

Abstract
Objective
Evaluate the role of p38 p38 mitogen activated protein kinase (MAPK) signaling pathway in lipopolysaccharide (LPS)-induced receptor activator of nuclear factor-κB ligand (RANKL) expression by murine periodontal ligament (PDL) cells.
Background
LPS from Gram-negative bacteria is one of the microbial-associated molecular patterns that initiate the immune/inflammatory response leading to tissue destruction observed in periodontitis.
Methods
Expression of RANKL and osteoprotegerin (OPG) mRNA was studied by reverse transcription polymerase chain reaction (RT-PCR) upon stimulation with LPS from Escherichia coli and Aggregatibacter actinomycetemcomitans. The biochemical inhibitor SB203580 was used to evaluate the contribution of p38 MAPK signaling pathway to LPS-induced RANKL and OPG expression. Stable cell lines expressing dominant negative forms of MAKPK kinase (MKK)-3 and MKK6 were generated to confirm the role of p38 MAPK pathway. An osteoclastogenesis assay using a co-culture model of the murine monocytic cell line RAW 264.7 was used to determine if osteoclast differentiation induced by LPS-stimulated PDL was correlated with RANKL expression.
Results
Inhibiting p38 MAPK previous to LPS stimulation resulted in a significant decrease of RANKL mRNA expression. OPG mRNA expression was not affected by LPS or p38 MAPK. LPS-stimulated PDL cells increased osteoclast differentiation, an effect that was completely blocked by OPG and significantly decreased by inhibition of MKK3 and MKK6, upstream activators of p38 MAPK. Conditioned medium from mPDL cultures did not increase osteoclast differentiation, indicating that PDL cells produced membrane-bound RANKL.
Conclusion
LPS resulted in a significant increase on RANKL in PDL cells. p38 MAPK pathway is required for LPS-induced membrane-bound RANKL expression in these cells.
Keywords: osteoclastogenesis, RANKL, PDL cells, p38 MAPK
Introduction
Coupled bone turnover is a tightly regulated process balanced by osteoblasts and osteoclasts. Uncoupling leads towards net bone loss associated with several inflammatory bone diseases including periodontal diseases. Initiated by proinflammatory stimuli originating from dental biofilm-associated bacteria (including lipopolysaccharide), molecular events are set in motion in the periodontal microenvironment leading towards periodontal tissue and bone destruction. Fundamental aspects of the bone resorption process have been appreciated over recent years following identification of receptor activator of nuclear factor-κB ligand (RANKL) and its decoy receptor osteoprotegerin (OPG). These cytokines constitute the current paradigm for the processes of osteoclast differentiation and activation. The induction of coupled bone resorption by inflammatory cytokines involves increased RANKL expression on various cell types, including osteoblastic cells, bone marrow stromal cells, endothelial cells, mononuclear cells and periodontal ligament fibroblasts which activate RANK on preosteoclastic cells to induce osteoclastogenesis and subsequent bone resorption (1–5).
The unique location of the periodontal ligament (PDL) between two mineralized tissues, cementum and alveolar bone, suggests that it may have an essential role in periodontal homeostasis. This assumption is supported by classical histological studies on periodontal regeneration(6). Thus, alveolar bone formation and resorption in the periodontium seems to be largely controlled by cells in the periodontal ligament, as also supported by studies on orthodontic movement of teeth where RANKL/OPG expression was shown to be involved (7). Even though PDL cells were initially considered as inhibitors of osteoclastogenesis and shown to express OPG (8) these cells were demonstrated to express RANKL in situations associated with resorption of mineralized tissue, including physiologic permanent tooth eruption and deciduous tooth root resorption (9–11). Specifically, PDL cells were shown to form bone-like tissues (12, 13) and also to express RANKL and OPG mRNA after stimulation with di-hydroxyl vitamin D3, prostaglandin (PG)E2, interleukin (IL)-1β, IL-1α, and mechanical stress (10, 14–18).
