

Proteins are subject to numerous post-translational modifications (PTMs) that can alter the chemical structure, and hence function, of the molecule.[1] The astonishing diversity of PTMs possible on proteins is exemplified by histones, nuclear proteins that form the protein core of the nucleosome particle.[2] Histones can be modified in a variety of ways including acetylation, phosphorylation, methylation, ADP-ribosylation and ubiquitylation.[3] Moreover, many, if not all, of these modifications can occur in combination.[4, 5] Indeed, there is growing evidence that functional cross-talk between histone PTMs is essential for the regulation of gene expression[3] and ultimately cell fate and identity.[6] Biochemical studies into the role of histone PTMs are often confounded by the difficulty associated with obtaining large quantities of homogeneously modified proteins. For this reason chemical approaches to obtaining post-translationally modified histones have received considerable attention in recent years.[7–9] Among the available strategies, the protein ligation approach, expressed protein ligation (EPL), offers the most flexibility in terms of the number and type of PTMs that can be incorporated.[10] To date, EPL has been used to generate phosphorylated,[8] acetylated, and methylated forms of histone H3,[7] acetylated H4,[11] and ubiquitylated H2B.[12] Nonetheless, many modified histones have yet to be accessed using semi-synthesis. A notable case in point is the N-terminal region of H2B, which has been described to possess several PTMs, including (poly)lysine acetylation and serine 14 phosphorylation, which have been implicated in transcription[13] and apoptotic chromatin compaction,[14] respectively. Differentially modified semi-synthetic H2B analogs would be useful to assess the affect of acetylation on both antibody recognition as well as on the efficiency of phosphorylation. In this report, we describe a general semi-synthetic route to H2B that allows the installation of PTMs into an otherwise native polypeptide background.
Previous efforts to generate semi-synthetic H2B have focused on C-terminal modifications.[12] Therefore, to obtain N-terminally modified H2B analogs we first needed to develop a suitable ligation strategy. In designing a useful semi-synthetic route, we wanted to employ a traceless-ligation method (i.e. one that would ultimately yield the native H2B amino acid sequence) that would be compatible with the most common PTMs, and allow synthetic access to several known modifications, especially phospho-serine 14 and neighboring Nε-acetyllysines. EPL is a semi-synthetic version of native chemical ligation (NCL) and as such requires the presence of a cysteine residue at the desired ligation junction.[15] However, H2B lacks any native cysteine residues, regardless of the species of origin. Thus, it is necessary to introduce a non-native cysteine for the purpose of ligation and thereafter remove it. We have previously shown that chemical desulfurization[16] is an efficient way to convert a non-native cysteine to a native alanine in the context of H2B.[12] Conveniently, H2B contains an alanine at position 17, suggesting that the native H2B sequence could be generated by desulfurization of the product of a ligation between a synthetic peptide, corresponding to H2B residues 1–16, and a recombinant polypeptide corresponding to residues 18–125 of H2B preceded by a cysteine (Scheme 1).
Scheme 1.
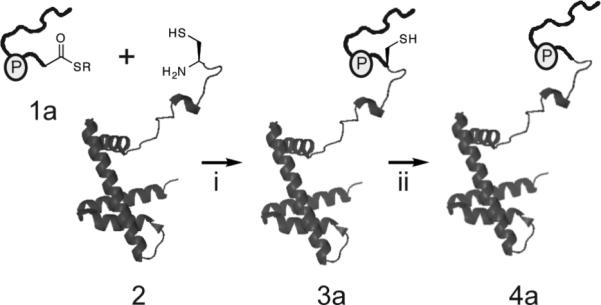
Ligation strategy used to generate semi-synthetic H2B. (i) EPL between a synthetic peptide α-thioester, corresponding to the N-terminal tail of H2B, and a recombinant protein bearing an N-terminal cysteine and corresponding to the remainder H2B. (ii) Raney-nickel reduction to convert the cysteine residue at the ligation site back to a native alanine.
