

Abstract
TRA-8, a monoclonal antibody to death receptor 5 induces apoptosis in various cancer cells; however the degree of sensitivity varies from highly sensitive to resistant. We have previously shown resistance to TRA-8 can be reversed using chemotherapeutic agents, but the mechanism underlying this sensitization was not fully understood. Here, we examined the combination of TRA-8 with doxorubicin or bortezomib in breast cancer cells. In TRA-8 resistant BT-474 and T47D cells, both chemotherapy agents synergistically sensitized cells to TRA-8 cytotoxicity with enhanced activation of apoptosis demonstrated by cleavage of caspases and PARP, reduced Bid, increased pro-apoptotic Bcl-2 proteins, and increased mitochondrial membrane depolarization. Doxorubicin or bortezomib combined with TRA-8 also reduced Bcl-XL and XIAP in treated cells. Furthermore, targeting these proteins with pharmacological modulators, AT-101, BH3I-2′ and AT-406, produced sensitization to TRA-8. TRA-8 combined with AT-101 or BH3I-2′, inhibitors of anti-apoptotic Bcl-2 proteins, produced synergistic cytotoxicity against ZR-75-1, BT-474, and T47D cells. The IAP targeting compound, AT-406, was synergistic with TRA-8 in BT-474 cells and to a lesser extent T47D cells. Activation of the intrinsic apoptotic pathway was a common mechanism associated with sensitization of TRA-8 resistant breast cancer cell lines. Collectively, these studies show that the Bcl-2 and IAP families of proteins are involved in TRA-8 and chemotherapy resistance via their modulation of the intrinsic apoptotic pathway. Targeting these proteins with novel agents sensitized TRA-8 resistant breast cancer cells, suggesting this approach may represent a potent therapeutic strategy in the treatment of breast cancer.
Keywords: breast neoplasms, TRAIL, death receptor antibody
Introduction
Limitations in the treatment of breast cancer include drug resistance, poor response rates, and drug toxicity. The first-line therapeutic regimens for metastatic breast cancer include chemotherapeutic agents and biological therapies, used alone or in combination (1). However, these therapeutic approaches are not sufficient for many patients, and metastatic breast cancer has a 5-year survival rate of only 26% (2). The current targeted treatments for breast cancer include Tamoxifen or aromatase inhibitors for estrogen receptor positive tumors (~60%) and Herceptin for Her-2/Neu positive tumors (20–25%) (3, 4). Targeted therapies may be given systemically with less toxicity than conventional chemotherapy, and have the potential to impact metastatic disease. However, some patients have innate or acquired resistance and a percentage of patients are left without any effective targeted treatment options.
One agent which is being investigated for the targeted treatment of breast cancer is tumor necrosis factor-related apoptosis-inducing ligand (TRAIL/Apo2 Ligand) (5). TRAIL is a member of the TNF superfamily and has been shown to induce apoptosis via a caspase dependent mechanism in many human breast cancer cell lines by binding to death receptors 4 and 5 (DR4, DR5) (5–8). TRAIL activates both extrinsic and intrinsic apoptosis through molecular crosstalk between these pathways (5, 9, 10). Moreover, while the ligand induces apoptosis in cancer cells, it lacks cytotoxicity against normal cells (5, 11). Harnessing this ability to stimulate both apoptotic pathways, monoclonal antibodies targeting individual TRAIL death receptors have been developed to provide longer half-lives in vivo and better specificity. TRA-8, which binds directly to DR5, is an agonistic antibody (12) that has been shown to have therapeutic potential in preclinical studies against a variety of cancer types, including breast cancer (12, 13).
As described by Rahman et al. (5, 14), breast cancer can be classified into different subtypes, which respond differently to TRAIL or agonistic death receptor antibodies. The majority of breast cancers are of the luminal subtype, which are hormone receptor positive (i.e. they express the estrogen receptor (ER) and/or progesterone receptor (PR)). The subtype with amplified HER-2 expression may be hormone receptor negative or positive. The basal or triple-negative subtype is comprised of tumors lacking ER and PR expression with no amplification of HER-2. Among nine breast cancer cell lines of various subtypes that were examined by our laboratory, each was shown to express DR5; however, only the basal cell lines were sensitive to TRA-8 induced cytotoxicity (13). The five luminal breast cancer cell lines were less sensitive or resistant to TRA-8. To overcome cellular resistance, we found that chemotherapeutic agents such as doxorubicin or paclitaxel used in combination with TRA-8 produced synergistic cytotoxicity. TRA-8 treatment inhibited the growth of 2LMP (subclone of MDA-MB-231) basal-type breast cancer xenografts in vivo (13). In other studies, the proteasome inhibitor, bortezomib, was shown to sensitize breast cancer cells to TRAIL-induced cytotoxicity (15, 16) and reduce the metastatic potential of 4T1 murine breast cancer cells in combination with MD5-1, a murine DR5 agonistic antibody (15). However, additional molecular markers for TRA-8 response and the underlying mechanisms of sensitization by these chemotherapeutic agents are not fully understood. Various regulatory molecules in the apoptotic pathways have been implicated in TRAIL sensitivity and sensitization by chemotherapy, including members of the Bcl-2 and inhibitors of apoptosis (IAP) families (5), but further characterization of the mechanisms would be useful for developing more efficient means of sensitizing resistant breast cancers.
In the current study, we investigated the mechanisms of sensitization of breast cancer cells (1 basal and 3 luminal cell lines) to TRA-8-induced cytotoxicity by doxorubicin, bortezomib and the small molecule apoptotic modulators, AT-101, BH3I-2′ and AT-406. Doxorubicin and bortezomib sensitized breast cancer cells to TRA-8-induced apoptosis, which was associated with intrinsic pathway activation and reductions in the anti-apoptotic proteins Bcl-XL or XIAP. Small molecule apoptotic modulators were used to investigate the importance of the Bcl-2 and IAP families of proteins in TRA-8 sensitization. AT-101 is a derivative of gossypol, a natural product of cottonseeds, which acts as a BH3-mimetic by binding to Bcl-2, Bcl-XL, Bcl-w and Mcl-1 (17, 18). BH3I-2′ is another BH3-mimietic, which binds to Bcl-2 and Bcl-XL. AT-406, a Smac-mimetic, binds to cellular inhibitor of apoptosis 1 and 2 (c-IAP-1/2), XIAP and livin (19, 20). These agents provide specific targeting of Bcl-2 and IAP families of proteins, and sensitized breast cancer cells to TRA-8-induced apoptosis via induction of the intrinsic apoptotic pathway. These results suggest that targeting of anti-apoptotic proteins may be valuable for enhancing the efficacy of TRAIL-targeted therapies for the treatment of breast cancer.
Results
Differential cytotoxicity of human breast cancer cell lines after treatment with anti-DR5 antibody in combination with chemotherapy
Sensitivity to TRA-8 anti-DR5 antibody-induced cytotoxicity alone or in combination with doxorubicin or bortezomib was examined in six human breast carcinoma cell lines. 2LMP cells treated with TRA-8 resulted in a dose-dependent decrease in cell viability with an IC50 concentration of 1.08 ng/ml (Fig. 1A). In contrast, the ZR-75-1 cell line had a TRA-8 IC50 of 387.7 ng/ml. The BT-474, T47D, MDA-MB-453, and ZR-75-30 cell lines were resistant to TRA-8 with no IC50 observed up to 1,000 ng/ml. This differential response to TRAIL receptor targeted therapy is consistent with previously reported results (13, 21). 2LMP and ZR-75-1 cells showed similar sensitivities to TRAIL ligand as TRA-8, while BT-474, T47D, MDA-MB-453, and ZR-75-30 cells were similarly TRAIL resistant (Supplementary Fig. S1). Flow cytometry analysis showed that DR5 expression on the surface of these breast cancer cell lines was variable (Supplementary Fig. S2), but the mean fluorescent intensity did not correlate with TRA-8 IC50 values (r = −0.309, p = 0.551).
Figure 1.