The RANKL/RANK/OPG system as key mediators of bone loss in periodontal disease comes from recent in vivo studies, both in animal models (19, 20) and in humans (21, 22), demonstrating the expression of higher levels of RANKL and lower levels of OPG in the presence of alveolar bone resorption (23). Even though numerous studies have demonstrate that bacterial LPS has bone-resorbing capacity in vitro and in vivo (24–27), controversy still exists regarding the potency of the different pathogen-associated molecular patterns in inducing RANKL expression. Recently, PDL fibroblasts have been shown to produce RANKL mRNA induced by IL-1α indirectly through PGE2 induction, which involved extracellular signal-regulated kinase (ERK) MAPK signaling(15). Also recently, all three MAPKs (ERK, c-Jun N-terminal kinase (JNK) and p38) were shown to be involved in IL-1β-induced RANKL expression by human periodontal ligament fibroblasts (28). Conversely, RANKL expression by P. gingivalis-infected osteoblasts did not involve p38 or ERK MAP Kinases as well as phosphoinositol-3 (PI-3) kinase pathways (29). In fact, these authors (29) reported that LPS from P. gingivalis was not responsible for the induction of RANKL in infected osteoblasts, which suggests that TLR-2 signaling pathway may not be involved in RANKL expression by these cells.
There is a lack of information on the signaling pathways involved in LPS-induced RANKL expression by PDL fibroblasts. Since these cells may play an important role on alveolar bone resorption, both during periodontal disease and orthodontic movement, understanding the signaling pathways involved may provide critical information towards alternative therapeutic strategies for the control of alveolar bone resorption process. Recent data from our group supports the role of novel therapeutics which blocks p38 signaling in preventing alveolar bone loss induced by LPS in vivo (3). Considering that RANKL expression may require different signaling pathways depending on the nature of extracellular stimulation and also on the cell type, in this manuscript we studied the role of p38 MAPK signaling on LPS-induced RANKL expression by PDL cells.
Materials and Methods
Cells and materials
Mouse periodontal ligament (PDL) fibroblasts immortalized with SV40 large T antigen were obtained from Dr. Martha Somerman (University of Washington, Seattle, WA). These cells were cultured in DMEM supplemented with 100 IU/mL penicillin, 100 μg/mL streptomycin and 10% heat-inactivated fetal bovine serum and maintained in a humidified atmosphere at 37°C and 5% CO2. Mouse PDL cells used were previously characterized for expression of genes normally expressed by primary PDL cells, including bone sialoprotein, osteopontin, osteocalcin and type I collagen (30). Unless noted otherwise all tissue culture reagents were obtained from Invitrogen. LPS from Escherichia coli (serotype 0127:B8) was purchased from Sigma and Aggregatibacter actinomycetemcomitans (formerly known as Actinobacillus actinomycetemcomitans) LPS was extracted from A. actinomycetemcomitans strain Y4 (serotype B) by the hot phenol-water method as described (23, 31). A. actinomycetemcomitans LPS used in the present study was recently characterized as part of other studies from our lab group (23). Both E.coli and A. actinomycetemcomitans LPS were diluted in serum-free defined culture medium (Opti-MEM, Invitrogen) at 1mg/mL. The biochemical inhibitor SB203580 was from Calbiochem and RANKL and OPG recombinant proteins were from R&D systems. Mouse RANKL monoclonal antibody was purchased from StressGen, and monoclonal GAPDH antibody was from Chemicon. The absence of protein in A. actinomycetemcomitans LPS preparations was confirmed by polyacrylamide gel electrophoresis of extract samples and subsequent staining with Silver Nitrate and Comassie blue and confirmed by spectrophotometry (<0.001% nucleic acid) and by a microassay for protein quantitation (Bio-Rad Lab., cat # 500-0002) based on the Bradford method (lower limit of detection: 1.2 μg/mL). Dominant negative genetic constructs of mutated MKK3 and MKK6 were obtained from J. Han (Scripps Institute, La Jolla, CA). Stable cell lines were prepared as described previously(3). Briefly, after co-transfection of the overexpression construct and of an empty vector including resistance to gentamycin, selection was carried out for several weeks in medium containing 800 μg/mL Geneticyn (Invitrogen Corp.) and a number of clones was screened by Western Blot to analyse the expected changes on expression of the signaling proteins.