NCL, and by extension, EPL requires the presence of an α-thioester group on the N-terminal peptide fragment. Typically, these α-thioesters are incorporated into synthetic peptides through Boc-based solid-phase peptide synthesis (SPPS).[17] Problematically, serine/threonine phosphate esters are labile to the HF cleavage conditions used in of Boc-SPPS.[18] To address this incompatibility, we turned to two recently described peptidyl-resin linker systems that allow the synthesis of peptide α-thioesters by Fmoc-based SPPS. To avoid the rapid hydrolysis of thioesters during the basic conditions of Fmoc-deprotection, these new linkers are based on the concept of a latent thioester that can be unmasked following chain elongation. One linker, 2-hydroxy-3-mercaptopropionic acid, was developed by Botti et al.[19] and involves chain-assembly off a stable peptide-(oxy)ester that can later be converted to the corresponding α-thioester under reducing conditions. Previously we employed this methodology to synthesize cyclic thioester peptide inhibitors of transmembrane receptors involved in S. aureus virulence.[20] The second linker, diaminobenzoic acid, was developed by Dawson and colleagues.[21] In this case, the linker is acylated post chain-assembly to give an active N-acylurea moiety, which, following cleavage, can be displace by thiols in a strategy analogous to the safety-catch linker developed by Kenner et al.[22] We successfully applied these two linker strategies to synthesize three differentially modified α-thioester peptides corresponding to residues 1–16 of H2B, 1a, 1b, and 1c (Figure S1). Of note, these linkers also bypass a complication associated with previously described syntheses of N-terminally modified histones,[7, 8, 11] namely the possibility of epimerization of C-terminal residues during solution phase α-thioester formation.
The C-terminal portion of H2B, 2, was obtained through recombinant expression of a fragment comprising residues 18–125, preceded by a methionine-cysteine dipeptide. Overexpression in E. coli resulted in the spontaneous removal of the initiator methionine and exposure of the N-terminal cysteine. Purification and characterization of this protein, however, revealed +28 and +72 mass shifts suggestive of the presence of thiazolidine adducts of the cysteine.[23] This was confirmed by in vitro methoxylamine treatment, which led to the clean conversion to a single species with mass consistent with the expected protein, 2 (Figure S2). With the synthetic and recombinant building blocks in hand, EPL was carried out through incubation of 1.5 equivalents of 1a, 1b or 1c with 1.0 equivalent of 2. The reactions were monitored by HPLC and ESI-MS, and were found to be >80% complete (based on consumption of 2) after 29–44 hours (Figure S3). Full length modified H2B proteins (3a, 3b, and 3c) were purified by RP-HPLC and their identities confirmed by MS. To obtain the native H2B sequence, in each case the purified material was treated with activated Raney-nickel as previously described.[12] This selective desulfurization converts the lone cysteine used for ligation to an alanine, rendering the ligation strategy traceless (Scheme 1). The desulfurization reaction was found to proceed smoothly for each of the H2B analogs, 3a–c, to give the final products, 4a–c (Figure 1). Ligations and desulfurizations typically generated 1.0–4.1 mg of 4a–c, corresponding to 25–45% overall isolated yield. Of note, the modified amino acids used here, namely phosphoserine (pSer) and Nε-acetyllysine (Lys(Ac)) are stable to the desulfurization procedure as no mass shift below that of expected was observed. Thus, we have established a strategy to generate H2B that is amenable to synthetic installation of phosphoryl and acetyl groups. In an effort to improve the yield of the final desulfurization step of our synthesis we compared a radical initiated homogeneous desulfurization procedure, described by Danishefsky and collegues,[24] to the Raney nickel method. Comparative desulfurization of 3a revealed the radical initiated approach was amenable to phospho-proteins (Figure S4) and also resulted in a significant twofold increase in isolated yield of homogenous product (Table 1). The decreased yield with the Raney nickel procedure presumably arises from insoluble protein aggregation,[7] which is lost during removal of nickel. These findings suggest that desulfurization of aggregation prone proteins, including histones, may be better suited to the radical initiated strategy.
