

Abstract
Multiwalled carbon nanotubes have been entrapped in monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary columns to afford stationary phases with enhanced liquid chromatographic performance for small molecules in the reversed phase. While the column with no nanotubes exhibited an efficiency of only 1800 plates/m, addition of a small amount of nanotubes to the polymerization mixture increased the efficiency to over 15,000 and 35,000 plates/m at flow rates of 1 and 0.15 μL/min, respectively. Alternatively, the native glycidyl methacrylate-based monolith was functionalized with ammonia and, then, shortened carbon nanotubes, bearing carboxyl functionalities, were attached to the pore surface through the aid of electrostatic interactions with the amine functionalities. Reducing the pore size of the monolith enhanced the column efficiency for the retained analyte, benzene, to 30,000 plates/m at a flow rate of 0.25 μL/min. Addition of tetrahydrofuran to the typical aqueous acetonitrile eluents improved the peak shape and increased the column efficiency to 44,000 plates/m calculated for the retained benzene peak.
Keywords: Porous polymer monolith, Carbon nanotubes, Poly(glycidyl methacrylate-co-ethylene dimethacrylate), Reversed phase chromatography, Small molecules
1. Introduction
Since the inception of rigid organic polymer monolithic columns in the early 1990s [1], their use as chromatographic separation media has continued to grow. The popularity of monolithic columns is fueled by their high permeability, which enables excellent performance in the fast separation of large molecules such as peptides, proteins, nucleic acids, and synthetic polymers at high flow velocities using gradient elution [2–13]. The high speed achieved in these separations results from the rapid convective mass transport in the large through pores — the only pores that are present in the un-modified monoliths. In this instance, the lack of small pores in the monolithic structure avoids the normally slow diffusional mass transport. However, this comes at the cost of surface area, thus making the monoliths unsuitable for the separation of small molecules in an isocratic mode due to the absence of the numerous interaction sites required for sufficient sample loading capacity. In our initial experiments, we found that a poor efficiency of only 18,000 plates/m for benzene could be achieved with the first generation of monolithic poly(styrene-co-divinylbenzene) columns [3]. Since the original development of rigid monoliths, several groups have attempted to increase the column efficiency for small molecules. For example, a recent optimization of the polymerization conditions for methacrylate-based monoliths has afforded capillary columns with 35,000–50,000 plates/m for retained benzene [14–18]. Other groups have sought alternative processes such as the polymerization of a single crosslinker [19–22], the termination of the polymerization reaction at an early stage [23–25], and the use of polymerizations at high temperature [26–28]. We have recently introduced a new modification reaction, hypercrosslinking, which enables a significant increase in the efficiency of monolithic columns [29]. While this reaction works well with monoliths prepared form styrene, chloromethylstyrene, and divinylbenzene, it is not readily applied to methacrylate-based monolithic columns. Even as all these methods have led to porous polymer monolithic columns with efficiencies exceeding those of our early columns [3], the preparation of highly efficient polymer-based monolithic columns for the isocratic separation of small molecules that perform as well as their silica-based monolithic counterparts [30, 31] remains a challenge.
Due to unique characteristics of nanoparticles, such as their large surface-to-volume ratio and their properties that differ from those of corresponding bulk materials, the use of nanomaterials in separation science is growing rapidly [32–34]. For example, nanostructures, such as polymer latex nanoparticles, fullerene derivatives, metal oxides, and carbon nanotubes have been used for the modifications of separation media for application in gas and liquid chromatography, capillary electrophoresis, and electrochromatography [32–43]. In the field of polymer monoliths, methacrylate columns with attached functionalized polymer nanoparticles were introduced first and these columns were used for the separation of saccharides [44] and in ion chromatography [45–48]. Monoliths with pores coated with gold nanoparticles have recently been prepared [49–51] and used for the pre-concentration of thiol containing peptides and the separation of proteins [49, 50], while monoliths with embedded hydroxyapatite nano-needles proved useful in the extraction of phosphorylated peptides from complex protein digests [52]. Li et al. entrapped carbon nanotubes into a poly(chloromethylstyrene-co-ethylene dimethacrylate) monolith to afford capillary columns for HPLC and capillary electrochromatography [53].
Thus, nanostructures hold a great potential for achieving efficient separations of small molecules. This article demonstrates the use carbon nanotubes entrapped within or attached to the pore surface of poly(glycidyl methacrylate-co-ethylene dimethacrylate) monoliths in order to improve the performance of the monolithic capillary columns in the isocratic separation of small molecules.