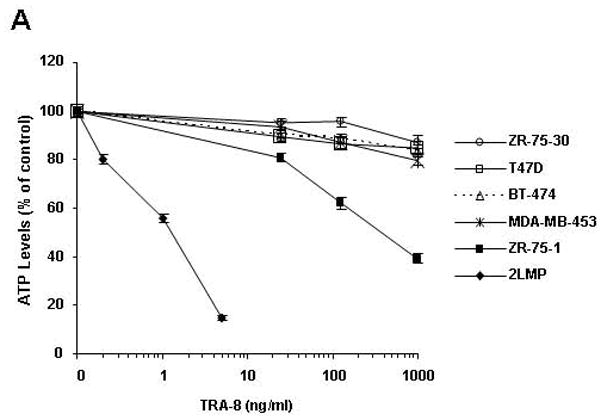
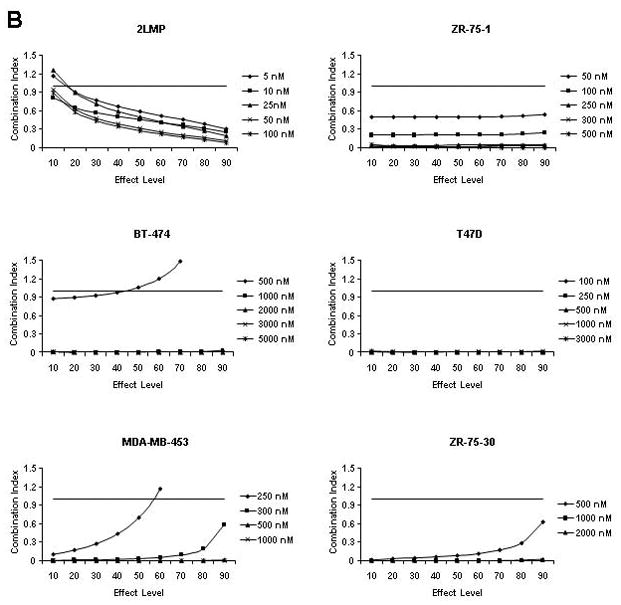
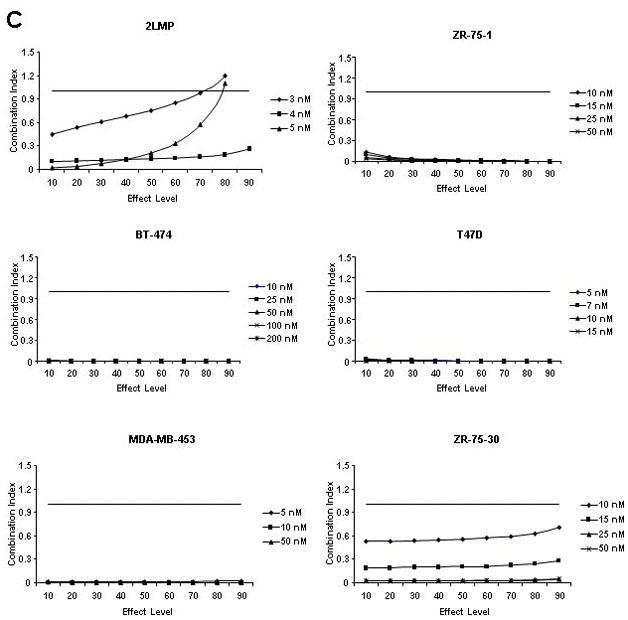
TRA-8-induced cytotoxicity against breast cancer cell lines was enhanced by doxorubicin or bortezomib pretreatment. Cells (1000/well) were plated in 96 well plates and incubated at 37 °C for 24 h. (A) Cells were then exposed to varying concentrations of TRA-8 for 24 h. For combination treatment, cells were pretreated with various doses of doxorubicin (B) or bortezomib (C) for 24 h, followed by TRA-8 treatment for an additional 24 h. Cell viability was determined using the ATPLite assay. Data obtained was analyzed to determine the combination index (CI) as a measure of drug interaction. CI values < 1 indicate synergism, CI values = 1 indicate additive effect, and CI values > 1 indicate antagonism.
Fig. 1B shows the interaction of TRA-8 and doxorubicin in each cell line expressed as a combination index (CI) where CI values < 1 indicate synergy, CI values = 1 indicate an additive effect, and CI values > 1 indicate antagonism. The combination of TRA-8 with varying concentrations of doxorubicin produced synergistic cytotoxicity against 2LMP, ZR-75-1, BT-474, T47D, MDA-MB-453, and ZR-75-30 cell lines. These results are particularly striking in the BT-474 cell line, as these cells are resistant to both doxorubicin and TRA-8 when used alone (less than 8% and 20% cytotoxicity, respectively), but the combination of these two agents resulted in up to 75% cytotoxicity. To examine the caspase dependence of the combined treatment with TRA-8 and doxorubicin, 2LMP and BT-474 cells were pretreated with the general caspase inhibitor, Z-VAD-FMK. In both 2LMP and BT-474 cells, the caspase inhibitor diminished the cytotoxicity of the combination of TRA-8 and doxorubicin (Supplementary Fig. S3), indicating that sensitization was caspase-dependent. As shown in Figure 1C, the addition of TRA-8 to bortezomib pre-treated cells produced synergistic cytotoxicity against 2LMP, ZR-75-1, BT-474, T47D, MDA-MB-453, and ZR-75-30 cell lines at all doses shown. Due to the synergistic cytotoxicity demonstrated in the 2LMP, ZR-75-1, BT-474 and T47D cell lines, these cells were chosen to further investigate the molecular mechanisms underlying the sensitization of cells to apoptosis by the combination treatments. 2LMP are of the basal subtype, while the other cell lines are of the luminal subtype, but have different receptor (PR and HER2) status, variable sensitivity to chemotherapy alone but all exhibited sensitization to treatment with the combination of chemotherapy and TRA-8.
Combining chemotherapy with TRA-8 induces caspase activation with Bid and PARP degradation
The TRAIL receptor pathway activated by TRA-8 involves binding to DR5, caspase cleavage and the subsequent induction of apoptosis. Despite the lack of correlation between TRA-8 sensitivity and surface DR5 expression, reports have shown that chemotherapy agents such as doxorubicin and etoposide can increase DR5 expression, which may relate to TRA-8 sensitization (22, 23). In BT-474 and T47D cells, doxorubicin produced an increase in DR5 expression, while bortezomib did not alter DR5 expression (Supplementary Fig. S4A and B). There was a positive correlation between DR5 expression and combination cytotoxicity in BT-474 cells (r = 0.9374, p = 0.0067); however, there was an inverse correlation between these variables in T47D cells (r = −0.9748, p = 0.0009). This indicates that alterations in DR5 expression by chemotherapy agents do not always predict sensitization to TRA-8.
To investigate the differential activation of caspases by TRA-8 in sensitive and resistant breast cancer cell lines, various apoptotic proteins were analyzed by Western Blot. In 2LMP cells, TRA-8 decreased the levels of pro-caspases and induced the cleavage of caspases-8, -9, and -3 after 3 h of treatment (Fig. 2). In addition, the pro-form of Bid was decreased and PARP was cleaved. Doxorubicin alone did not produce caspase cleavage, and the combination of doxorubicin and TRA-8 produced cleavage of caspases similar to that observed with TRA-8 alone in these cells. In ZR-75-1 cells, TRA-8 alone induced cleavage of caspases-8, -9, -3 and PARP in a dose-dependent manner, but did not change Bid levels. Doxorubicin combined with TRA-8 produced cleavage of caspases to a greater extent than TRA-8 alone and decreased Bid levels and induced PARP cleavage. In the TRA-8 resistant BT-474 and T47D cells, neither TRA-8 nor doxorubicin alone induced caspase activation. Only combined treatment with these two agents decreased Bid and produced cleavage of caspases-8, -9, -3 and PARP.
Figure 2.

Caspase-8, -9, -3 and PARP cleavage and decreased Bid levels were induced by combination treatment in breast cancer cell lines. 2LMP, ZR-75-1, BT-474 and T47D cells were pretreated for 24 h with doxorubicin (DOX; 50, 500, 5000, and 1000 nM, respectively) or bortezomib (BTZ; 5, 50, 100, and 10 nM, respectively) before the addition of TRA-8 (ng/ml) for 3 h. Whole cell lysates were analyzed by Western blotting. β-Actin was used as a loading control.
Next, we examined the effects of TRA-8 in combination with bortezomib on breast cancer cell lines. In 2LMP cells, bortezomib alone produced no activation of caspases, but when combined with TRA-8 there was cleavage of caspases-8, -9 and -3 (Fig. 2). In ZR-75-1 cells, bortezomib combined with TRA-8 produced increased caspase-8, -9 and -3 cleavage compared to TRA-8 alone. The bortezomib and TRA-8 combination also reduced the level of Bid and produced PARP cleavage. Similar to our observations with the combination of doxorubicin and TRA-8, only the combination of bortezomib and TRA-8 resulted in caspase cleavage in BT-474 and T47D cells. These results demonstrate that activation of apoptosis in TRA-8 resistant luminal cell lines occurs only after combined treatment with chemotherapy and TRA-8, and supports the hypothesis that the increased cytotoxicity observed with combination treatment occurs as a result of increased apoptosis.