Semi Quantitative RT-PCR
Reverse transcription-PCR was used to evaluate mRNA expression as described recently(3). Briefly, total RNA was harvested using Trizol (Invitrogen) reagent according to the manufacturer’s instructions. Complementary DNA was synthesized by reverse transcription of 500 ng of total RNA using 2.5 μM Oligo (dT) 16 primers and 1.25 U/uL Moloney murine leukemia virus reverse transcriptase in the presence of 5.5 mM MgCl2, 2 mM dNTPs and 0.4 U/μL of RNAse inhibitor, according to the manufacturer’s protocol (Applied Biosystems). 2 μL of the RT reaction product were used on a 25 μL total volume PCR reaction mix. The primer pair used for RANKL (acession# AF019048): sense 5′-CAGCACTCACTGCTTTTATAGAATCC-3′; antisense 5′-AGCTGAAGATAGTCTGTAGGTACGC-3′; for OPG (accession# NM008764) was: sense 5′-TGTAGAGAGGATAAACGG-3′; antisense 5′-CTAGTTATAAGCAGCT-TAT-3′; whereas the primer pair for GAPDH (acession# NM002046) was: sense 5′-CACCATGGAGAAGGCCGGGG-3′; antisense 5′-GACGGACACATTGGGGTAG-3′. 50 pmol/μL of each primer were used in the PCR reactions, yielding products of 467, 503 and 418bp for RANKL, OPG and GAPDH, respectively. Taq DNA polymerase and other PCR reagents were purchased from Invitrogen and the conditions for RANKL and OPG were 35 cycles (32 cycles for OPG) of 94°C for 1 min, 56°C for 1 min, 72°C for 2 min, and a final extension step at 72°C for 10 min in the presence of 2.5 mM MgCl2, whereas for GAPDH the conditions were 25 cycles of 94°C for 1 min, 52°C for 1 min, 72°C for 2 min, and a final extension step at 72°C for 10 min in the presence of 1.5 mM MgCl2. PCR products were resolved in 1.5% agarose gels, stained with 0.5 μg/mL ethidium bromide. The images and densitometric measurements were obtained with a digital documentation system (Gel Doc XR, Bio-Rad). RANKL and OPG mRNA quantity in each sample was subsequently normalized to the quantity of GAPDH mRNA and expressed as fold change over unstimulated control using Molecular Analyst Software (version 4.5.2, BioRad). The fold change on the expression of RANKL was divided by the fold change on the expression of OPG to obtain the RANKL: OPG ratio.
Western Blot analysis
5×104 PDL cells were grown for 24 hours in each well of 6-well plates, de-induced in culture medium containing 0.3% FBS for 12 hours and stimulated with either E. coli or A. actinomycetemcomitans LPS for 72 hours both with and without a 30-minute pre-treatment with 10μM of SB203580. Whole cell lysates were harvested by scraping the PDL cells with SDS sample buffer (62.5 mM Tris HCl buffer pH 6.8, 10% glycerol, 50 mM DTT, 2% SDS, 0.01% bromophenol blue) on ice, followed by sonication for 10 seconds and heat-denaturation at 95°C for 5 minutes. Total protein content was quantitated by a Bradford-based microassay. For western blotting, 30 μg of total proteins were separated on 10% Tris-Cl polyacrylamide gels run at 100 V for 60 min and subsequently electro-transferred for another 60 min at 110 mA in a semi-dry apparatus to nitrocellulose membranes. The membranes were blocked (Tris-buffered saline with 5% non-fat dry milk, 0.1% Tween-20) for 1 hour at room temperature and then probed overnight at 4°C with primary antibodies. The presence of the primary antibodies was detected by using HRP-conjugated secondary antibody and a chemiluminescence system (SuperSignal West Pico Chemiluminescent Substrate, Pierce). Digital images and quantitation of the membranes were obtained and analyzed on a chemiluminescent documentation system (ChemiDoc XRS, Bio-Rad).