Figure 1.

Characterization of semisynthetic proteins. Analytical RP-HPLC (top) and ESI-MS (bottom) of modified H2B analogs, 4a–c. The sequence of N-terminal region of each construct is shown beneath the ESI-MS with modified residues highlighted in bold and the underlined alanine representing site of desulfurization. ESI-MS: 4a (M+H)+ observed: 13,900 ± 4 Da; expected: 13,897 Da; 4b (M + H)+ observed: 14,069 ± 5 Da; expected: 14,066 Da; 4c (M + H)+ observed: 13,988 ± 3 Da; expected: 13,986 Da).
Table 1.
Comparison of yields from Raney nickel and radical initiated desulfurization of 3a
| Method | Ave. yield[a] (%) | Yield range (%) |
|---|---|---|
| Raney nickel | 32 ± 4 | 30–35 |
| Radical initiated | 70 ± 2 | 68–71 |
yields are isolated yields from HPLC purification of two independent experiments
Antibodies are a ubiquitous tool in the field of epigenetics and are commonly used to detect the presence of specific modifications from cell extracts. An important consideration when using these tools, however, is the effect of other modifications on antigen recognition. We decided to test a commercial polyclonal antibody (Upstate, 07–191), raised against a synthetic peptide corresponding to residues 6–16 of H2B with a pSer residue 14, against semi-synthetic H2B-pS14 (4a) and H2B-pS14-4xAc (4b). To assess the affect of acetylation on antigen recognition, 4a and 4b were resolved by SDS-PAGE, transferred to PVDF and probed with the α-H2B-pS14 antibody. As expected, semi-synthetic H2B-pS14, as well as endogenous H2B-pS14 obtained from apoptotic cells, was detected at the appropriate molecular weight (Figure 2A and 2B). However, introduction of acetyl groups at neighboring residues completely ablated all recognition of the phosphorylated protein (Figure 2A). This result highlights the limitations of antibodies in broad genome screens, such as ChIP on chip experiments, where histone proteins are heterogenous and often bear multiple PTMs. Recent mass-spectrometry based proteomic efforts have shown that H2B exists in several polyacetylated states.[25, 26] This MS data, together with our findings, complicates antibody dependent efforts to quantify endogenous levels of H2B-pS14.
Figure 2.

Antibody and enzyme recognition of modified H2Bs. A) Coomassie stained SDS-PAGE gel and α-H2B-pS14 western blot of semi-synthetic modified H2B analogs, H2B-pS14 (4a) and H2B-pS14-4×Ac (4b). B) Coomassie stained SDS-PAGE gel and α-H2B-pS14 western blot of histones isolated from healthy HEK-293 cells (−), or HEK-293 cells treated with 1 mM EtOH to induce apoptosis (+). C) Coomassie stained SDS-PAGE gel and 32P radiograph of unmodified H2B (H2B), polyacetylated H2B analog 4c (4×Ac) and H2B-S14E mutant (S14E) treated with either wild-type Mst1 kinase (WT) or an inactive mutant (K59R) in the presence of 32P-labeled ATP. D) Quantification of phosphorylation levels from radiography data of the type shown in panel C (n = 3).
To assess the effect of these acetyl groups on enzyme-substrate recognition we tested H2B-4xAc (4c) against human mammalian sterile twenty-like (Mst1) kinase. Mst1 has been demonstrated to phosphorylate H2B both in vivo[14] and in vitro.[27] The kinase domain (residues 1–330)[28] of either wild-type or inactive (K59R) Mst1 with an N-terminal FLAG-tag was transfected into HEK-293 cells. Cells were then harvested and Mst1 was efficiently immunoprecipitated from the cytosolic fraction using α-FLAG agarose gel (Figure S5). Enriched Mst1 was then assayed against recombinant wild-type H2B and 4c substrates using 32P labeled ATP. Unlike the antibody, Mst1 is tolerant of the acetyl marks. The enzyme is able to recognize and phosphorylate both polyacetylated and unmodified H2B (Figure 2C and 2D). Phosphorylation is only observed in samples treated with active Mst1 and not the inactive K59R mutant enzyme (Figure 2C) suggesting the activity being detected is from Mst1 and not a low abundance contaminant from our pulldown. Furthermore, because a recombinant H2B-S14E mutant substrate displayed minimal phosphorylation (Figure 2C and 2D), the modification appears to be predominantly at serine 14.