Increased activation of the intrinsic apoptotic pathway after combination treatment with TRA-8 and chemotherapy
The combination of TRA-8 and chemotherapy produced cleavage of caspase-9 in 2LMP, ZR-75-1, BT-474 and T47D cells, which is downstream of the mitochondria and suggests the involvement of the intrinsic mitochondrial apoptotic pathway in the induction of cytotoxicity. Figure 3A shows that there was a significant reduction in mitochondrial membrane potential (ΔΨm) in TRA-8 sensitive 2LMP cells treated with TRA-8 alone and in combination with doxorubicin or bortezomib. In ZR-75-1 cells, TRA-8 alone and in combination with doxorubicin or bortezomib and bortezomib alone produced mitochondrial membrane depolarization, while doxorubicin alone had no effect. In BT-474 cells, TRA-8 or doxorubicin alone did not alter the ΔΨm, but bortezomib, or combination treatment with TRA-8 and either chemotherapeutic agent produced a significant decrease in ΔΨm. In T47D cells, only doxorubicin + TRA-8 or bortezomib + TRA-8 significantly reduced ΔΨm.
Figure 3.
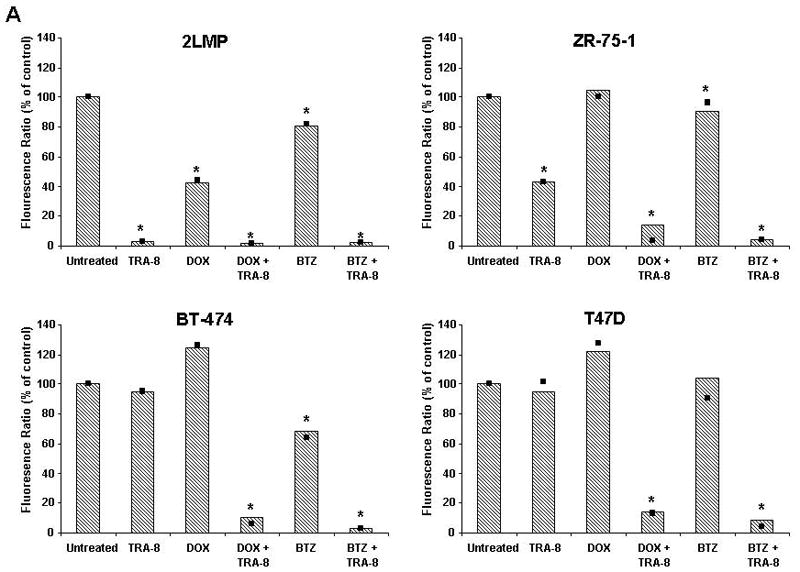
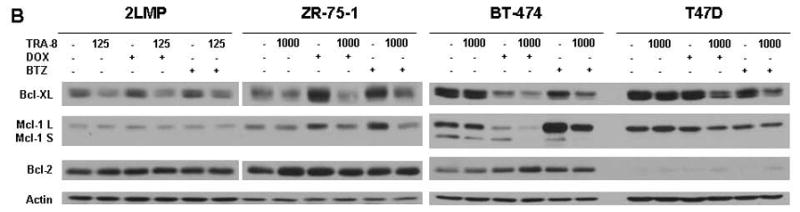
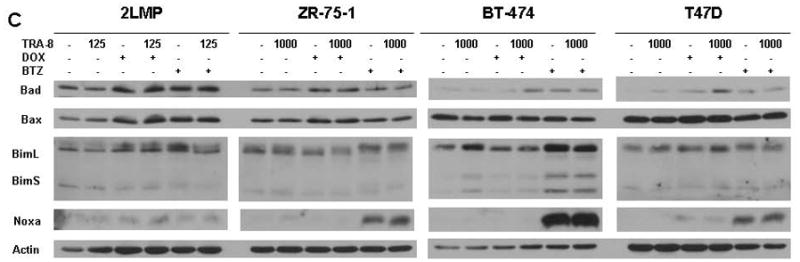
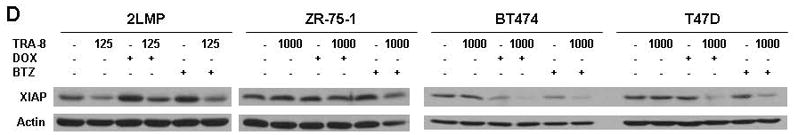
TRA-8 in combination with chemotherapy reduces mitochondrial membrane potential and decreases anti-apoptotic proteins. (A) 2LMP, ZR-75-1, BT-474 and T47D cells were treated with doxorubicin (DOX; 50, 500, 5000, and 1000 nM, respectively) or bortezomib (BTZ; 5, 50, 100, and 10 nM, respectively) for 12 h prior to the addition of TRA-8 at 125 ng/ml in 2LMP cells and 1000 ng/ml in the remaining cell lines. Mitochondrial membrane potential was assayed via JC-1 staining and flow cytometry after 24 h of TRA-8 treatment. The fluorescence ratio is reported as the median of 3 independent experiments relative to untreated controls. *, p < 0.05. (B–D) Cells were exposed to DOX or BTZ for 24 h prior to TRA-8 (1,000 ng/ml) treatment for 3 h. Cells were then harvested, and whole cell lysates were analyzed via Western blotting using antibodies against Bcl-XL, Bcl-2, Mcl-1 (B); Bad, Bax, Bim, Noxa (C); and XIAP (D).
To further investigate the impact of combination treatment on the intrinsic apoptotic pathway and to identify specific proteins involved in the chemotherapy-induced sensitization, the modulation of members of the Bcl-2 family was examined. In 2LMP cells, the anti-apoptotic protein Bcl-XL was reduced by treatment with TRA-8 alone and in combination with doxorubicin or bortezomib (Fig. 3B). In ZR-75-1 cells, the individual chemotherapy agents increased Bcl-XL, but combined with TRA-8 the levels of Bcl-XL were reduced to basal levels. In BT-474 cells, doxorubicin alone and in combination with TRA-8 reduced the levels of Bcl-XL, while only combination treatment reduced the levels in T47D cells. Bcl-XL was also reduced by combination treatment with bortezomib and TRA-8 in BT-474 and T47D cells. Another anti-apoptotic protein, Mcl-1, was decreased in BT-474 cells with doxorubicin alone and in combination with TRA-8, while bortezomib alone and in combination with TRA-8 increased Mcl-1 expression. In 2LMP and T47D cells, there was little or no change in Mcl-1 following any treatments. In ZR-75-1 cells, both doxorubicin and bortezomib increased Mcl-1, while the combination treatments with TRA-8 reduced the protein to basal levels. The levels of Bcl-2 were not altered by any treatment. These results indicate that the intrinsic pathway was activated, possibly due to a decrease in Bcl-XL, and that Mcl-1 does not play a role in this effect.
We also examined the levels of pro-apoptotic Bcl-2 family members Bad, Bax, Bim, and Noxa (Fig. 3C). In 2LMP cells, treatment with TRA-8, doxorubicin, or bortezomib, or TRA-8 in combination with these drugs did not alter the expression of these proteins. Bad levels were increased by doxorubicin or combination treatment with doxorubicin and TRA-8 in ZR-75-1 cells, and by bortezomib alone, and the combination of doxorubicin or bortezomib with TRA-8 in BT-474 and T47D cell lines. Noxa, a protein whose degradation is regulated by the proteasome (24), was increased by bortezomib treatment alone and in combination with TRA-8 in ZR-75-1, BT-474, and T47D cells. Bim was increased in BT-474 cells by bortezomib alone and in combination with TRA-8. No common modulation of pro-apoptotic proteins appears to account for TRA-8 sensitization; however, an overall increase in pro-apoptotic Bcl-2 molecules supports the observation that chemotherapy enhanced intrinsic pathway activation. Given the changes in Bcl-2 family members induced by chemotherapy agents in TRA-8 resistant breast cancer cell lines, we examined the basal levels of Bcl-2 family members to determine if expression of these proteins correlated with sensitivity to TRA-8. However, the basal levels of these proteins did not correlate with cell line TRA-8 sensitivity (Supplementary Fig. S5 and Fig. 1). Thus, chemotherapeutic agents may decrease modulators of intrinsic resistance to TRAIL-mediated apoptotic signaling and enhance the response to TRA-8 via an increase in pro-apoptotic molecules.
Also involved in the regulation of TRAIL-mediated apoptosis is the IAP family of proteins, which negatively regulate caspase activation. Basal levels of IAP proteins did not appear to correlate with TRA-8 sensitivity (Supplemental Fig. S6 and Fig. 1). Yet, XIAP protein levels were decreased following treatment with TRA-8 alone and in combination with doxorubicin or bortezomib in 2LMP cells (Fig. 3D). In T47D cells, neither doxorubicin nor TRA-8 alone produced a change in XIAP levels, while the combination produced a decrease in XIAP levels. In BT-474 cells, doxorubicin reduced XIAP levels alone, but led to a greater reduction when used in combination with TRA-8. The combination of bortezomib and TRA-8 also reduced XIAP in BT-474 and T47D cells. These results demonstrate that XIAP may be involved in the chemotherapy-induced enhancement of TRA-8-mediated apoptosis.