2. Experimental part
2.1. Materials
Glycidyl methacrylate (GMA), ethylene dimethacrylate (EDMA), cyclohexanol, 1-dodecanol, azobisisobutyronitrile (AIBN), 3-(trimethoxysilyl)propyl methacrylate, nitric acid, hydrochloric acid, sodium hydroxide, isopropanol, uracil, benzene, toluene, ethylbenzene, propylbezene, butylbenzene, pentylbenzene, pyrenecarboxylic acid, didodecyldimethyl ammonium bromide, sodium dodecylsulfate, and HPLC grade solvents acetonitrile, methanol, and acetone were obtained from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade tetrahydrofuran and concentrated sulfuric acid were obtained from EMD Chemicals (Gibbstown, NJ, USA). All chemicals were used as received with the exception of EDMA and GMA, which were first purified by passing them through activated alumina (activated, basic, Brockman I, 150 mesh). Water was purified by a Nanopure Water System (Barnstead, Chicago, IL, USA) and filtered through 0.20 μm nylon membrane filters (Millipore, Bedford, MA, USA) prior to use.
Polyimide-coated coated fused silica capillaries (365 μm o.d. × 100 μm i.d.) were purchased from Polymicro Technologies (Phoenix, AZ, USA). Multiwalled carbon nanotubes (5–10 nm i.d., 10–30 nm o.d., 1–2 μm length, batch SN2303) were purchased from Sun Innovations Inc. (Fremont, CA, USA).
2.2. Instrumentation
A nanoAcquity UPLC system (Waters, Milford, MA, USA) consisting of a binary solvent pump, sample manager, autosampler, and TUV detector equipped with a 10 nL cell was used for the separations. An external 10 nL injector with an electric actuator (CN4, Vici Valco Instruments, Houston, TX, USA) was used for sample injections.
IR spectra were acquired using a Spectrum One IR (Perkin Elmer, Waltham, MA, USA) with a horizontal attenuated total reflectance (HATR) assembly. Ten scans were typically carried out with a film prepared from 2 mg/mL dispersion of MWNT in isopropanol. Thermogravimetry-mass spectrometry (TGA-MS) analyses were performed in aluminum pans using a Q5000IR (TA Instruments, New Castle, DE, USA) equipped with a Thermostar Mass Spectrometer (Pfeiffer Vacuum, Nashua, NH, USA). Nitrogen adsorption/desorption isotherms were measured using a Micromeritics ASAP 2010 surface area and porosimetry analyzer (Norcross, GA, USA) and used for the calculation of the surface areas. An ICnano system (Ionscope, Melbourn, UK) was used to obtain scanning ion conductance images. A Gemini Ultra Field-Emission Scanning Electron Microscope (SEM, Zeiss, Peabody, MA, USA) was used for all SEM imaging.
2.3. Preparation of monolithic capillary columns
The capillary was rinsed with acetone and water, flushed with 0.2 mol/L sodium hydroxide for 30 min at a flow rate of 0.25 μL/min using a syringe pump, and then rinsed with water. Next, 0.2 mol/L hydrochloric acid was pumped through the capillary for 30 min at a flow rate of 0.25 μL/min, followed by water and ethanol. A 20% w/w solution of 3-(trimethoxysilyl)propyl methacrylate in 95% ethanol with an apparent pH adjusted to 5 using acetic acid, was pumped through the capillary at a flow rate of 0.25 μL/min for 1 h. The capillary was then washed with acetone, dried in a stream of nitrogen, and left at room temperature overnight before use.
Standard monoliths were prepared using the procedure and the conditions we developed previously [54]. In brief, a polymerization mixture comprising glycidyl methacrylate (24 wt%), ethylene dimethacrylate (16 wt%), and cyclohexanol (54 wt%) with 1-dodecanol (6 wt%) as porogens and azobisisobutyronitrile (1% w/w with respect to monomers) as the initiator was purged with nitrogen for 5 min. The solution was then sonicated for another 10 min to remove oxygen and introduced into the vinylized capillaries. Both ends of the capillary were sealed and the capillary was placed in a thermostated water bath. Following polymerization at 55 or 70 °C for 24 h, a few centimeters were cut from both ends of the capillary, the monolithic column was flushed with acetonitrile, and used for separations.
2.4. Entrapment of nanotubes in monolith
A specific amount of pristine MWNT was dispersed in the polymerization mixture comprised of glycidyl methacrylate (24 wt%), ethylene dimethacrylate (16 wt%), and cyclohexanol (54 wt%) with 1-dodecanol (6 wt%) and azobisisobutyronitrile (1% w/w with respect to monomers). This dispersion was then filled in capillaries and polymerized as described in section 2.3.
2.5. Oxidative cutting of carbon nanotubes
Cutting of the MWNT was performed using an established oxidative cutting procedure [55, 56]. In brief, 560 mg of pristine MWNT were mixed with 84 mL of 3:1 sulfuric acid/nitric acid and sonicated in a water bath for 24 h at 35 °C. Then, 500 mL of water were added slowly to the mixture and the suspension was centrifuged. The supernatant was removed and the shortened MWNT were repeatedly re-suspended in water and centrifuged until the supernatant remained clear, colorless, and pH neutral. This washing process was then repeated with acetone, tetrahydrofuran, and methanol. The resulting oxidized carbon nanotubes had an average length of 130±30 nm as determined by scanning electron microscopy.