In conclusion, we have developed a novel traceless ligation strategy for histone H2B that provides synthetic access to acetylated and phosphorylated analogs of the protein. To our knowledge, this is the first reported semi-synthetic strategy for N-terminally modified H2B. This strategy could be modified and applied to study other histone phosphorylation events including H2A-pS1,[29] H3-pS10,[30] and H4-pS1.[29] The proteins synthesized here were then used to demonstrate that a commercial antibody raised against H2B-pS14 did not recognize this modification in the presence of acetylated lysine 5, 11, 12, and 15. This differed from in vitro kinase activity assays, where Mst1 was able to phosphorylate serine 14 on both unmodified and polyacetylated H2B. The work herein also highlights the importance of understanding the potential shortcomings inherent to antibody-generated data sets. The use of modification specific antibodies in the area of epigenetic research are of great value but the data generated should be considered in the context of broader studies that are beginning to elucidate the density of histone modifications.[4, 5, 31]
Experimental Section
General Methods
Amino acid derivatives and coupling reagents were purchased from Novabiochem. Rink Amide resin was purchased from ChemMatrix. E. coli BL21(DE3) cells was purchased from Novagen. Sephacryl S-200 resin was obtained from GE Healthcare. Restriction enzymes and T4 ligase were obtained from New England Biolabs. Criterion 15% and 4–20% Tris-HCl gels were purchased from Biorad. PCR purification and gel extraction kits were purchased from Qiagen. All other chemical reagents were purchased from Sigma-Aldrich or Fisher Scientific. Analytical and semi-preparative scale RP-HPLC were performed on a Hewlett-Packard 1100 series instrument using Vydac C18 columns (4 mm × 150 mm; 10 mm × 250 mm) at 1 and 4 mL minute−1, respectively. HPLC buffer A consisted of 0.1% trifluoroacetic acid (TFA) in water and buffer B consisted of 90% acetronitrile, 0.1% TFA in water. Preparative and process scale RP-HPLC were performed on a Waters DeltaPrep 4000 system connected to a Waters 486 tunable detector using Vydac C18 columns (22 × 250 mm; 50 × 250 mm) at 15 and 30 mL minute−1, respectively. Size-exclusion and ion-exchange chromatography were performed on an AKTA FPLC system from GE Healthcare equipped with a P-920 pump and a UPC-900 monitor. ESI-MS was performed on a Sciex API-100 single quadrupole mass spectrometer. Primer synthesis and DNA sequencing were performed by Integrated DNA Technologies and Genewiz, respectively.