Inhibition of Bcl-2 and IAP proteins sensitize breast cancer cells to TRA-8-induced cytotoxicity
To confirm that the effects of chemotherapy on the expression of Bcl-XL and XIAP were important determinants of TRA-8 sensitization, we examined whether other compounds directly targeting these families of proteins would sensitize breast cancer cells to TRA-8. The 2LMP, BT-474, T47D and ZR-75-1 breast cancer cell lines were exposed to increasing doses of AT-101 or AT-406 alone or in combination with TRA-8 (Fig. 4A). The BH3-mimetic, AT-101, sensitized the 2LMP, ZR-75-1, BT-474 and T47D cell lines to TRA-8 in a synergistic manner. To provide further confirmation of the importance of Bcl-XL, BH3I-2′, a BH3-mimetic that selectively targets Bcl-2 and Bcl-XL, was used to treat cells prior to TRA-8 treatment (Fig. 4B). This agent synergistically sensitized the ZR-75-1, BT-474 and T47D cell lines, similar to AT-101, indicating that the mechanism of sensitization in these cell lines involve Bcl-2 and/or Bcl-XL.
Figure 4.

Targeting of the Bcl-2 or IAP families of proteins sensitizes breast cancer cells to TRA-8. (A) Cells (1000/well) were exposed to various concentrations of AT-101 for 24 h and treated for an additional 24 h with TRA-8. (B) Cells were treated for 24 h with BH3I-2′ prior to the addition of TRA-8 (25 ng/ml) for 24 h. (C) Cells were exposed to AT-406 for 24 h, then TRA-8 for an additional 24 h. Cell viability was determined using the ATPLite assay. Data obtained was analyzed to determine the combination index (CI) as a measure of drug interaction. CI values < 1 indicate synergism, CI values = 1 indicate an additive effect, and CI values > 1 indicate antagonism. (D) Cells were transfected for 24 h with 100 nM XIAP siRNA or transfection agent (Mock) prior to TRA-8 (1000 ng/ml) treatment for an additional 24 h. XIAP expression was determined by western blot. (E) Cells (1000/well) were transfected with 100 nM XIAP siRNA or a non-specific control siRNA for 24 h and treated with TRA-8 for an additional 24 h. ATP levels were determined relative to untreated control cells and represent the mean of samples run in quadruplicate from at least 3 independent experiments; error bars represent SE. ***, p < 0.001.
The IAP targeting Smac mimetic, AT-406, sensitized the 2LMP, BT-474 and T47D cell lines to TRA-8 in a synergistic manner (Fig. 4C), while the combination index could not be calculated in the ZR-75-1 cells because the combination did not produce any cytotoxicity above that of TRA-8 alone. Although the interaction between AT-406 and TRA-8 in the T47D cells was synergistic, the cells were resistant to both agents alone and combined there was never more than 40% cytotoxicity (Supplementary Fig. S7). To extend these observations, siRNA was used to knock down XIAP. In BT-474 cells, the addition of XIAP siRNA for 48 h greatly decreased the level of XIAP protein (Fig. 4D) and decreased gene expression (data not shown). Knockdown of XIAP sensitized BT-474 cells to TRA-8, leading to a significant increase in cytotoxicity compared to TRA-8 or XIAP siRNA alone or a non-specific siRNA (alone or in combination with TRA-8) (Fig. 4E). In the T47D cell line, anti-XIAP siRNA did not significantly affect the response to TRA-8 compared to non-specific siRNA, indicating that XIAP knockdown was not sufficient to sensitize these cells to TRA-8-induced cytotoxicity. Thus, it appears that neither the Bcl-2 nor IAP families are exclusively responsible for the sensitization effect. However, sensitization to TRA-8-induced apoptosis was achieved in each breast cancer cell line by targeting at least one of these families of proteins.
To investigate whether there is activation of apoptosis in cells treated with TRA-8 in combination with AT-101 or AT-406, and to determine which apoptotic mechanisms are involved, alterations in apoptotic proteins and the mitochondrial membrane potential in the various cell lines were examined. AT-101 or AT-406 alone did not produce changes in apoptotic protein levels in any of the cell lines tested. The TRA-8 sensitive cell line, 2LMP, showed dose-dependent cleavage of caspase-8, -9, -3, PARP and reduced Bid levels with TRA-8 alone or in combination with AT-101 or AT-406 (Fig. 5A). In ZR-75-1 cells, TRA-8 alone and in combination with AT-101 or AT-406 produced caspase and PARP cleavage, and reduced Bid. However, the combination of TRA-8 with AT-101 led to more prominent caspase cleavage compared to TRA-8 alone or combined with AT-406. BT-474 cells were sensitized to TRA-8 by both AT-101 and AT-406 with the induction of caspase-8, -9, -3 and PARP cleavage. In contrast, when AT-101 was used in combination with TRA-8 there was no effect on the Bid level, whereas AT-406 in combination with TRA-8 produced a slight decrease in Bid. In T47D cells, the combination of AT-101 with TRA-8 also produced activation of apoptotic proteins with cleavage of caspases and PARP, and a reduction in Bid. TRA-8 alone in this cell line reduced Bid levels and produced only the largest cleavage product of caspase-3, p20. Further supporting the lack of IAP importance in T47D sensitization, there was little cleavage of caspase-8 and no cleavage of caspase-9. Minimal cleavage of caspase-3 and PARP, and no alterations in the level of Bid, were observed in T47D cells treated with the combination of TRA-8 and AT-406. The lack of caspase-9 activation following AT-406 and TRA-8 combination treatment is in agreement with the lower cytotoxicity observed in T47D cells. However, other breast cancer cell lines showed sensitization following treatment with AT-406, supporting the importance of the IAP family in TRA-8 resistance.
Figure 5.

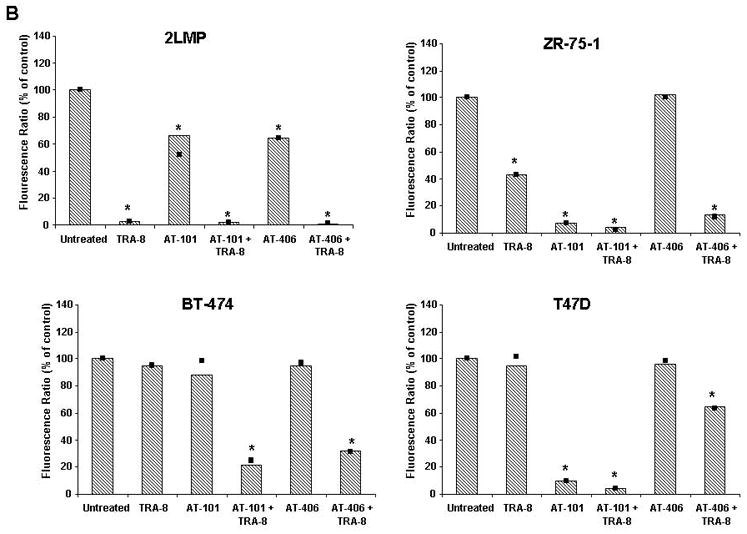
Activation of apoptotic pathways in breast cancer cells following treatment with small molecule modulators in combination with TRA-8. (A) 2LMP, ZR-75-1, BT-474 or T47D cells were treated with AT-101 (2.5, 15, 5, or 10 μM, respectively) or AT-406 (3, 20, 10, or 20 μM, respectively) for 24 h followed by treatment with TRA-8 (25 or 125 ng/ml) for an additional 5 h. Lysates were analyzed by immunoblotting with antibodies against the indicated proteins. (B) Cells were treated for 24 h with AT-101 or AT-406 before the addition of 125 ng/ml (2LMP) or 1000 ng/ml (ZR-75-1, BT-474, T47D) of TRA-8. Mitochondrial membrane potential was assayed via JC-1 staining and flow cytometry after TRA-8 treatment. The fluorescence ratio is reported as the median of 3 independent experiments relative to untreated controls. *, p < 0.05.
We then determined the effect of Bcl-2 and IAP inhibition on the intrinsic apoptotic pathway by determining ΔΨm. In 2LMP cells, AT-101 or AT-406 alone produced a decrease in ΔΨm, but there was a significantly larger decrease in the potential with TRA-8 alone or in combination with either compound (Fig. 5B). In ZR-75-1 and T47D cells, AT-101 alone produced a decrease in ΔΨm, as did the combination of AT-101 and TRA-8. In contrast, AT-406 alone did not alter the ΔΨm; however there was a decrease in the ΔΨm following the combined exposure to AT-406 and TRA-8. In BT-474 cells, only the combination of TRA-8 with AT-101 or AT-406 produced mitochondrial membrane depolarization.