Osteoclastogenesis Assays
5 × 104 PDL cells were plated on 6-well dishes and 24 h later the medium was changed to 0.2% FBS-containing medium and the cells stimulated with E. coli and A. actinomycetemcomitans LPS for 72 h. Since preliminary experiments indicated similar ability of both E. coli and A. actinomycetemcomitans LPS to induce RANKL mRNA in PDL cells as well as in their capacity to support osteoclastogenesis, only LPS from periopathogenic A. actinomycetemcomitans was used to stimulate the stable cell lines MKK3dn-PDL and MKK6dn-PDL. LPS was added to the co-cultures at 0 and 32 h. Conditioned medium was removed at 72 h, the cell layer washed with PBS twice and scraped off the wells and resuspended in 0.5 mL of fresh 0.2% FBS-containing medium. 3 × 104 cells were then plated in each well of a 6-well plate. These cells were plated onto RAW 264.7 cells, that were growing these 6-well plates (initial plating density: 5 × 104 cells/well) for 24 h. 0.5 mL of conditioned medium was added to RAW 264.7 cells to determine the contribution of soluble RANKL (sRANKL) by the PDL cells. RANKL (50 ng/mL) and OPG (100 ng/mL) were used as controls for the capacity of RAW 264.7 cells to differentiate into osteoclasts in response to RANKL, as well as for the ability of OPG to block this process. The co-cultures were maintained for 6 days, with one change of medium on day 3, and then submitted to the TRAP staining, as described on the Technical Bulletin #445, BD BioSciences. All reagents used for this assay were from Sigma. Briefly, culture medium was removed and the cells washed twice with PBS. Cells were then fixed with 10% glutaraldehyde for 15 minutes (37°C), washed twice with pre-warmed PBS and stained for 5–10 minutes at 37°C with the staining buffer, pH 5.0 (50 mM Sodium Acetate buffer, 30 mM Sodium Tartrate, 0.1 mg/ml Naphtol AS-MX phosphate, 0.1% Triton X-100 and 0.3 mg/ml of Fast Red Violet LB stain). After removal of the staining buffer, the cells were washed 3 times with PBS. These cells were observed on an inverted microscope (at 40X) magnification, depending on cell density) using bright field and the numbers of stained cells containing three or more nuclei were counted.
Statistical analysis
Pairwise comparisons between experimental groups were performed using the t-test with Welch’s correction for unequal variances. Comparison between fold changes on mRNA expression between LPS-stimulated and untreated cells was performed with the one-sample t-test. Significance level was set to 5% and all calculations were performed using Prism 4 software (GraphPad, Inc).
Results
p38 MAPK regulates preferentially RANKL mRNA expression in LPS-stimulated PDL cells
Preliminary experiments indicated that maximum expression of RANKL mRNA occurred after an 18 to 24 h stimulation with either LPS (Figure 1, panels A and B). The 18 h period of LPS stimulation was thereafter chosen to enable the study of LPS on RANKL gene expression in PDL cells. Semi-quantitative RT-PCR indicated that inhibition of p38 MAPK with SB203580 prior to stimulation with LPS from E. coli or A. actinomycetemcomitans resulted in a significant decrease on the expression of RANKL mRNA (Figure 2A). Interestingly, OPG mRNA expression was not consistently affected by LPS stimulation, and also not significantly changed by inhibiting p38 MAPK (Figure 2B). Nevertheless, the decrease on RANKL expression achieved with SB203580 was of sufficient magnitude to reduce the RANKL:OPG ratio (Figure 2C). Since it is this ratio that will ultimately indicate the net effects on osteoclast differentiation and activation, these results indicate that blocking p38 MAPK might impair osteoclastogenesis induced by LPS-stimulated PDL cells. However, in spite of being significant the LPS-induced stimulation on RANKL expression by PDL cells was somewhat modest, represented by a 40% increase over the level observed with unstimulated cells. On the other hand, the decrease on RANKL mRNA expression after blocking p38 MAPK activity with SB203580 was clear when E.coli LPS or A. actinomycetemcomitans Y4 LPS was used for stimulation.
Figure 1. Time course of RANKL mRNA expression induced by E.coli and A. actinomycetemcomitans LPS in PDL cells.