2.6. Attachment of carbon nanotubes to pore surface
Ammonium hydroxide was pumped through a GMA-EDMA monolith until the effluent was basic. The capillary was capped at both ends and held at room temperature for 75 min to afford a monolith with primary amine functionalities. The column was then flushed with water until the effluent had a neutral pH. This monolith contains 0.9 atomic % of nitrogen as determined by X-ray photoelectron spectroscopy.
An aqueous dispersion 1 mg/mL of the shortened MWNT was pumped through the monolith at a flow rate of 0.25 μL/min until an initial breakthrough was observed. The columns were then flushed for 10 min with the mobile phase before being attached to the chromatographic system and used.
3. Results and discussion
3.1. Entrapment of carbon nanotubes in methacrylate monoliths
In order to better monitor any changes in the reversed phase chromatographic performance of monoliths after incorporation of the carbon nanotubes, we prepared the monoliths from two relatively polar monomers: glycidyl methacrylate and ethylene dimethacrylate. Glycidyl methacrylate is particularly convenient as it provides the reactive functionalities that can be used to modify the pore surface chemistry of the monoliths as needed.
Our first approach to the incorporation of multiwall carbon nanotubes in the monolith involved their direct addition to the polymerization mixture, followed by polymerization. However, due to the hydrophobic nature of MWNT, aggregation and sedimentation of the pristine nanotubes is observed when they are added to mixtures of the neat monomers without porogenic solvents. Addition of the porogenic solvents, 1-dodecanol and cyclohexanol, to the monomers and MWNT followed by mixing leads to a homogeneous black mixture as the MWNT remain dispersed for several days. This demonstrates the surfactant-like properties of the porogens, enabling homogeneous dispersion of the MWNT in the polymerization mixture prior to its polymerization.
Larger scale batches of a standard GMA-EDMA monolith and its counterpart containing 0.1 wt% MWNT were first prepared in vials and used for measurement of the surface areas. The white-colored GMA-EDMA monoliths prepared at 55 and 70 °C exhibit surface areas of 38 and 42 m2/g, respectively. Addition of MWNT to the polymerization mixtures affords grey colored monoliths, with no appreciable change in surface areas given the very small amount of added nanotubes. This finding is corroborated with a negligible change in the backpressure of the monolithic capillary columns that remains within ±5% at all tested flow rates.
While polymerization temperature is well known to control the pore size of monolithic columns [54, 57], it was observed that it also affects the entrapment of MWNT in the polymer matrix. MWNT form larger aggregates that partly segregate from the monolith during the polymerization reaction carried out at a temperature of 55 °C as a result of incompatibility with the methacrylate polymer. These partly embedded tubes then protrude from the surface of the monolith. Fig. 1 shows a scanning ion conductance micrograph of the internal structure of a monolith that clearly shows nanotubes crossing the through pores. By increasing the polymerization temperature to 70 °C, the polymerization proceeds faster. Since the segregation of the MWNT from the polymerizing mixture is a slow process which is controlled by the length of the tubes, the MWNT do not have enough time to phase separate and remain mostly contained within the polymer matrix without a significant effect on the surface chemistry of the monolith. Therefore, all further polymerizations were carried out at a temperature of 55 °C. A complete dispersion of MWNT is only observed up to 0.25 wt% MWNT (with respect to monomers). At higher MWNT content, the MWNT do not fully disperse and the monoliths tend to crack with the formation of voids.
Fig. 1.

Scanning ion conductance microscopy image of MWNT bridging the through pore of a poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith containing entrapped nanotubes.
The parent GMA-EMDA monolithic column prepared at a temperature of 55 °C affords only 1,800 plates/m for benzene. Entrapment of pristine MWNT into a monolith results in an increase in both efficiency and resolution. A column efficiency of 15,400 plates/m determined at a flow rate of 1 μL/min is achieved at a content of 0.25 wt% MWNT (with respect to the monomers) as demonstrated by the separation of alkylbenzenes shown in Fig. 2. However, at the minimum of the van Deemter curve, represented with a flow rate of 0.15 μL/min, a column containing 0.25% MWNT exhibits a decent efficiency of 35,000 plates/m for benzene, a significant increase compared to the bare column. This maximizing of efficiency increases the time required for the separation of all six alkylbenzenes to greater than 1 hour. Fig. 3 shows that increasing the percentage of organic solvent in the mobile phase to 55%, significantly reduces the separation time. However, this decrease in retention time is accompanied by a decrease in resolution. These results are similar to those observed by Li et al. for a monolithic column containing entrapped oxidized MWNT [53]. Li et al. also found a significant increase in the retention of aromatic analytes, which was attributed to the specific structure, size, and charge characteristics of the nanotubes. However, no mention was made of any change in column efficiency.