Differential regulation of caspase-3 in human breast cancer cells treated with TRA-8, chemotherapy or apoptotic modulators
To further investigate differences in cytotoxicity in the breast cancer cell lines with combination treatment, we monitored the cleavage of caspase-3 to its active form by flow cytometry. TRA-8 alone and in combination with doxorubicin, bortezomib, AT-101 or AT-406 induced cleavage of caspase-3 in 2LMP cells (Fig. 6). In ZR-75-1 cells, TRA-8 alone produced an increase in caspase-3 cleavage. However, the combination of TRA-8 with doxorubicin, bortezomib, or AT-101 produced greater cleavage of caspase-3 than TRA-8 alone. AT-406 combined with TRA-8 induced a modest increase in caspase-3 (~8%) compared to TRA-8 alone. In T47D and BT-474 cells, TRA-8 alone did not induce changes in caspase-3, nor did doxorubicin, AT-101 or AT-406 alone. In contrast, bortezomib alone in T47D cells induced cleavage of caspase-3. In both T47D and BT-474 cells, doxorubicin, bortezomib, AT-101 and AT-406 increased caspase-3 cleavage when combined with TRA-8.
Figure 6.
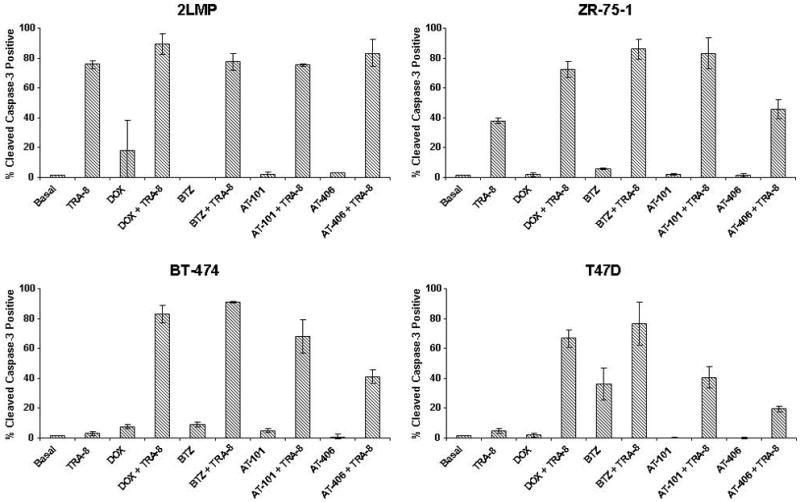
Cleaved caspase-3 levels following combination drug and TRA-8 treatment. 2LMP, ZR-75-1, BT-474 and T47D cells were treated for 24 h with DOX (50, 500, 5000, and 1000 nM, respectively), BTZ (5, 50, 100, and 10 nM, respectively), AT-101 (2.5, 15, 5, or 10 μM, respectively), or AT-406 (3, 20, 10, or 20 μM, respectively) before the addition of TRA-8 (125, 1000, 1000, or 1000 ng/ml, respectively, for the different cell lines) for 12 h. Cells were harvested, stained with cleaved caspase-3 antibody, and analyzed by flow cytometry. Values represent the percentage of cells positive for cleaved caspase-3 relative to untreated control cells and are reported as an average of 3 independent experiments; error bars represent SD.
Discussion
Over the past several years, we and others have examined the effects of TRAIL and TRAIL receptor-targeting antibodies against a variety of human cancer cell lines both in vitro and in vivo (6, 11–13, 21). Many reports have demonstrated the activity of TRAIL or TRA-8, an agonistic monoclonal antibody to DR5, used as single agents against certain human breast cancer cell lines; however other breast cancer cell lines were resistant to these treatments. It was later discovered that TRAIL and TRA-8 sensitive breast cancer cell lines were primarily those with a triple negative basal phenotype, while ER+ (luminal phenotype) or HER2 over-expressing cell lines were predominantly resistant (5, 13). We examined whether other cellular markers predicted sensitivity to TRA-8 in resistant breast cancer cell lines and showed that surface expression of DR5, and basal levels of Bcl-2 and IAP proteins did not correlate with sensitivity to TRA-8 (Supplementary Fig. S2, S5, and S6). There have been reports that the innate resistance of the luminal cells can be reversed by combination treatment with chemotherapeutic agents or various other agents, such as histone deacetylase inhibitors (12, 25). We have shown sensitization with doxorubicin and bortezomib in the current study (Fig. 1). While the luminal subtype clinically has the best prognosis, the development of resistance is still a common problem with node-positive patients having a 10-year overall survival rate of 65% (1, 26). Enhancing the efficacy of initial chemotherapy with the addition of targeted therapies would be beneficial.
Numerous mechanisms have been proposed for the enhanced efficacy between TRAIL and other agents, many of which involve activation of apoptotic pathways (12, 21, 27, 28). TRA-8 sensitization by doxorubicin and bortezomib was related to increased caspase activation and intrinsic pathway involvement, as evidenced by the results in Fig. 2 and 3A. We hypothesize that this chemotherapy-induced sensitization of breast cancer cell lines to TRA-8 anti-DR5 antibody involves the modulation of apoptotic proteins, such as Bcl-XL and XIAP. To investigate this hypothesis, we examined protein expression of members of the Bcl-2 family to determine if regulation of these proteins would explain the marked increase in cytotoxic response to combination treatment. Bcl-XL, an anti-apoptotic member of the Bcl-2 family, was shown to be over-expressed in human breast cancer tissue specimens as well as in a TRAIL-resistant breast cancer cell line (29). In the current study, all of the breast cancer cell lines expressed Bcl-XL and the levels were reduced by TRA-8 treatment in the TRA-8-sensitive 2LMP cell line (Fig. 3B). The reduction in Bcl-XL levels by combination treatment in the TRA-8 resistant luminal cells (BT-474, T47D, ZR-75-1) demonstrated the possible involvement of Bcl-XL in the mechanism of sensitization. Mcl-1, another anti-apoptotic Bcl-2 family member, was decreased by doxorubicin and TRA-8 combination treatment in BT-474 cells, but increased by bortezomib treatment in this cell line suggesting it does not play a primary role in sensitization. In examining pro-apoptotic Bcl-2 family members, there was no common modulation of Bad, Bax, Bim, or Noxa by both chemotherapy agents; however, bortezomib alone and in combination with TRA-8 as well as doxorubicin combined with TRA-8 did increase certain pro-apoptotic Bcl-2 proteins in TRA-8 resistant cell lines (Fig. 3C). Thus, the overall effect with both chemotherapy agents (doxorubicin or bortezomib) combined with TRA-8 was increased activation of the intrinsic apoptotic pathway with Bcl-XL playing a role in sensitization of luminal cell lines.
In addition to the Bcl-2 family, the IAP family of proteins also regulates activation of apoptosis (30). One member of this family, XIAP, has three baculoviral IAP repeat (BIR)-binding domains. BIR1 and 2 are known to bind and inhibit caspases-3 and -7, while BIR3 inhibits caspase-9 allowing XIAP to impact both the intrinsic and extrinsic apoptotic pathways. The reduction in XIAP by doxorubicin treatment alone in BT-474 cells, and by the combination of doxorubicin or bortezomib with TRA-8 in 2LMP, BT-474 and T47D cells highlight its importance in sensitization. Other investigators have shown that XIAP inhibition enhances TRAIL or Fas-induced apoptosis in pancreatic and other cancer cell lines (31–33). Recent reports described the use of a synthetic Smac peptide or XIAP siRNA to sensitize breast cancer cell lines to TRAIL (34, 35). Sun et al. reported the development of a series of Smac mimetics designed to improve the oral bioavailability while maintaining affinity for IAP proteins and cytotoxicity against MDA-MB-231 breast cancer cells, which lead to the development of the AT-406 compound used in these studies (20, 36, 37). These results, combined with our present findings, suggest that XIAP modulation may not only be a mechanism for TRA-8 sensitization, but also an important pharmacological target for inducing apoptosis in cancer cells.