Synthesis of modified H2B peptides
The sequence corresponding to residues 1–16 of X. laevis H2B was synthesized using manual solid-phase peptide synthesis with an Fmoc Nα protection strategy and using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) for amino-acid activation. Standard tbutyl side-chain protection was used throughout except for modified amino acids: acetyl lysine at 5, 11, 12, and 15, for 1b and 1c, and N-α-Fmoc-O-benzyl-L-phosphoserine, for 1a and 1b peptides. Peptide 1a was assembled on a Rink-amide resin that had previously been loaded with the linker 2-(t-butyl-dimethyl-silanyloxy)-3-t-butyldisulfanylpropionic acid.[20] Following chain assembly, the peptide was cleaved from the resin with TFA:TIS:H2O (95:2.5:2.5) for 3.5 hours. The crude peptide was then incubated in reducing buffer [100 mM phosphate buffer, pH 7.0, 100 mM tris(2-carboxyethyl)phosphine (TCEP), 150 mM 2-mercaptoethanesulfonate (MES)] for 1 hour at room temperature to generate the desired α-thioester prior to purification. Peptides 1b and 1c were synthesized using a diaminobenzoic acid linker.[21] In both cases, the C-terminal residue Fmoc-Lys(Boc)-OH was coupled to diaminobenzoic acid in solution as described.[21] The resulting amino-acyl-linker was then loaded on to Rink-amide resin using HBTU and the remainder of the sequence assembled using Fmoc-protocols. Following chain assembly, the linker was activated on the solid phase by acylation with p-nitrophenylchloroformate and the subsequent o-aminoanilide was transformed to an N-acylurea moiety as described.[21] The peptides were cleaved from the resin with TFA:TIS:H2O (95:2.5:2.5) for 3.5 hours and the crude peptides were then incubated in MES buffer (150 mM phosphate buffer, pH 6.8 and 150 mM MES) for 1 hour at room temperature to generate α-thioesters. All peptides were purified by semi-preparative RP-HPLC using a gradient of 7.5–22.5% HPLC buffer B over 45 minutes. Peptides 1a–c were characterized by ESI-MS (1a (M + 2H)+ observed: 914 Da; expected: 913 Da; 1b (M + 2H)+ observed: 998 Da; expected: 997 Da; 1c (M + 2H)+ observed: 957 Da; expected: 957 Da).
Expression of H2B, H2B-S14E, and 2
The H2B-S14E point mutant was generated by site-directed mutagenesis with primers xH2BS14E-F (5'-CCA GCC CCG AAG AAA GGC GAG AAG AAA GCG GYG ACC AAG-3') and xH2B-S14E-R (5'-CTT GGT CAC CGC TTT CTT CTC GCC TTT CTT CG GGG CTG G-3') using an H2B expression plasmid as a template. QuikChange was carried out with a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) as per manufacturer's recommendations. Recombinant H2B and H2B-S14E were generated by overexpression in E. coli as previously described.[12] X. laevis H2B, residues 18–125, were PCR amplified from an H2B expression plasmid using xH2B-MC18-F (5'-G GAA TTC CAT ATG TGT GTG ACA AGA CTC AGA AGA AAG ACG G-3') and xH2B-125-R (5'-CG GGA TCC TTA CTT GGC GCT GGT GTA CTT G-3') to insert a methionine-cysteine dipeptide upstream of valine-18. The PCR product was digested with NdeI and BamHI and inserted into the pET3 vector (Novagen). Protein expression proceeded with transformation of BL21(DE3)pLysS E. coli cells (Novagen). Cultures were inoculated with a single colony and grown at 37 °C in the presence of ampicillin and chloramphenicol. When the cultures reached an optical density of 0.6 (A600) they were induced with IPTG (0.5 mM) and grown for an additional 3 hours. Cells were then pelleted by centrifugation (6,800 × g, 15 minutes, 4 °C), resuspended in lysis buffer [10 mM Tris-HCl, pH 7.2, 200 mM NaCl, 1 mM EDTA, 1 mM PMSF, and 10 µg/mL DNase (150 mL / 6 L culture)] and lysed using a French Press. Inclusion bodies were isolated from cell lysates by centrifugation (26,000 × g, 20 minutes, 4 °C) and resuspended with wash buffer [20 mM Tris-HCl, pH 7.2, 200 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, and 1 % Triton X-100 (60 mL / 6 L culture)]. The suspension was centrifuged again to re-isolate insoluble material for a second wash in wash buffer, and a third wash in wash buffer with no Triton X-100. The final pellet was then solubilized with 1:1 DMSO:extraction buffer [20 mM Tris-HCl, pH 7.5, 6 M guanidinium chloride, 200 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol (4 mL / 6 L culture)] by nutating for 15 minutes at room temperature. 10 mL of extraction buffer was then added and allowed to nutate for an additional 60 minutes. The solution was cleared by centrifugation (26,000 × g, 20 minutes, 4 °C) and the supernatant was syringe filtered (Pall, 0.45 μM GHP membrane) and purified on a Sephacryl S-200 column (GE Healthcare), eluting with extraction buffer. Purified fractions were combined, dialyzed into water containing 2 mM DTT, and lyophilized. To resolve +28 and +72 Da mass shifts corresponding to thiazolidine adducts of the cysteine, lyophilized H2B was resuspended in 100 mM sodium acetate, pH 5, 100 mM NaCl, and 250 mM methoxylamine and incubated at 37 °C, nutating, for 60 minutes. The final product, 2, was purified using process RP-HPLC with a 40–55% gradient of buffer B over 60 min and characterized by ESI-MS ((M + H)+ observed: 12,248 ± 3 Da; expected: 12,247 Da). The final yield of protein 2 was 18.7 mg/L.