Fig. 2.

Separation of uracil and alkylbenzenes using a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary column (a) and its counterpart containing 0.25 wt% entrapped MWNT (with respect to the monomers) (b), both prepared at a temperature of 55 °C. Conditions: Column, 180 mm × 100 μm i.d., mobile phase 45% acetonitrile-5% THF-50% water, flow rate 1.00 μL/min, back pressure 16 MPa, UV detection at 254 nm; Peaks: uracil (1), benzene (2), toluene (3), ethylbenzene (4), propylbenzene (5), butylbenzene (6), and amylbenzene (7).
Fig. 3.

Separation of uracil and alkylbenzenes using a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary column with entrapped 0.25 wt% MWNT (with respect to the monomers) prepared at 55 °C. Conditions: Column, 180 mm × 100 μm i.d., mobile phase 55% acetonitrile-5% THF-40% water, flow rate 1.00 μL/min, back pressure 17 MPa, UV detection at 254 nm; For peaks assignment see Fig. 2.
3.2. Surface attachment of the MWNT
An alternative approach to the incorporation of MWNT into the monolithic columns involves their direct attachment to the pore surface. Initial attempts to modify the GMA-EDMA monolith surface with MWNT involved the circulation of dispersions of pristine MWNT through the monolith. However, as a result of their hydrophobic nature, the native MWNT immediately aggregate and sediment when immersed in aqueous or polar solvents, even at very low concentrations of less than 0.1 mg/mL. As surfactants are known to facilitate the dispersion of carbon nanotubes in water [58, 59], several were tested including didodecyldimethylammonium bromide, sodium dodecylsulfate, and pyrene carboxylic acid (PCA). Only PCA enabled the dispersion of up to 2 mg of MWNT per mL. However when these dispersions were used, the columns became plugged within a few seconds at flow rates of 0.1–1.0 μL/min and with any MWNT concentration in the range of 0.2–2.0 mg/mL. Clearly, the length and morphology of the native 1–2 μm MWNT (Fig. 4) prevents the MWNT from perfusing through the tortuous pores of the monoliths, which have pore sizes not exceeding 1.9 μm.
Fig. 4.

SEM image of pristine 1–2 μm long MWNT aggregated after exposure to water (top) and SEM micrograph of oxidatively cut MWNT.
Shorter carbon nanotube fragments can be obtained by oxidizing the nanotubes in a mixture of sulfuric and nitric acids, a process that introduces functionalities such as carboxylic acids at the tips of the oxidized nanotube fragments [60, 61]. Using such an oxidation procedure Samori et al. found that the loading of carboxyl groups onto the oxidized MWNT was 1.7–2.0 mmol/g [56]. The SEM image shown in Fig. 4 shows that after 24 h of oxidization the length of the cut MWNT is only 130±30 nm. While thermogravimetric analysis (TGA) of the pristine MWNT shows a weight loss of less than 2% at 600 °C, the loss after 24 h of oxidation amounts to 10% (Fig. 5). Part of this loss (ca. 2.5%) is attributed to water absorbed from the air, confirming the highly hygroscopic nature of the oxidized tubes. Monitoring the mass spectrum of the gaseous products released during the thermogravimetric analysis shows peaks characteristic of carbon dioxide (m/z = 44) and methyl ions (m/z = 15), accounting for over 70% of the species released. This finding supports the presence of carboxylic acid groups at the surface of the oxidized MWNT. Similarly, while the IR spectrum of the original MWNT shown in Fig. 6 is almost featureless in the range of 1500–1720 cm−1, the IR of the oxidized MWNT exhibits strong adsorptions at 1583, 1710, and 3000 cm−1, indicating the presence of carboxylic acid groups. In contrast, the IR spectrum of the oxidized MWNT, heated to 600 °C during the TGA analysis, no longer includes the characteristic carboxyl bands, as a result of a thermally induced decarboxylation process.
Fig. 5.