To further investigate the hypothesis that modulation of Bcl-XL and XIAP is a mechanism contributing to TRA-8 sensitization, we employed AT-101, BH3I-2′, and AT-406 small molecule inhibitors to selectively target the Bcl-2 and IAP families of proteins. AT-101 has shown preclinical activity against a variety of human tumor cell lines, including lymphoma (38) and prostate cancer (39, 40). Synergistic interactions between AT-101 and chemotherapy agents have also been observed, for example, with 4-hydroxycyclophosphamide against mantle cell lymphoma lines (17) or with docetaxel against PC-3 prostate cancer cells (40). Clinical trials are ongoing with AT-101 alone or in combination with chemotherapy in several cancer types (18, 41). Other Bcl-2 targeting molecules have been successfully combined with TRAIL preclinically. For instance, BH3I-2′ (which binds both Bcl-2 and Bcl-XL) produced synergistic cytotoxicity after combination treatment with TRAIL against C4-2 prostate cancer cells (42). Also, ABT-737, which binds Bcl-2, Bcl-XL, and Bcl-w, produced synergistic cytotoxicity with TRAIL against Panc-1 pancreatic cancer cells (43). However, BH3I-2′ and ABT-737 do not bind to Mcl-1. Mcl-1 levels did not correlate with drug sensitization in the breast cancer cell lines, but high Mcl-1 levels have been shown to contribute to resistance to BH3 mimetics (44, 45). The advantage of using AT-101 instead of other Bcl-2 inhibitors currently in development is its ability to target Mcl-1 directly, as well as, upregulate pro-apoptotic Puma and Noxa as reported by Meng et al. (40). In the current study, combination treatment with either AT-101 or BH3I-2′ and TRA-8 produced synergistic cytotoxicity, enhanced activation of caspases and intrinsic pathway activation in TRA-8 resistant luminal breast cancer cell lines (Fig. 5 and 6). To our knowledge, this study is the first to combine AT-101 with a TRAIL receptor-targeted therapy in breast cancer. These findings provide further support the proposed role of Bcl-XL in chemotherapy-induced sensitization of breast cancer cells and the targeting of the Bcl-2 family to enhance TRAIL receptor-mediated therapies.
Apoptosis-driven therapeutics have also focused on the IAP family of proteins. AT-406, a novel Smac mimetic which binds c-IAP-1/2, livin, and XIAP, was recently shown to synergistically inhibit the growth of 2LMP human breast cancer xenografts when combined with TRAIL (19). However, as illustrated in the current study, 2LMP basal-genotype cells were sensitive to death receptor-induced apoptosis by TRA-8 alone. The effect of combining TRAIL receptor targeted treatment with AT-406 has not been studied previously in resistant luminal breast cancer cells lines. In this study, AT-406 sensitized the TRA-8 resistant BT-474 cell line and to some extent the T47D cell line, but not the ZR-75-1 cell line (Fig. 4C and S7). Knockdown of XIAP with siRNA and measurements of caspase-3 cleavage confirmed the role of XIAP in the sensitization of BT-474 cells. These results show that targeting of the IAP family of proteins sensitizes certain breast cancer cell lines to TRAIL-induced apoptosis with activation of the intrinsic apoptotic pathway.
Another important observation is that AT-101 and AT-406 in combination with TRA-8 induced cleavage and activation of caspase-8 (Fig. 5A), which acts upstream of the mitochondria. These combinations could be affecting the activation of caspase-8 via regulation of the death inducing signaling complex (DISC) that forms at the death receptor. One factor known to regulate DISC formation is cellular FLICE-inhibitory protein (c-FLIP). c-FLIP is generally considered an anti-apoptotic protein, which inhibits caspase-8 activation via binding of homologous domains within FADD and caspase-8 (46). We found that AT-101 and AT-406 did not change c-FLIP levels (data not shown). The lack of regulation of c-FLIP by AT-101 and AT-406 suggests c-FLIP is not involved in the mechanism of TRA-8 sensitization. There may be additional mechanisms of DISC regulation by these agents. Li et al. (47) and Sun et al. (48) reported the formation of an anti-apoptotic complex associated with DR5. The complex contained DDX3, c-IAP-1, and GSK3. c-IAP-1 is a target of AT-406 and removal of this protein from the DISC would allow caspase-8 activation seen with AT-406 and TRA-8 treatment in resistant breast cancer cell lines. This preliminary data suggests that more research investigating death receptor associated-proteins is warranted and may reveal additional mechanisms by which the Bcl-2 and IAP families of proteins and novel agents targeting these proteins regulate death receptor-mediated apoptosis.
For TRAIL-receptor targeted therapies to be successful in the clinical setting, innate or acquired resistance will need to be overcome. Our studies show that agents capable of sensitizing breast cancer cells to the anti-DR5 antibody, TRA-8, include chemotherapy agents (doxorubicin or bortezomib) and novel small molecule apoptotic modulators (AT-101 or AT-406) that target the Bcl-2 or IAP families of proteins either directly or indirectly. We are the first to show that the AT compounds were effective in promoting apoptosis and sensitizing breast cancer cells to TRA-8. Current breast cancer regimens in combination with TRA-8 and small molecule apoptotic modulators could provide a promising direction for the treatment of breast cancer.
Materials and Methods
Cell Lines and Reagents
The 2LMP subclone of the human basal breast cancer cell line MDA-MB-231 was obtained from Dr. Marc Lippman (University of Miami, Miami, FL) and was grown in improved MEM supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA). The T47D luminal human breast cancer cell line was obtained from Dr. Andra Frost (University of Alabama at Birmingham, Birmingham, AL) and was grown in RPMI 1640 supplemented with 10 μg/ml insulin and 10% FBS. The BT-474, ZR-75-1, ZR-75-30, and MDA-MB-453 luminal human breast cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA). The ZR-75-30 cell line was maintained in RPMI with 4.5 g/liter glucose, 10 mM HEPES, 1 mM sodium pyruvate and 10% FBS. The 2LMP, BT-474, ZR-75-1, and MDA-MB-453 cell lines were maintained as previously described (13). All cell lines were maintained in antibiotic-free medium at 37°C in a 5% CO2 atmosphere and routinely screened for Mycoplasma contamination.
Purified monoclonal TRA-8 IgG1 antibody was provided by Dr. Tong Zhou (University of Alabama at Birmingham, Birmingham, AL). SuperKiller TRAIL and TRAIL dilution buffer were purchased from Axxora (San Diego, CA). Doxorubicin was purchased from Sigma-Aldrich (Saint Louis, MO) and prepared as a 10 mM stock solution in distilled water. Bortezomib was obtained from the University of Alabama at Birmingham Hospital Pharmacy (Birmingham, AL) and reconstituted in PBS as a 2.63 mM stock solution. AT-101 and AT-406 were kindly provided by Ascenta Therapeutics (Malvern, PA).
Cell Viability Assay
Cells were trypsinized and seeded at 1000 cells/well in Costar 96-well plates in complete media and incubated overnight at 37°C. Cells were pretreated with various doses of drug for 24 h prior to the addition of TRA-8 and incubated for an additional 24 h before assessment of cell viability by measurement of cellular ATP levels using the ATPLite luminescence-based assay (Perkin Elmer, Boston, MA) and a TopCount Luminescence Reader (Packard Instruments, Meriden, CT).
Western Blot Analysis
Cells were plated at 5 × 106 cells/ml in complete media and incubated overnight. After treatment, cells were washed with PBS once and lysed with RIPA (radio-immunoprecipitation assay) buffer with 150 mmol/L NaCl, 50 mmol/L Tris (pH, 7.4), 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 10 mmol/L sodium orthovandate, and 1:100 Protease Inhibitor Cocktail (Sigma), then sonicated on ice once for 15 sec and centrifuged for 10 min at 4°C. The protein concentration of each sample was determined using a Lowry detergent compatible assay (Bio-Rad Laboratories, Inc., Hercules, CA). Samples (15–25 μg of protein) were resolved by SDS-polyacrylamide gel and transferred onto PVDF (polyvinylidene fluoride) membranes. Membranes were incubated with primary antibodies to caspase-3 (Stressgen, Ann Arbor, MI), XIAP (R&D Systems, Minneapolis, MN), caspase-8, PARP (BD PharMingen, San Diego, CA), caspase-9, Bcl-XL, Bid, Bad, Bak, Bik, Bim, Puma (Cell Signaling Technology, Beverly, MA), Noxa (EMD, Darmstadt, Germany), Livin (Imgenex, San Diego, CA), Bcl-2 (ThermoScientific, Fremont, CA), and Mcl-1 (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C, then horseradish peroxidase-labeled secondary antibodies (Bio-Rad Laboratories) for 1 h. Primary antibodies to c-IAP-1 and -2 were gifts from Dr. Tong Zhou (Birmingham, AL). Proteins were visualized using chemiluminescence reagents (GE Healthcare, Piscataway, NJ) according to manufacturer’s instructions.
