiorin (+)-2 (217 mg, 0.59 mmol) in THF (6 mL) was added ammonium acetate (54.8 mg, 0.71 mmol). The reaction mixture was stirred at room temperature until TLC indicated completion of the reaction. The mixture was diluted with water, extracted three times with ethyl acetate, washed with water, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to yield sclerotiorinamine 22 as an orange oil (229 mg, 100%) which was used directly in the next step without further purification.
N-Methylsclerotiorinamine (+)-25
Sclerotiorinamine (+)-22 (4.3 mg, 0.01 mmol) in CH2Cl2 (200 μL) was cooled to 0°C. Trimethyloxonium tetrafluoroborate (3.6 mg, 0.24 mmol) was added, followed by the slow addition of diisopropylamine (4.2 μL, 0.02 mmol). The reaction was stirred at 0°C until completion of the reaction was observed (TLC analysis). Purification over silica (EtOAc) provided N-methylsclerotiorinamine (+)-25 as a red oil (3.4 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 7.76 (1H, s), 7.04 (1H, s), 6.97 (1H, d, 15.6 Hz), 6.12 (1H, d, 15.6 Hz), 5.72 (1H, d, 9.8 Hz), 3.62 (3H, s), 2.46(1H, m), 2.17 (3H, s), 1.86 (3H, s), 1.54 (3H, s), 1.43 (1H, ovrlp), 1.34 (1H, ovrlp), 1.01 (3H, d, 6.9 Hz), 0.88 (3H, t, 7.5 Hz); 13C NMR (75.0 MHz, CDCl3) δ194.0, 184.6, 170.4, 148.7, 146.3, 145.3, 145.0, 142.1, 131.9, 114.8, 114.7, 111.4, 102.4, 85.0, 42.1, 35.3, 30.3, 23.5, 20.6, 20.5, 12.8, 12.3; IR (thin film) 2961, 2922, 2360, 1734, 1703, 1593, 1500, 1253, 1201cm−1 CIHRMS M+ calculated for C22H26NO4Cl 404.1629, found 404.1625. [α]D22 = +114 (lit. reported = +112)5d (c = 0.1, CH3OH).
(+)-8-O-Methylsclerotiorinamine (+)-3
To sclerotiorinamine 22 (25 mg, 0.064 mmol) in CH2Cl2:MeOH 9:1 (5 mL) was added trimethylsilyldiazomethane (2.0M in diethylether) (160 μL, 0.3 mmol). The reaction was stirred at rt for 1 h. Purification using silica gel chromatography (ether:hexanes 2:3) provided (+)-8-O-methylsclerotiorinamine 3 as an orange gum (26 mg, >95%.). 1H NMR (400 MHz, CDCl3) δ 9.05 (1H, s), 7.55 (1H, s), 7.5 (1H, d, J = 15.9 Hz), 6.60 (1H, d, 16 Hz), 5.71 (1H, d, 9.6 Hz), 4.01 (3H, s), 2.49 (1H, m), 2.10 (3H, s), 1.84 (3H, s), 1.55 (3H, s), 1.43 (2H, m), 1.34 (1H, m), 0.99 (3H, d, J=9.6 Hz), 0.87 (3H, t, J = 7.5 Hz); 13C NMR (75.0 MHz, CDCl3) δ192.7, 170.1, 162.4, 159.6, 149.5, 146.6, 143.1, 132.5, 130.5, 124.6, 119.1, 115.5, 111.5, 80.7, 61.9, 34.9, 30.2, 23.1, 20.4, 20.4, 12.6, 12.0; IR (thin film) 2961, 2871, 1742, 1696, 1611, 1579, 1279, 1248 cm−1 CIHRMS M+ calculated for C22H26NO4Cl 404.1629, found 404.1635. [α]D22 = +104 (lit. reported = +102) (c = 0.1, CH3OH). All spectroscopic data matched those reported in the literature.5d
7-epi-8-O-Methylsclerotiorinamine 29
Starting from (−)-sclerotiorinamine 30 the same procedure to synthesize (+)-22 and (+)-3 was followed to produce 29. All spectroscopic data was indistinguishable from (+)-3. ([α]D22 = −80 (c = 0.1, CH3OH).
Supplementary Material
Acknowledgments
This work was generously supported by the National Institutes of Health (GM-073855), Wyeth Research, and Merck Research Laboratories. We also thank the NSF (CHE-0443618) for the high resolution mass spectrometer used in this work.
Footnotes
Supporting Information Available: Complete experimental procedures and compound characterization data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Osmanova N, Schultze W, Ayoub N. Phytochem Rev. 2010;9:3115–342. [Google Scholar]
- 2.Zhu J, Grigoriadis N, Lee JP, Porco JA., Jr J Am Chem Soc. 2005;127:9342. doi: 10.1021/ja052049g. [DOI] [PubMed] [Google Scholar]
- 3.Dong S, Zhu J, Porco JA., Jr J Am Chem Soc. 2008;130:2738–2739. doi: 10.1021/ja711018z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Funahashi Y, Nakaya K, Hirota S, Yamauchi O. Chem Lett. 2000:1172–1173. [Google Scholar]
- 5.Sclerotiorin: MacCurtin T, Reilly J. Nature. 1940;146:335.Eade RA, Page H, Robertson A, Turner K, Whalley WB. J Chem Soc. 1957:4913.Whalley WB, Ferguson G, Marsh WC, Restivo RJ. J Chem Soc, Perkin I. 1976;13:1366.8-O-methylsclerotiorinamine: Nam JY, Kim HK, Kwon J-Y, Han MY, Son K-H, Lee UC, Choi JD, Kwon BM. J Nat Prod. 2000;63:1303. doi: 10.1021/np0001169.