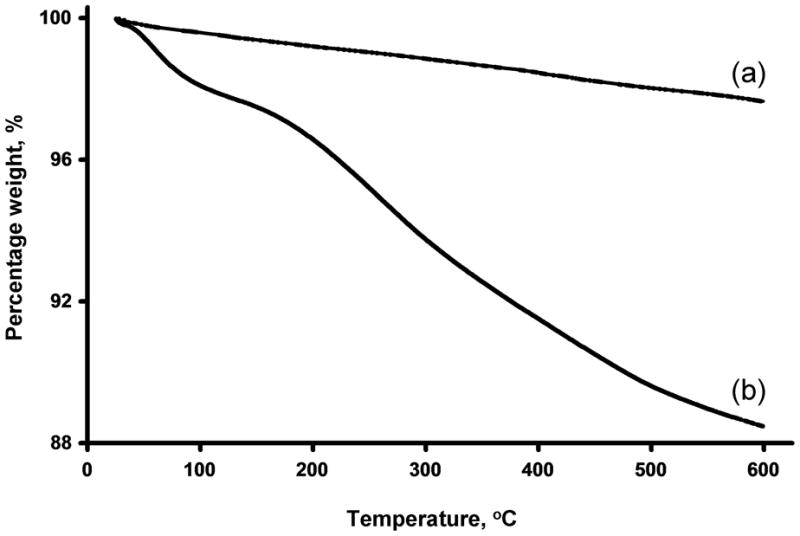
Thermogravimetric analysis of pristine MWNT (a) and their oxidatively cut counterparts (b) using a heating rate of 20 °C/min.
Fig. 6.

FT-IR spectra of prisine MWNT (a), MWNT after oxidative cutting (b), and after heating the oxidized MWNT to 600 °C.
The short oxidized carbon nanotubes disperse readily in aqueous solvents and can be easily passed through the pores of the monoliths. Therefore, pumping a 1 mg/mL solution of oxidized MWNT through the poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith results in both column darkening and in a 10% increase in back pressure. These observations suggest that some tubes remain retained in the pores of the monolith, most likely through a combination of partial entanglement and hydrophobic interactions, though reaction of the carboxylic acid moieties of the nanotubes with the epoxy groups present on the polymer monolith is also possible. However, upon flushing with acetonitrile, the column returns to its original white color as oxidized nanotubes are eluted and the back pressure returns to its original value. Therefore, no reaction between carboxylic acid and epoxy moieties seems to have taken place and relying on hydrophobic interactions for retention of the tubes in the column is not sufficient. Thus, it is necessary to explore a different mechanism for attachment of the nanotubes to the monolith.
The epoxy groups of the poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith readily react with ammonia to afford primary amine functionalities [62]. While this treatment renders the monolith useless for separations in the reversed phase mode, as demonstrated by the lack of separation ability shown in Fig. 7, it provides a means to retain the oxidized nanotubes within the monolith through interactions between their carboxylic acid moieties and the surface amino groups of the modified monolith. The oxidized nanotubes are now retained within the monolith and do not elute even when flushed with pure acetonitrile. The electrostatic nature of this interaction is readily confirmed as a subsequent treatment of the monolith containing oxidized MWNT with 0.15 mol/L hydrochloric acid releases again all of the MWNT from the column.
Fig. 7.

Separation of uracil and alkylbenzenes using a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary column prepared at 55 °C and modified with ammonia. Conditions: Column, 200 mm × 100 μm i.d., mobile phase 50% acetonitrile-50% water, flow rate 1.00 μL/min, back pressure 17 MPa, UV detection at 254 nm; For peaks assignment see Fig. 2.
Fig. 8 illustrates the separation of six alkylbenzenes using a monolithic amine-modified column incorporating immobilized oxidized MWNT. Although these oxidized MWNT are more hydrophilic than their pristine counterparts are, they still provide sufficient hydrophobicity to the column, thus enabling separation. In contrast to the poor performance of the column treated with ammonia, the efficiency of the column containing oxidized MWNT increases to 23,000 plates/m for benzene at 0.25 μL/min and its selectivity is increased. This experiment confirms that the improvements in both selectivity and retention both result from the incorporation of oxidized MWNT. The mechanism responsible for these effects is not completely known. Previous literature describing experiments with oxidized MWNT deposited within porous silica beads suggests that a high affinity of the immobilized nanotubes for aromatic compounds is the cause [63], however, a reduction in the column efficiency was also noted. Andre et al. compared carbon nanotubes to a graphite sheet (sp2 carbon) rolled in a tube [42]. The π–π interaction at the large contact area are thought to be responsible for high retention of benzene derivatives. Since the oxidation modifies only the tips of the nanotubes, all the area along the length the tube remains available for the interactions.
Fig. 8.

Separation of uracil and alkylbenzenes using a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary column prepared at 55 °C and modified with ammonia followed by attachment of oxidized MWNT. Conditions: Column, 160 mm × 100 μm i.d., mobile phase 45% acetonitrile-55% water, flow rate 0.25 μL/min, back pressure 8 MPa, UV detection at 254 nm; For peaks assignment see Fig. 2.
A reduction in the pore size of the parent monolith to 0.47 μm, using a polymerization temperature of 70 °C, followed by amination and attachment of oxidized MWNT, afforded a column with an increased efficiency of 30,000 plates/m. Most likely, this results from the enhanced surface area of the column, which accommodates more nanotubes for improved chromatographic performance. Although this simple approach of reducing pore size improved the efficiency by 24%, this was accompanied by a 3.8 fold increase in back pressure, which in turn limits the applicable flow rates to 0.25 μL/min or less in our chromatographic system.