RT-PCR shows that RANKL mRNA expression induced by LPS is biphasic, reaching an early peak after 4 h of stimulation and reached the maximum after 18 to 24 hours (A and B). E. coli LPS was a more potent inducer of OPG expression (A), which was already noticeable after 2 h of stimulation and remained essentially constant throughout the 24 h period. A. actinomycetemcomitans Y4 LPS increased OPG mRNA modestly, and this effect was delayed, since it took 18 hours of stimulation to be more evident. Stimulation for periods longer than 24 h did not induce further increases on either RANKL or OPG mRNA expression (data not shown). Also, we did not observe noticeable levels of RANKL mRNA expression by PDL cells in the absence of any stimulation (data not shown). Representative images of three independent experiments.
Figure 2. p38 MAPK regulates preferentially RANKL in LPS-stimulated PDL cells.

mPDL cells were grown to near confluency and de-induced in media containing 0.3% FBS for 8 h. The specific inhibitor for p38 MAPK (SB203580) was added to the culture medium at 10 μM 30 minutes before the 18 h stimulation with 1 μg/mL of LPS from E.coli and A. actinomycetemcomitans LPS. Total RNA was harvested and RT-PCR was performed and quantitated using GelDoc System. Results indicate that inhibition of p38 MAPK reduced LPS-induced RANKL mRNA expression, especially after E.coli LPS stimulation (A). No significant regulation of OPG mRNA expression was observed following p38 inhibition (B). The decrease on RANKL expression was sufficient to reduce the RANKL:OPG ratio (C). Figures are representative of three independent experiments and bar graphs indicate mean ± standard deviation of normalized fold changes of normalized gene expression. *Indicates significant difference (p<0.05) on mRNA expression in comparison to unstimulated cells. # indicates significant difference (p<0.05) on mRNA expression in comparison to LPS treated cells.
RANKL protein expression induced by LPS also requires p38 MAPK activity
Expression of RANKL at the protein level after stimulation with either LPS confirmed the relevance of p38 MAPK, since blocking this pathway with the biochemical inhibitor SB203580 resulted in a significant decrease on protein expression (Figure 3, A and B). Also, protein expression is most probably indirect since we could not find detectable levels of RANKL after 24 and 48 hours of stimulation (data not shown). We also could not detect RANKL protein on concentrated cell culture supernatants.
Figure 3. RANKL protein expression by LPS-stimulated PDL cells is dependent on p38 MAPK activity.

PDL cells grown on 6-well plates were de-induced for 12 hours in culture medium containing 0.3% FBS and then stimulated with LPS from either E.coli or A. actinomycetemcomitans for 72 hours. The specific inhibitor for p38 MAPK (SB203580; 10 μM) was added to the culture medium 30 minutes before stimulation with LPS (1 μg/mL). (A) Western blot analysis of RANKL expression from PDL whole cell lysates. Positive control for RANKL expression is cell lysates from the human prostate cancer cell line (PC-3). (B) Density analysis of data from three independent experiments shows significant inhibition of LPS-induced RANKL protein by SB203580 (* p<0.05).
LPS-stimulated PDL cells support osteoclastogenesis by membrane-bound RANKL expression
Morphological changes (size and multinucleation) and expression of tartrate resistant acid phosphatase (TRAP) are indicative of terminal osteoclast differentiation. RAW 264.7 cell line is capable of differentiation into osteoclasts in the presence of exogenous RANKL (Figure 4A and Figure 5, (32)). The osteoclastogenesis assay indicates the functionality of RANKL produced by LPS-stimulated PDL cells (Figure 5). The results mimicked the findings for RANKL mRNA expression, as a significant (p<0.05) increase on the number of TRAP+ multinucleated cells was observed after co-culture with PDL cells stimulated with LPS (Figure 4B). A reduction on the number of osteoclasts occurred when OPG (100 ng/mL) was added to the co-cultures, indicating that osteoclast differentiation was associated with RANKL expression by PDL cells. This decrease was significant (p<0.05) for E.coli LPS stimulation and nearly reached statistical significance (p=0.056) after A. actinomycetemcomitans Y4 LPS stimulation. Since conditioned medium from LPS-stimulated PDL cells had no effect on osteoclast formation (data not shown), it is concluded residual LPS used for stimulating the PDL cells was not a confounding factor, as well as that LPS-stimulated cells did not produce soluble RANKL.