Mitochondrial Membrane Potential
The mitochondrial membrane potential (ΔΨm) was assessed using JC-1 (5, 5, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazolylcarbocyanine iodide), which is a cationic dye that accumulates in the mitochondrial membrane to form aggregates that fluoresce red. When the ΔΨm is lost in apoptotic cells, the dye cannot aggregate and remains in monomeric form that fluoresces green. Cells were plated at 3 × 106 cells/ml in complete media, incubated overnight and then either left untreated or treated with drug for 24 h, then TRA-8 was added for an additional 12 h prior to cell harvest. Cells were stained using a JC-1 mitochondrial membrane potential detection kit (Cell Technology, Mountain View, CA) according to manufacturer’s instructions and analyzed via a FACScan flow cytometer and CellQuest software (BD Biosciences, San Jose, CA). To determine ΔΨm changes in vitro produced by TRA-8 and drugs, fluorescence ratios were calculated as the median red fluorescence value divided by the green fluorescence value in treated cells as a percentage of the ratio in untreated control cells.
Flow Cytometry Analysis
Cells were plated at 3 × 106 cells/ml in complete media, incubated overnight and then either left untreated or treated with drug for 24 h. TRA-8 was then added for an additional 12 h prior to cell harvest with trypsin. Cells were washed with FACS buffer (PBS with 1% bovine serum albumin (BSA), 0.1% saponin, and 0.01% sodium azide), then fixed in 1% paraformaldehyde on ice for 15 min. Cells were resuspended in blocking buffer (PBS with 3% BSA, 0.1% saponin, and 0.01% sodium azide) for 15 min on ice, after which, cells were stained with cleaved caspase-3 (Asp175) antibody (Cell Signaling Technology) for 30 min on ice while shaking. Cells were washed once in FACS buffer, then incubated with secondary antibody, Alexa Fluor 488 (Invitrogen, Carlsbad, CA) for 30 min on ice, washed again in FACS buffer and resuspended in 200 μl of FACS buffer. Samples were analyzed on a FACScan flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Ashland, OR).
Statistics
The combination index (CI) for dose-effect relationships of TRA-8 and drugs in combination was calculated based on the multiple drug-effect equation of Chou-Talalay (49). CI = DA/ICX,A + DB/ICX,B where CI is combination index, ICX,A and ICX,B are the concentrations of drugs to result in X% inhibition for each respective drug alone, and DA and DB are concentrations of each drug in the mixture that yield X% inhibition. The combination index curve or modified isobologram is generated by plotting CI vs. X, ranging from 0% to 100%. The drug interaction is readily identified at any level of inhibition. The resulting combination index (CI) theorem of Chou-Talalay offers quantitative definition for additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) in drug combinations. The quantitative diagnostic plot was generated with Statistical Analysis Software (SAS) version 9.1. The synergism effect was further confirmed with concentration-effective curve with nonlinear regression method (50) and Isobologram methods (data not shown).
Mitochondrial membrane potential was measured as a JC-1 fluorescence ratio, then compared to untreated control cells (% of control). Individual drug and drug plus antibody fluorescence ratios were compared between treated cells and untreated control cells using a nonparametric Kruskal Wallis test.
For siRNA knockout experiments, a two-group student t-test was used to compare the differences in cytotoxicity between treated and untreated control cells.
Supplementary Material
TRAIL sensitivity varies amongst human breast cancer cell lines. Cells (1000/well) were plated in 96 well plates and incubated at 37°C for 24 h. Cells were then exposed to varying concentrations of TRAIL for 24 h prior to the determination of ATP levels as a measure of cell viability.
Analysis of DR5 surface expression in a panel of human breast cancer cell lines. Cells were harvested using EDTA and stained with TRA-8 for 1 h at 4 °C followed by Alexa-conjugated goat anti-mouse IgG1, then analyzed by flow cytometry. Black histograms, TRA-8 staining; grey histograms, control mouse IgG1 isotype antibody.
A general caspase inhibitor prevents cytotoxicity due to TRA-8 and combination treatment, but not doxorubicin-induced cytotoxicity. Cells were exposed to Z-VAD-FMK, a general caspase inhibitor (CI), for 2 h prior to the addition of DOX for 24 h; then cells were incubated with TRA-8 for an additional 24 h. Cell viability is expressed as the ATP level as a percentage of the untreated controls. Values are means and SE from a representative experiment with 4 replicates in each assay.
Supplementary Figure 4. Analysis of DR5 surface expression following chemotherapy treatment in human breast cancer cell lines. BT-474 and T47D cells were treated with (A) doxorubicin (5,000 nM or 1,000 nM, respectively) or (B) bortezomib (200 nM or 10 nM, respectively) for 24 h. Cells were harvested using EDTA and stained with TRA-8 for 1 h at 4 °C followed by Alexa-conjugated goat anti-mouse IgG1, then analyzed by flow cytometry. Grey histograms, TRA-8 staining; black histograms, TRA-8 staining in chemotherapy treated cells.
Basal expression of Bcl-2 family members in a panel of breast cancer cell lines. Cells were plated and incubated overnight. Whole cell lysates were analyzed by Western blot using antibodies against the indicated proteins.
Basal expression of IAP family members in a panel of breast cancer cell lines. Cells were plated, incubated overnight, and then whole cell lysates were harvested. Western blot analysis was preformed to detect the designated proteins.
Targeting of the IAP family of proteins sensitizes certain breast cancer cell lines to TRA-8. Cells were exposed to various concentrations of AT-406 for 24 h, then TRA-8 for an additional 24 h. ATP levels were determined relative to untreated control cells and the means and SE of quadruplicate samples from at least 3 independent experiments are shown.
Acknowledgments
We thank Dr. Patsy G. Oliver for technical assistance and helpful discussions, Dr. Elizabeth R. Rayburn for helpful comments on the article, Dr. Albert F. LoBuglio for reviewing the revised manuscript, and Sally Lagan for assistance in preparation of the manuscript.
Supported by DOD grant W81XWH-06-1-0706, DOD grant BCDAMD17-00-1-0119, and NIH grant 2 P50 CA089019.
Footnotes
Disclosure of Potential Conflicts of Interest
Dr. Buchsbaum has intellectual property related to the TRA-8 anti-DR5 antibody.
References
- 1.Guarneri V, Conte P. The curability of breast cancer and the treatment of advanced disease. Eur J Nucl Med Mol Imaging. 2004;31 (Suppl 1):S149–61. doi: 10.1007/s00259-004-1538-5. [DOI] [PubMed] [Google Scholar]
- 2.Bernard-Marty C, Cardoso F, Piccart M. Facts and controversies in systemic treatment of metastatic breast cancer. Oncologist. 2004;9:617–32. doi: 10.1634/theoncologist.9-6-617. [DOI] [PubMed] [Google Scholar]
- 3.Nicolini A, Giardino R, Carpi A, Ferrari P, Anselmi L, Colosimo S, et al. Metastatic breast cancer: an updating. Biomed Pharmacother. 2006;60:548–56. doi: 10.1016/j.biopha.2006.07.086. [DOI] [PubMed] [Google Scholar]
- 4.Giacinti L, Claudio P, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11:1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- 5.Rahman M, Davis S, Pumphrey J, Bao J, Nau M, Meltzer P, et al. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat. 2009;113:217–30. doi: 10.1007/s10549-008-9924-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 7.Pan G, Ni J, Wei Y, Yu G, Gentz R, Dixit V. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 8.Walczak H, Degli-Esposti M, Johnson R, Smolak P, Waugh J, Boiani N, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–97. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao C, Song J, Vilimanovich U, Kneteman N. Modulation of TRAIL signaling complex. Vitam Horm. 2004;67:81–99. doi: 10.1016/S0083-6729(04)67006-3. [DOI] [PubMed] [Google Scholar]
- 10.Ashkenazi A, Herbst R. To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest. 2008;118:1979–90. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashkenazi A, Pai R, Fong S, Leung S, Lawrence D, Marsters S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchsbaum D, Zhou T, Lobuglio A. TRAIL receptor-targeted therapy. Future Oncol. 2006;2:493–508. doi: 10.2217/14796694.2.4.493. [DOI] [PubMed] [Google Scholar]
- 13.Buchsbaum D, Zhou T, Grizzle W, Oliver P, Hammond C, Zhang S, et al. Antitumor efficacy of TRA-8 anti-DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin Cancer Res. 2003;9:3731–41. [PubMed] [Google Scholar]
- 14.Rahman M, Pumphrey J, Lipkowitz S. The TRAIL to targeted therapy of breast cancer. Adv Cancer Res. 2009;103:43–73. doi: 10.1016/S0065-230X(09)03003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shanker A, Brooks AD, Tristan CA, Wine JW, Elliott PJ, Yagita H, et al. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J Natl Cancer Inst. 2008;100:649–62. doi: 10.1093/jnci/djn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks A, Ramirez T, Toh U, Onksen J, Elliott P, Murphy W, et al. The proteasome inhibitor bortezomib (Velcade) sensitizes some human tumor cells to Apo2L/TRAIL-mediated apoptosis. Ann N Y Acad Sci. 2005;1059:160–7. doi: 10.1196/annals.1339.042. [DOI] [PubMed] [Google Scholar]