By decreasing the pore size of the monolith, tailing of the peaks became more prevalent, showing an asymmetry of at least 1.8 for the alkylbenzenes. Reduction in the analytes concentration in the injected sample below 5 μL/mL did not lead to an improvement of the peak tailing. Adding tetrahydrofuran (THF) to the mobile phase has been shown to reduce peak tailing on polystyrene-based columns [64, 65] leading to the expectation that addition of some THF would reduce tailing since the carbon nanotubes are also comprised of a multiplicity of aromatic rings. As shown in Fig. 9, addition of up to 5% THF reduces the tailing. For example, a mobile phase containing 2.5% of THF reduces the tailing factor to less than 1.3 and affords a good column efficiency of 44,000 plates/m for benzene. The retention times of all alkylbenzenes are stable with a RSD < 1.6%, even after passing more than 6,000 column volumes through the column. This confirms that there is no leaching of the MWNT from the monolith.
Fig. 9.

Separation of uracil and alkylbenzenes using a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) capillary column prepared at 70 °C and modified with ammonia followed by attachment of oxidized MWNT. Conditions: Column, 170 mm × 100 μm i.d., mobile phase 50% acetonitrile-50% water (A) and a 47.5% acetonitrile-2.5% THF-50% water-mixture (B); flow rate 0.25 μL/min, back pressure 30 MPa; UV detection at 254 nm; For peaks assignment see Fig. 2.
4. Conclusions
This study demonstrates that the attachment of a very small amount of MWNT onto the pore surface or MWNT entrapment into poly(glycidyl methacrylate-co-ethylene dimethacrylate) monoliths, significantly increases both retention and column efficiency of the capillary columns. Optimization of the porous structure of the monolith, MWNT attachment, and the mobile phase, produced monolithic capillary columns exhibiting an efficiency of 44,000 plates/m for retained benzene. This efficiency is significantly higher than that achieved by other groups via optimization of the polymerization conditions [14, 15]. Our results clearly demonstrate the ability of carbon nanostructures to significantly affect the separation performance of monolithic columns. Our current experiments aim at determining a mechanism that would explain a substantial effect on monolithic column efficiency with a very small amount of nanostructures incorporated. We are also expanding the repertoire of nanostructures incorporated into the polymer to modify both efficiency and selectivity of the monolithic capillary columns.
Acknowledgments
Financial support of S.D.C. and J.M.J.F by a grant of the National Institute of Health (GM48364) is gratefully acknowledged. All experimental and characterization work performed at the Molecular Foundry, Lawrence Berkeley National Laboratory and F.S. were supported by the Office of Science, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract No. DE-AC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Svec F, Fréchet JMJ. Anal Chem. 1992;54:820. [Google Scholar]
- 2.Wang Q, Svec F, Fréchet JMJ. Anal Chem. 1993;65:2243. doi: 10.1021/ac00065a013. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Svec F, Fréchet JMJ. J Chromatogr A. 1994;669:230. doi: 10.1016/0021-9673(94)80352-8. [DOI] [PubMed] [Google Scholar]
- 4.Petro M, Svec F, Gitsov I, Fréchet JMJ. Anal Chem. 1996;68:315. doi: 10.1021/ac950726r. [DOI] [PubMed] [Google Scholar]
- 5.Petro M, Svec F, Fréchet JMJ. J Chromatogr A. 1996;752:59. doi: 10.1016/s0021-9673(96)00510-9. [DOI] [PubMed] [Google Scholar]
- 6.Fréchet JMJ, Svec F, Petro M. Abstracts of Papers of the American Chemical Society. 1997;214:217-MSE. [Google Scholar]