Figure 4. LPS-stimulated PDL cells stimulate osteoclastogenesis, which is regulated by p38 MAPK pathway.

Stimulation with RANKL induces RAW 264.7 cells to differentiate into multinucleated TRAP+ cells, whereas pre-treatment with OPG inhibits this effect *Indicates significant (p<0.05) difference from unstimulated cells and #indicates a significant decrease on the number of osteoclasts with OPG treatment (A). mPDL cells were stimulated with LPS from E. coli and A. actinomycetemcomitans Y4 and cultured 3 days. These cells were then co-cultured with RAW 264.7 cells for an additional 6 days and the number of multinucleated, TRAP+ cells was counted. Stimulation with LPS increased significantly the number of TRAP+ cells and this effect was inhibited by OPG *Indicates significant (p<0.05) difference from unstimulated cells and #indicates a significant decrease on the number of osteoclasts with OPG treatment. !Indicates p=0.056 for the significance of the decrease on the number of osteoclasts with OPG treatment in A. actinomycetemcomitans Y4 LPS-stimulated cells. (B). Co-culture of RAW cells with stable PDL cell lines over-expressing dominant negative mutants of MKK3 (MKK3dn-PDL) and MKK6 (MKK6dn-PDL), upstream activators of p38 MAPK, significantly decreased the number of TRAP+ cells *Indicates significant (p<0.05) difference from non-transfected PDL cells (C). Bar graphs indicate mean ± standard deviation of number of TRAP+, multinucleated cells counted in each well.
Figure 5. RAW 264.7 differentiated into TRAP+, multinucleated osteoclastic-like cells in response to exogenous RANKL or co-cultured with LPS-stimulated PDL cells.
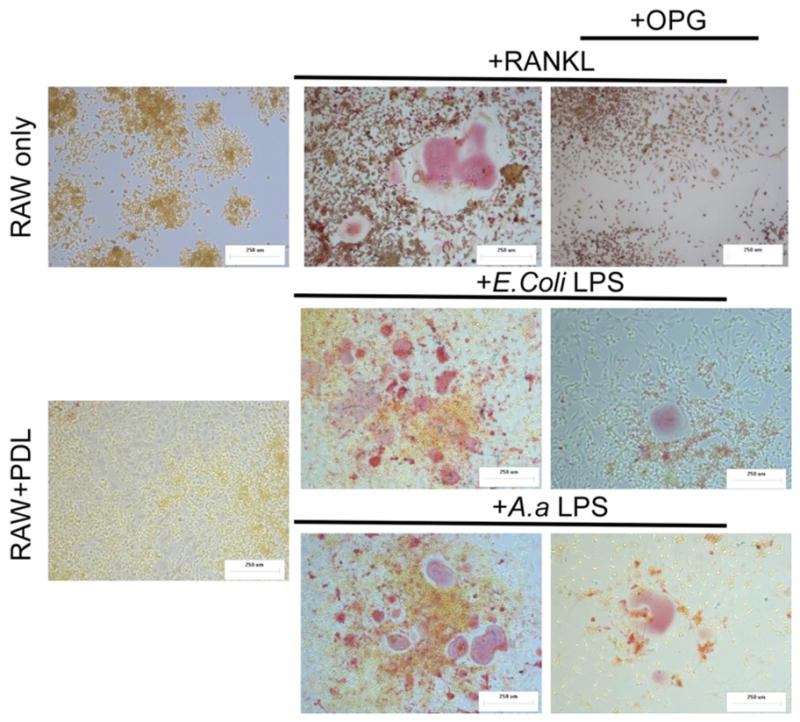
Top panel depicts RAW 264.7 cells only (5 × 104) cultured for 6 days without or with RANKL (50ng/mL) and RANKL/OPG (100ng/mL). Middle panel indicate co-culture experiments where vehicle control or LPS (1 μg/mL) treated mPDL cells were stimulated for 72h, then 5 × 104 cells were added to each well containing 5 × 104 RAW 264.7 cells. Co-cultures were maintained for 6 days, both in the presence and absence of OPG (100ng/mL) (right panels). All images are representative of three independent experiments.