- 17.Paoluzzi L, Gonen M, Gardner J, Mastrella J, Yang D, Holmlund J, et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111:5350–8. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- 18.Kang M, Reynolds C. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–32. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Wang G, Wang J, Min P, Ni X, Zhang Y, et al. Synergisitc effect of AT-406, a small molecule inhibitor of XIAP, with chemotherapy and tyrosine kinase inhibitors. Proc Am Assoc Cancer Res; 100th Annual Meeting of the American Association for Cancer Research; 2009; Denver, CO. 2009. p. 1917. [Google Scholar]
- 20.Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, et al. SM-406 (AT-406): A potent and orally active IAP antagonist in clinical development. J Med Chem. 2011 doi: 10.1021/jm101505d. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S. Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999;59:734–41. [PubMed] [Google Scholar]
- 22.Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20:205–12. doi: 10.1128/mcb.20.1.205-212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Kakehi Y, Mizutani Y, Nishiyama H, Kamoto T, Megumi Y, et al. Enhancement of TRAIL/Apo2L-mediated apoptosis by adriamycin through inducing DR4 and DR5 in renal cell carcinoma cells. Int J Cancer. 2003;104:409–17. doi: 10.1002/ijc.10948. [DOI] [PubMed] [Google Scholar]
- 24.Baou M, Kohlhaas S, Butterworth M, Vogler M, Dinsdale D, Walewska R, et al. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2010 doi: 10.3324/haematol.2010.022368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulda S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr Cancer Drug Targets. 2008;8:132–40. doi: 10.2174/156800908783769355. [DOI] [PubMed] [Google Scholar]
- 26.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 27.Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–400. [PubMed] [Google Scholar]
- 28.Voortman J, Resende T, Abou El Hassan M, Giaccone G, Kruyt F. TRAIL therapy in non-small cell lung cancer cells: sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Mol Cancer Ther. 2007;6:2103–12. doi: 10.1158/1535-7163.MCT-07-0167. [DOI] [PubMed] [Google Scholar]
- 29.Kim I, Jung Y, Noh D, Song Y, Choi C, Oh B, et al. Functional screening of genes suppressing TRAIL-induced apoptosis: distinct inhibitory activities of Bcl-XL and Bcl-2. Br J Cancer. 2003;88:910–7. doi: 10.1038/sj.bjc.6600795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckelman B, Salvesen G, Scott F. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–94. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogler M, Walczak H, Stadel D, Haas T, Genze F, Jovanovic M, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425–34. doi: 10.1158/0008-5472.CAN-08-2436. [DOI] [PubMed] [Google Scholar]
- 32.Jost P, Grabow S, Gray D, McKenzie M, Nachbur U, Huang D, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–9. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varfolomeev E, Alicke B, Elliott J, Zobel K, West K, Wong H, et al. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J Biol Chem. 2009;284:34553–60. doi: 10.1074/jbc.M109.040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fandy TE, Shankar S, Srivastava RK. Smac/DIABLO enhances the therapeutic potential of chemotherapeutic drugs and irradiation, and sensitizes TRAIL-resistant breast cancer cells. Mol Cancer. 2008;7:60. doi: 10.1186/1476-4598-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foster F, Owens T, Tanianis-Hughes J, Clarke R, Brennan K, Bundred N, et al. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 2009;11:R41. doi: 10.1186/bcr2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun H, Stuckey JA, Nikolovska-Coleska Z, Qin D, Meagher JL, Qiu S, et al. Structure-based design, synthesis, evaluation, and crystallographic studies of conformationally constrained Smac mimetics as inhibitors of the X-linked inhibitor of apoptosis protein (XIAP) J Med Chem. 2008;51:7169–80. doi: 10.1021/jm8006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun H, Lu J, Liu L, Yi H, Qiu S, Yang CY, et al. Nonpeptidic and potent small-molecule inhibitors of cIAP-1/2 and XIAP proteins. J Med Chem. 2010;53:6361–7. doi: 10.1021/jm100487z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohammad RM, Wang S, Aboukameel A, Chen B, Wu X, Chen J, et al. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(−)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther. 2005;4:13–21. [PubMed] [Google Scholar]
- 39.Loberg R, McGregor N, Ying C, Sargent E, Pienta K. In vivo evaluation of AT-101 (R-(−)-gossypol acetic acid) in androgen-independent growth of VCaP prostate cancer cells in combination with surgical castration. Neoplasia. 2007;9:1030–7. doi: 10.1593/neo.07778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng Y, Tang W, Dai Y, Wu X, Liu M, Ji Q, et al. Natural BH3 mimetic (−)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol Cancer Ther. 2008;7:2192–202. doi: 10.1158/1535-7163.MCT-08-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu G, Kelly W, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. 2009;15:3172–6. doi: 10.1158/1078-0432.CCR-08-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ray S, Bucur O, Almasan A. Sensitization of prostate carcinoma cells to Apo2L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis. 2005;10:1411–8. doi: 10.1007/s10495-005-2490-y. [DOI] [PubMed] [Google Scholar]
- 43.Huang S, Sinicrope F. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res. 2008;68:2944–51. doi: 10.1158/0008-5472.CAN-07-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Delft M, Wei A, Mason K, Vandenberg C, Chen L, Czabotar P, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–37. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Wang H, Wang Z, Makhija S, Buchsbaum D, Lobuglio A, et al. Inducible resistance of tumor cells to tumor necrosis factor-related apoptosis-inducing ligand receptor 2-mediated apoptosis by generation of a blockade at the death domain function. Cancer Res. 2006;66:8520–8. doi: 10.1158/0008-5472.CAN-05-4364. [DOI] [PubMed] [Google Scholar]
- 48.Sun M, Song L, Li Y, Zhou T, Jope R. Identification of an antiapoptotic protein complex at death receptors. Cell Death Differ. 2008;15:1887–900. doi: 10.1038/cdd.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enz Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 50.Zhao L, Feng SS. Effects of lipid chain length on molecular interactions between paclitaxel and phospholipid within model biomembranes. J Colloid Interface Sci. 2004;274:55–68. doi: 10.1016/j.jcis.2003.12.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TRAIL sensitivity varies amongst human breast cancer cell lines. Cells (1000/well) were plated in 96 well plates and incubated at 37°C for 24 h. Cells were then exposed to varying concentrations of TRAIL for 24 h prior to the determination of ATP levels as a measure of cell viability.
Analysis of DR5 surface expression in a panel of human breast cancer cell lines. Cells were harvested using EDTA and stained with TRA-8 for 1 h at 4 °C followed by Alexa-conjugated goat anti-mouse IgG1, then analyzed by flow cytometry. Black histograms, TRA-8 staining; grey histograms, control mouse IgG1 isotype antibody.
A general caspase inhibitor prevents cytotoxicity due to TRA-8 and combination treatment, but not doxorubicin-induced cytotoxicity. Cells were exposed to Z-VAD-FMK, a general caspase inhibitor (CI), for 2 h prior to the addition of DOX for 24 h; then cells were incubated with TRA-8 for an additional 24 h. Cell viability is expressed as the ATP level as a percentage of the untreated controls. Values are means and SE from a representative experiment with 4 replicates in each assay.
Supplementary Figure 4. Analysis of DR5 surface expression following chemotherapy treatment in human breast cancer cell lines. BT-474 and T47D cells were treated with (A) doxorubicin (5,000 nM or 1,000 nM, respectively) or (B) bortezomib (200 nM or 10 nM, respectively) for 24 h. Cells were harvested using EDTA and stained with TRA-8 for 1 h at 4 °C followed by Alexa-conjugated goat anti-mouse IgG1, then analyzed by flow cytometry. Grey histograms, TRA-8 staining; black histograms, TRA-8 staining in chemotherapy treated cells.
Basal expression of Bcl-2 family members in a panel of breast cancer cell lines. Cells were plated and incubated overnight. Whole cell lysates were analyzed by Western blot using antibodies against the indicated proteins.
Basal expression of IAP family members in a panel of breast cancer cell lines. Cells were plated, incubated overnight, and then whole cell lysates were harvested. Western blot analysis was preformed to detect the designated proteins.
Targeting of the IAP family of proteins sensitizes certain breast cancer cell lines to TRA-8. Cells were exposed to various concentrations of AT-406 for 24 h, then TRA-8 for an additional 24 h. ATP levels were determined relative to untreated control cells and the means and SE of quadruplicate samples from at least 3 independent experiments are shown.