- 7.Gusev I, Huang X, Horváth C. J Chromatogr. 1999;855:273. doi: 10.1016/s0021-9673(99)00697-4. [DOI] [PubMed] [Google Scholar]
- 8.Premstaller A, Oberacher H, Huber CG. Anal Chem. 2000;72:4386. doi: 10.1021/ac000283d. [DOI] [PubMed] [Google Scholar]
- 9.Eder K, Reichel E, Schottenberger H, Huber CG, Buchmeiser MR. Macromolecules. 2001;34:4334. [Google Scholar]
- 10.Gu B, Chen Z, Thulin CD, Lee ML. Anal Chem. 2006;78:3509. doi: 10.1021/ac060284r. [DOI] [PubMed] [Google Scholar]
- 11.Trojer L, Lubbad SH, Bisjak CP, Wieder W, Bonn GK. J Chromatogr A. 2007;1146:216. doi: 10.1016/j.chroma.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Levkin PA, Eeltink S, Stratton TR, Brennen R, Robotti K, Yin H, Killeen K, Svec F, Fréchet JMJ. J Chromatogr A. 2008;1200:55. doi: 10.1016/j.chroma.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Tolley HD, Lee ML. Anal Chem. 2009;81:9416. doi: 10.1021/ac9020038. [DOI] [PubMed] [Google Scholar]
- 14.Coufal P, Čihák M, Suchánková J, Tesařová E, Bosáková Z, Štulík K. J Chromatogr A. 2002;946:99. doi: 10.1016/s0021-9673(01)01570-9. [DOI] [PubMed] [Google Scholar]
- 15.Moravcová D, Jandera P, Urban J, Planeta J. J Sep Sci. 2003;26:1005. [Google Scholar]
- 16.Moravcová D, Jandera P, Urban J, Planeta J. J Sep Sci. 2004;27:789. doi: 10.1002/jssc.200401778. [DOI] [PubMed] [Google Scholar]
- 17.Huo Y, Schoenmakers PJ, Kok WT. J Chromatogr A. 2007;1175:81. doi: 10.1016/j.chroma.2007.10.048. [DOI] [PubMed] [Google Scholar]
- 18.Aoki H, Kubo T, Ikegami T, Tanaka N, Hosoya K, Tokuda D, Ishizuka N. J Chromatogr A. 2006;1119:66. doi: 10.1016/j.chroma.2006.01.133. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Yang L, Wang Q. J Chromatogr A. 2009;1216:3098. doi: 10.1016/j.chroma.2009.01.089. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Tolley DH, Lee ML. J Chromatogr A. 2010;1217:4934. doi: 10.1016/j.chroma.2010.05.048. [DOI] [PubMed] [Google Scholar]
- 21.Lubbad SH, Buchmeiser MR. J Sep Sci. 2009;32:2521. doi: 10.1002/jssc.200900188. [DOI] [PubMed] [Google Scholar]
- 22.Lubbad SH, Buchmeiser MR. J Chromatogr A. 2010;1217:3223. doi: 10.1016/j.chroma.2009.10.090. [DOI] [PubMed] [Google Scholar]
- 23.Trojer L, Bisjak CP, Wieder W, Bonn GK. J Chromatogr A. 2009;1216:6303. doi: 10.1016/j.chroma.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Greiderer A, Trojer L, Huck CW, Bonn GK. J Chromatogr A. 2009;1216:7747. doi: 10.1016/j.chroma.2009.08.084. [DOI] [PubMed] [Google Scholar]
- 25.Nischang I, Teasdale I, Bruggemann O. J Chromatogr A. 2010;1217:7514. doi: 10.1016/j.chroma.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 26.Peters EC, Svec F, Fréchet JMJ, Viklund C, Irgum K. Macromolecules. 1999;32:6377. [Google Scholar]
- 27.Viklund C, Irgum K, Svec F, Fréchet JMJ. Macromolecules. 2001;34:4361. [Google Scholar]
- 28.Meyer U, Svec F, Fréchet JMJ, Hawker CJ, Irgum K. Macromolecules. 2000;33:7769. [Google Scholar]
- 29.Urban J, Svec F, Fréchet JMJ. Anal Chem. 2010;82:1621. doi: 10.1021/ac100008n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minakuchi H, Nakanishi K, Soga N, Ishizuka N, Tanaka N. Anal Chem. 1996;68:3498. doi: 10.1021/ac960281m. [DOI] [PubMed] [Google Scholar]
- 31.Guiochon G. J Chromatogr A. 2007;1168:101. [Google Scholar]
- 32.Zhang ZX, Wang ZY, Liao YP, Liu HW. J Sep Sci. 2006;29:1872. doi: 10.1002/jssc.200600154. [DOI] [PubMed] [Google Scholar]
- 33.Nilsson C, Nilsson S. Electrophoresis. 2006;27:76. doi: 10.1002/elps.200500535. [DOI] [PubMed] [Google Scholar]
- 34.Nilsson C, Birnbaum S, Nilsson S. J Chromatogr A. 2007;1168:212. doi: 10.1016/j.chroma.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 35.Guihen E, Glennon JD. Anal Lett. 2003;36:3309. [Google Scholar]