Expressed protein ligation
Purified peptides 1a–c (1–2 mg, 0.6–1.0 µmol, 1.5 eq.) and protein 2 (4–9 mg, 0.4–0.7 µmol, 1 eq.) were dissolved in 200 µL of ligation buffer (300 mM sodium phosphate, pH 7.8, 3 M guanidinium chloride, 100 mM MES), adjusted to pH 7.8 with NaOH, and incubated at room temperature for 29–44 hours. The reaction was monitored by RP-HPLC and ESI-MS and quenched with 200 µL of 50% HPLC buffer B containing TCEP (100 mM). The ligation products, 3a–c, were then purified using semi-preparative RP-HPLC with a 42–55% B gradient over 45 minutes and confirmed by ESI-MS: 3a (2.8 mg, 50%), (M+H)+ observed = 13,929 ± 2 Da, expected = 13,929 Da; 3b (5.0 mg, 57%), (M+MES+H)+ observed = 14,244 ± 3 Da, expected = 14,097 Da; 3c (6.8 mg, 71%), (M+MES+H)+ observed = 14,161 ± 2 Da, expected = 14,017 Da.
Desulfurization
Raney-nickel reduction, as previously described,[12] was used to convert C17 of proteins 3a–c to the native alanine. In a typical reaction, 3a–c, were dissolved in 2 mL of desulfurization buffer (200 mM sodium phosphate, pH 7.0, 6 M guanidinium chloride, 35 mM TCEP). Raney-nickel was prepared by adding NaBH4 (200 mg, 5.287 mmol) to a stirred solution of nickel acetate (1.2 g, 4.822 mmol) in 3 mL of H2O. After 5 minutes, the Raney-nickel was filtered, washed with 200 mL of H2O and added to the protein solution. Fresh Raney-nickel preparations were added after 4, 6, 8, and 10 hours. Raney-nickel was pelleted by centrifugation (17,000 × g, 2 minutes) and washed twice with 0.75 mL of desulfurization buffer. The supernatant and washes containing protein were combined, added to an equivalent volume of 50% HPLC buffer B and the products, 4a–c, were purified using semi-preparative RP-HPLC with a 42–55% B gradient over 45 minutes and characterized by ESI-MS: 4a (1.0 mg, 33%), (M+H)+ observed = 13,900 ± 4 Da, expected = 13,897 Da; 4b (1.5 mg, 50%), (M+H)+ observed = 14,069 ± 5 Da, expected = 14,066 Da; 4c (4.1 mg, 60%), (M+H)+ observed = 13,988 ± 3 Da, expected = 13,986 Da. Radical initiated desulfurization, as previously described,[24] was used to convert C17 of protein 3a to the native alanine. In a typical reaction, 300 mg of 3a, were dissolved in 150 mL of desulfurization buffer 2 (200 mM sodium phosphate, pH 7.0, 6 M guanidinium chloride). To this solution was added 3 mL ethane thiol, 225 mL 0.5 M TCEP pH 7.0, 15 mL 2-methylpropane thiol, and 3.75 mL VA-061 (0.2 M stock in methanol). The resulting solution was incubated in a 37 °C water bath for 24 h allowing for complete desulfurization. The product was purified by semipreparative RP-HPLC using a 0–73% gradient of buffer B, yielding 4a (214 mg, 71%) (M+H)+ observed = 13,899 ± 4 Da, expected = 13,897 Da. Repeat radical initiated desulfurizations of 3a (n = 2) yielded 4a with an average yield of 70%. Repeat Raney nickel desulfurizations of 3a on comparable scale (n = 2) yielded 4a with an average isolated yield of 32%.