Inhibition of MKK3 and MKK6 decrease osteoclastogenesis induced by LPS-stimulated PDL cells
Co-culture experiments using stable cell lines over-expressing dominant negative forms of upstream activators of p38 MAPK, MKK3 (MKK3dn-PDL) and MKK6 (MKK6dn-PDL), resulted in a significant (p<0.05) decrease on the number of TRAP+ cells in comparison to non-transformed murine PDL cell line (Figure 4C). This indicates that upstream activators of p38 MAPK are involved in LPS-induced RANKL expression by PDL cells. Furthermore, MKK3dn-PDL and MKK6dn-PDL had the same effect magnitude on inhibition of osteoclast differentiation, suggesting that both kinases are important for LPS signaling through p38 MAPK pathway. This finding might also suggest that the β isoform of p38, which is activated only by MKK6, does not play a relevant role on LPS-induced RANKL expression.
Discussion
In periodontal disease pathogenesis, microbial virulence factors or microbial-associated molecular patterns (MAMPs) induce an immune/inflammatory response that will result in bone resorption (33, 34). Understanding the molecular mechanisms involved in coupled bone turnover can have a profound impact on the development of therapeutic approaches aiming at the control of bone resorption associated with infections. PDL cells were shown to express RANKL in response to IL-1α, IL-1β, Aa LPS, vitamin D3, dexamethasone, PGE2 and mechanical stress (10, 15, 16, 35, 36). These findings illustrate the role of PDL cells in alveolar bone resorption, whereas their relevance to bone formation during periodontal regenerative therapies is also well established (37–40).
Osteoclast differentiation was shown to be related with the activation of various intracellular signaling pathways, including JNK, p38 and ERK MAP Kinases, as well as PI-3-kinase and protein kinase A (PKA) pathway (41–43). However, the role of different signaling pathways on gene expression may differ according to the nature of extracellular stimulation, cell type and even cell differentiation state (44). Supporting this concept, bacterial-induced RANKL expression by osteoblasts was found not to require p38, ERK MAP Kinases or the PI-3-kinase pathway (29), whereas p38 MAPK was recently shown to regulate RANKL mRNA and protein expression by osteoblasts stimulated by adiponectin (45). In this study we show that p38 MAP Kinase and its upstream activators MKK3 and MKK6 are required for LPS-induced RANKL expression by PDL cells. Moreover, the results of the osteoclastogenesis assays support a role for the membrane-bound form of RANKL expressed by these cells in response to LPS stimulation.
We have previously shown that LPS induces activation of p38 MAP Kinase in PDL cells (3) and in this study we demonstrated that inhibiting p38 MAP Kinase pathway not only decreases LPS-induced RANKL expression and inhibits PDL cell-induced osteoclastogenesis. It is suggested that the decrease on RANKL expression caused by inhibiting p38 and the subsequent shift on the RANKL:OPG ratio affects the osteoclastogenesis process. These results also agree with our previous data regarding RANKL expression and induction of osteoclastogenesis by bone marrow stromal cells stimulated with IL-1β and TNFα (3). Importantly, we have also shown that the physiological cues of coupled bone, namely VitD3 and PTH-induced RANKL expression and not affected by p38 inhibition. Other investigators have reported decreased osteoclastogenesis induced by RANKL-stimulated bone marrow-derived cells with p38 inhibitors (46, 47), and recently all three MAP kinases were shown to be involved in IL-1β-induced RANKL expression by PDL fibroblasts (28).