- 36.Zhang ZX, Yan B, Liao YP, Liu HW. Anal Bioanal Chem. 2008;391:925. doi: 10.1007/s00216-008-1930-2. [DOI] [PubMed] [Google Scholar]
- 37.Qu QS, Shen F, Shen M, Hu XY, Yang GJ, Wang CY, Yan C, Zhang YK. Anal Chim Acta. 2008;609:76. doi: 10.1016/j.aca.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 38.Tran CD, Challa S. Analyst. 2008;133:455. doi: 10.1039/b716443b. [DOI] [PubMed] [Google Scholar]
- 39.Liu FK, Hsu YT, Wu CH. J Chromatogr A. 2005;1083:205. doi: 10.1016/j.chroma.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 40.Jimenez-Soto JM, Moliner-Martinez Y, Cardenas S, Valcarcel M. Electrophoresis. 2010;31:1681. doi: 10.1002/elps.200900628. [DOI] [PubMed] [Google Scholar]
- 41.Qu QS, Zhang XX, Zhao ZZ, Hu XY, Yan C. J Chromatogr A. 2008;1198–1199:95. doi: 10.1016/j.chroma.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 42.Andre C, Gharbi T, Guillaume YC. J Sep Sci. 2009;32:1757. doi: 10.1002/jssc.200800683. [DOI] [PubMed] [Google Scholar]
- 43.Zhong Y, Zhou W, Zhang P, Zhu Y. Talanta. 2010;82:1439. doi: 10.1016/j.talanta.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 44.Hilder EF, Svec F, Fréchet JMJ. J Chromatogr A. 2004;1053:101. [PubMed] [Google Scholar]
- 45.Hutchinson JP, Zakaria P, Bowie AR, Macka M, Avdalovic N, Haddad PR. Anal Chem. 2005;77:407. doi: 10.1021/ac048748d. [DOI] [PubMed] [Google Scholar]
- 46.Zakaria P, Hutchinson JP, Avdalovic N, Liu Y, Haddad PR. Anal Chem. 2005;77:417. doi: 10.1021/ac048747l. [DOI] [PubMed] [Google Scholar]
- 47.Glenn KM, Lucy CA, Haddad PR. J Chromatogr A. 2007;1155:8. doi: 10.1016/j.chroma.2007.01.098. [DOI] [PubMed] [Google Scholar]
- 48.Haddad PR, Hilder EF, Evenhuis C, Schaller D, Pohl C, Flook KJ. Abstracts of Papers of the American Chemical Society. 2009:236. [Google Scholar]
- 49.Xu Y, Cao Q, Svec F, Fréchet JMJ. Anal Chem. 2010;82:3352. doi: 10.1021/ac1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao Q, Xu Y, Liu F, Svec F, Fréchet JMJ. Anal Chem. 2010;82:7416. doi: 10.1021/ac1015613. [DOI] [PubMed] [Google Scholar]
- 51.Connolly D, Twamley B, Paull B. Chem Commun. 2010;46:2109. doi: 10.1039/b924152c. [DOI] [PubMed] [Google Scholar]
- 52.Krenkova J, Lacher NA, Svec F. Anal Chem. 2010;82 doi: 10.1021/ac1018815. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Chen Y, Xiang R, Ciuparu D, Pfefferle LD, Horvath C, Wilkins JA. Anal Chem. 2005;77:1398. doi: 10.1021/ac048299h. [DOI] [PubMed] [Google Scholar]
- 54.Viklund C, Svec F, Fréchet JMJ, Irgum K. Chem Mater. 1996;8:744. [Google Scholar]
- 55.Liu J, Rinzler AG, Dai H, Hafner JH, Bradley RK, Boul PJ, Lu A, Iverson T, Shelimov K, Huffman CB, Rodriguez-Macias F, Shon YS, Lee TR, Colbert DT, Smalley RE. Science. 1998;280:1253. doi: 10.1126/science.280.5367.1253. [DOI] [PubMed] [Google Scholar]
- 56.Samori C, Sainz R, Menard-Moyon C, Toma FM, Venturelli E, Singh P, Ballestri M, Prato M, Bianco A. Carbon. 2010;48:2447. [Google Scholar]
- 57.Svec F, Fréchet JMJ. Macromolecules. 1995;28:7580. [Google Scholar]
- 58.Moore VC, Strano MS, Haroz EH, Hauge RH, Smalley RE, Schmidt J, Talmon Y. Nano Lett. 2003;3:1379. [Google Scholar]
- 59.Simmons TJ, Bult J, Hashim DP, Linhardt RJ, Ajayan PM. ACS Nano. 2009;3:1865. doi: 10.1021/nn800860m. [DOI] [PubMed] [Google Scholar]
- 60.Liu P, Wang T. Appl Phys A, Mater Sci Process. 2009;97:771. [Google Scholar]
- 61.Forrest GA, Alexander AJ. J Phys Chem C. 2007;111:10792. [Google Scholar]
- 62.Svec F, Hrudková H, Horák D, Kálal J. Angew Macromol Chem. 1977;63:37. [Google Scholar]
- 63.Menna E, la Negra F, Prato M, Tagmatarchis N, Ciogli A, Gasparrini F, Misiti D, Villani C. Carbon. 2006;44:1609. [Google Scholar]
- 64.Ells B, Wang Y, Cantwell FF. J Chromatogr. 1999;835:3. [Google Scholar]
- 65.Bowers LD, Pedigo S. J Chromatogr A. 1986;371:243. [Google Scholar]