Western blot analysis
For cellular H2B-pS14, HEK-293 cells were treated with 1 mM ethanol for 6 hours then harvested by scraping. Histones were acid extracted as previously described.[32] Histones from cellular sources and chemically phosphorylated H2B analogs were resolved on a Criterion Tris-HCl 15% gel (BioRad) and transferred to PVDF. Importantly, efficient transfer of histone proteins requires Towbin's buffer supplemented with 0.02% SDS. Western blot analysis was performed with a polyclonal antibody raised against residues 6–16 H2B, phosphorylated at serine 14 (1:4,000, Upstate #07-191).
Cloning, transfection, and enrichment of Mst1
WT and K59R Mst1 kinase constructs were generated by PCR amplification with hMst1-F (5'-CG GGA TCC ATG GAT TAC AAG GAT GAC GAC GAT AAG GCG ATG GAG ACG GTA CAG CTG AGG-3') and hMst1-R (5'-GC TCT AGA CTA CAT CGT GCC AGA ATC CAT TTC ATC C-3') using pJ3H-Mst1 and pJ3M-Mst1 K59R as templates (Addgene). The PCR product was digested with BamHI and XbaI and inserted into pCDNA3 (Invitrogen). The sequenced constructs were then transfected into HEK-293 cells with Fugene 6 (Roche) as per manufacturer's recommendations. 48 hours after transfections cells were harvested by scraping and cell pellets were lysed by dounce homogenization and sonication. The insoluble proteome was then removed by centrifugation (100,000 × g, 30 minutes, 4 °C) and the supernatant (soluble proteome) was incubated with pre-washed M2-agarose affinity gel (Sigma) for 2 hours. Beads were collected by centrifugation (6,000 × g, 2 minutes, 4 °C), washed three times with PBS containing 0.1% Triton X-100 and once with PBS alone. Mst1 was finally eluted, twice, with 0.5 mg/mL triple-FLAG peptide (Sigma) in PBS. Enrichment was assessed by Western blot analysis against input, wash, and elution fractions (Figure S5). Briefly, samples were separated on a Criterion Tris-HCl 4–20% gel (BioRad), transferred to PVDF membrane and blotted against the FLAG epitope using the M2 antibody (1:5,000, Sigma).
Mst1 kinase activity assay
Mst1 was assayed against semi-synthetic and recombinant H2B substrates. The kinase assay consisted of 4.75 μM of H2B substrate and 3 μL of Mst1 kinase, WT or K59R, in reaction buffer [25 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 50 μM ATP, 2.5 μCi of γ-32P-ATP (Amersham)]. Assays proceeded for 5–20 minutes at which point reactions were quenched with 4 × SDS-PAGE loading buffer, boiled for 8 minutes, and separated on a Criterion Tris-HCl 15% gel (BioRad). Gels were stained with coomassie blue, destained, dried for 2 hours at 75 °C, and imaged with a Typhoon 8400 scanner (GE Healthcare).
Acknowledgements
We would like to thank M.K. O'Reilly, M.R. Pratt, S. Frutos, M. Vila-Perello, and S.W. Lockess for helpful discussion. This work was funded by the U. S. National Institutes of Health (GM055843 & EB001991) and the Starr Foundation. K.P.C. was supported by an American Cancer Society postdoctoral fellowship. R.K.M. was supported by the NIH MSTP grant GM07739.