- 6.(a) Ebner T, Eichelbaum M, Fischer P, Meese CO. Arch Pharm (Weinheim) 1989;322:399. doi: 10.1002/ardp.19893220704. [DOI] [PubMed] [Google Scholar]; (b) Smith BT, Wendt JA, Aube J. Org Lett. 2002;4:2577. doi: 10.1021/ol026230v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dearden MJ, Firkin CR, Hermet JP, O’Brien P. J Am Chem Soc. 2002;124:11870. doi: 10.1021/ja027774v. [DOI] [PubMed] [Google Scholar]; (b) Dearden MJ, McGrath MJ, O’Brien P. J Org Chem. 2004;69:5789. doi: 10.1021/jo049182w. [DOI] [PubMed] [Google Scholar]; (c) Ebner DC, Trend RM, Genet C, McGrath MJ, O’Brien P, Stoltz BM. Angew Chem Int Ed. 2008;47:6367. doi: 10.1002/anie.200801865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) O’Brien P. Chem Comm. 2008:655. doi: 10.1039/b711420f. [DOI] [PubMed] [Google Scholar]; (e) Bilke JL, Moore SP, O’Brien P, Gilday J. Org Lett. 2009;11:1935–1938. doi: 10.1021/ol900366m. [DOI] [PubMed] [Google Scholar]
- 8.See Supporting Information for complete experimental details.
- 9.For representative examples of internal oxidation of ligands in L2Cu2O2 complexes, see: Mahapatra S, Halfen JA, Tolman WB. J Am Chem Soc. 1994;116:9785.Mahadevan V, Hou Z, Cole AP, Root DE, Lal TK, Solomon EI, Stack TD. J Am Chem Soc. 1997;119:11996–11997.Itoh S, Nakao H, Berreau LM, Kondo T, Komatsu M, Fukuzumi S. J Am Chem Soc. 1998;120:2890–2899.
- 10.Lipoxygenase: Nam JY, Son KH, Kim HY, Han MY, Kim SU, Choi JD, Kwon BM. J Microbiol Biotechnol. 2000;10:544–546.Chidananda C, Rao L, Jagan M, Sattur AP. Biotechnol Lett. 2006;28:1633–1636. doi: 10.1007/s10529-006-9133-4.: Grb2-Shc: Chidananda C, Sattur AP. J Agric Food Chem. 2007;55:2879–2883. doi: 10.1021/jf062032x.: aldose reductase: cholesteryl ester transfer protein: Tomoda H, Matsushima C, Tabata N, Namatame I, Tanaka H, Bamberger MJ, Arai H, Fukazawa M, Inoue K, Omura S. J Antibiot. 1999;52:160–170. doi: 10.7164/antibiotics.52.160.
- 11.Zelle RE, Deninno MP, Selnick HG, Danishefsky SJ. J Org Chem. 1986;51:5032. [Google Scholar]
- 12.Ramamoorthy G, Acevedo CM, Alvira E, Lipton MA. Tet Asymm. 2008;19:2546–2554. doi: 10.1016/j.tetasy.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couladouros EA, Bouzas EA, Magos AD. Tetrahedron. 2006;62:5272–5279. [Google Scholar]
- 14.For a related route to the bromide congener of 19, see: Clark RC, Lee SY, Boger DL. J Am Chem Soc. 2008;130:12355–12369. doi: 10.1021/ja8012819.
- 15.Zhu J, Germain AR, Porco JA., Jr Angew Chem Int Ed. 2004;43:1239. doi: 10.1002/anie.200353037. [DOI] [PubMed] [Google Scholar]
- 16.Lang M, Steglich W. Synthesis. 2005:1019–1027. [Google Scholar]
- 17.Zhu J, Porco JA., Jr Org Lett. 2006;8:5169. doi: 10.1021/ol062233m. [DOI] [PubMed] [Google Scholar]
- 18.Natsume M, Takahashi Y, Marumo S. Agr Biol Chem. 1988;52:307–312. [Google Scholar]
- 19.(a) Fujimoto H, Matsudo T, Yamaguchi A, Yamazaki M. Heterocycles. 1990;30:607–616. [Google Scholar]; (b) Matsuzaki K, Tanaka H, Omura S. J Antibiot. 1995;48:708–713. doi: 10.7164/antibiotics.48.708. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki K, Ikeda H, Masuma R, Tanaka H, Omura S. J Antibiot. 1995;48:703–707. doi: 10.7164/antibiotics.48.703. [DOI] [PubMed] [Google Scholar]
- 21.For N- and O-methylation of pyridones see: Chung NM, Tieckelmann H. J Org Chem. 1970;35:2517.
- 22.For formation of vinylogous pyridones from azaphilones, see: Wei W-G, Yao Z-J. J Org Chem. 2005;70:4585–4590. doi: 10.1021/jo050414g.
- 23.Lei YX, Rappoport Z. J Org Chem. 2002;67:6971–6978. doi: 10.1021/jo0203802. [DOI] [PubMed] [Google Scholar]
- 24.(a) Aoyama T, Terasawa S, Sudo K, Shioiri T. Chem Pharm Bull. 1984;32:3759–3760. [Google Scholar]; (b) Kuhnel E, Laffan DDP, Lloyd-Jones GC, Martinez T, Shepperson IR, Slaughter JL. Angew Chem Int Ed. 2007;119:7205–7208. doi: 10.1002/anie.200702131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.