References
- [1].Walsh C. Posttranslational modification of proteins. Roberts and Company Publishers; Greenwood Village: 2006. [Google Scholar]
- [2].Luger K, Richmond TJ. Curr Opin Struct Biol. 1998;8:33. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- [3].Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- [4].Coon JJ, Ueberheide B, Syka JE, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Proc Natl Acad Sci U S A. 2005;102:9463. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Nat Methods. 2007;4:487. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- [6].Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Nature. 2006;441:349. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- [7].He S, Bauman D, Davis JS, Loyola A, Nishioka K, Gronlund JL, Reinberg D, Meng F, Kelleher N, McCafferty DG. Proc Natl Acad Sci U S A. 2003;100:12033. doi: 10.1073/pnas.2035256100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shogren-Knaak MA, Fry CJ, Peterson CL. J Biol Chem. 2003;278:15744. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- [9].Simon MD, Chu F, Racki LR, Cruz C. C. de la, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. Cell. 2007;128:1003. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Flavell RR, Muir TW. Acc Chem Res. 2009;42:107. doi: 10.1021/ar800129c. [DOI] [PubMed] [Google Scholar]
- [11].Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Science. 2006;311:844. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- [12].McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Nature. 2008;453:812. doi: 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Myers FA, Chong W, Evans DR, Thorne AW, Crane-Robinson C. J Biol Chem. 2003;278:36315. doi: 10.1074/jbc.M305822200. [DOI] [PubMed] [Google Scholar]
- [14].Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, Allis CD. Cell. 2003;113:507. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- [15].Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- [16].Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- [17].Camarero JA, Muir TW. Curr Protoc Protein Sci. 2001;Chapter 18(Unit18):4. doi: 10.1002/0471140864.ps1804s15. [DOI] [PubMed] [Google Scholar]
- [18].Williams W. a. W., DB . Biologically active peptides: Design, synthesis, and utilization. CRC Press; Boca Raton: 1993. [Google Scholar]
- [19].Botti P, Villain M, Manganiello S, Gaertner H. Org Lett. 2004;6:4861. doi: 10.1021/ol0481028. [DOI] [PubMed] [Google Scholar]
- [20].George EA, Novick RP, Muir TW. J Am Chem Soc. 2008;130:4914. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- [21].Blanco-Canosa JB, Dawson PE. Angew Chem Int Ed Engl. 2008;47:6851. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kenner GW, Mcdermot, Sheppard RC. Journal of the Chemical Society D-Chemical Communications. 1971:636. [Google Scholar]
- [23].Gentle IE, De Souza DP, Baca M. Bioconjug Chem. 2004;15:658. doi: 10.1021/bc049965o. [DOI] [PubMed] [Google Scholar]
- [24].Wan Q, Danishefsky SJ. Angew Chem Int Ed Engl. 2007;46:9248. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- [25].Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, Grauslund M, Hansen AM, Jensen ON. Mol Cell Proteomics. 2006;5:1314. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- [26].Pesavento JJ, Kim YB, Taylor GK, Kelleher NL. J Am Chem Soc. 2004;126:3386. doi: 10.1021/ja039748i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Anand R, Kim AY, Brent M, Marmorstein R. Biochemistry. 2008 doi: 10.1021/bi800309m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Creasy CL, Ambrose DM, Chernoff J. J Biol Chem. 1996;271:21049. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- [29].Barber CM, Turner FB, Wang Y, Hagstrom K, Taverna SD, Mollah S, Ueberheide B, Meyer BJ, Hunt DF, Cheung P, Allis CD. Chromosoma. 2004;112:360. doi: 10.1007/s00412-004-0281-9. [DOI] [PubMed] [Google Scholar]
- [30].Lim JH, Catez F, Birger Y, West KL, Prymakowska-Bosak M, Postnikov YV, Bustin M. Mol Cell. 2004;15:573. doi: 10.1016/j.molcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- [31].Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Nat Genet. 2008;40:897. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shechter D, Dormann HL, Allis CD, Hake SB. Nat Protoc. 2007;2:1445. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]