Since we evaluated only p38 MAPK, we cannot rule out a significant role for other signaling pathways in LPS-induced RANKL expression by PDL cells. Nevertheless, further confirmation for the role of p38 MAPK pathway on osteoclastogenesis supported by LPS-stimulated PDL cells was provided in this study by the use of genetic constructs for over-expression of dominant negative forms of MKK3 and MKK6, upstream activators of p38 MAPK. Interestingly, MKK3 and MKK6 were recently shown to have differential effects on osteoclast differentiation induced by RANKL in bone marrow stromal cells. Even though both MKK3 and MKK6 were important for p38 MAPK activation after RANKL stimulation, only MKK6 played a role on osteoclast differentiation induced by RANKL in bone marrow cells(48). Our finding of a similar role for MKK3 and MKK6 on osteoclast differentiation supported by LPS-stimulated PDL cells probably reflects the role of both kinases on RANKL expression by these cells, whereas RANKL signaling in osteoclast precursor cells may be affected primarily by MKK6.
Collectively, this information suggests that p38 signaling is important not only for RANKL gene expression but also for RANKL signaling. Regulation of OPG expression seems to be more complex, since we did not observe consistent changes associated with either E.coli or A. actinomycetemcomitans Y4 LPS stimulation. This agrees with the findings of Tiranathanagul et al.(36), which used human primary PDL fibroblasts and a non-commercial preparation of LPS from A. actinomycetemcomitans. Moreover, Okahashi et al. (29) also did not report regulation of OPG mRNA expression by osteoblasts after infection with P. gingivalis. On the other hand, Lossdorfer et al. (11) have shown increased OPG mRNA expression by PDL cells after stimulation with PTH, whereas RANKL mRNA expression was not detected.
The results of the osteoclastogenesis assay indicate that increase on RANKL expression induced by LPS was of sufficient magnitude to alter the RANKL: OPG ratio shifting the bone turnover towards resorption. This is in spite of no significant changes on OPG mRNA expression induced by LPS in these cells. However, the increase on osteoclast numbers induced by LPS-stimulated cells was somewhat discrete (40 and 20% for E.coli and A. actinomycetemcomitans Y4 LPS, respectively), paralleling our findings for regulation of RANKL expression at the mRNA and protein levels. Yet, these results indicate that PDL cells can have relevant consequences on bone turnover upon LPS stimulation. Wada et al.(8) have shown that unstimulated PDL cells inhibit osteoclastogenesis and attributed this effect to production of OPG. In this study we show that the osteoclastogenesis induced by LPS-stimulated PDL cells can be attributed to membrane-bound RANKL expression, since OPG blocked this effect completely and conditioned medium from LPS-stimulated cells did not induce osteoclastogenesis. This requirement of direct cell-to-cell contact between PDL and osteoclast precursor cells was first reported by Kanzaki et al. (14), however they did not use any extracellular stimulation to induce RANKL expression. There are other important methodological differences on the osteoclastogenesis model used that may account for differences between our results and those of others(17), including the nature of stimulation used to induce RANKL (mechanical compression versus LPS), the osteoclast precursor cells used (peripheral blood monocytes versus monocytic/macrophage cell line) and the time of co-culture (3 weeks versus 5–6 days). In our model, by scrapping off the PDL cells after stimulation with LPS we were able to adjust the number of cells plated onto the precursor cells to avoid influences on cell proliferation that the LPS stimulation might had on the PDL cells.
Our findings on the osteoclastogenesis assay are contrary to a report in which LPS-stimulated PDL cells did not induce osteoclast formation in a co-culture with precursor cells (49). This might be due to the different experimental models used. We used a continuous lineage of mouse PDL cells, whereas Wada et al. (49) used primary human PDL cells. In the primary cells, OPG expression was shown to be more affected than RANKL after LPS stimulation. Moreover, significant variability was shown on the level of basal and stimulated RANKL expression among the primary PDL cells, which might be responsible for the striking differences between the results of Wada et al.(49) and those of Tiranathanagul et al. (36), who reported increased RANKL expression induced by A. actinomycetemcomitans LPS in primary PDL fibroblasts, whereas OPG expression was not affected. Also note that we did experience some variability in osteoclastogenesis assays where PDL cells cultured with RAW macrophages did result in osteoclast formation on occasion. Most likely these data may be due to the passage number with the RAW cells rather than the PDL cell line. Despite this experimental baseline differences, significant stimulation of TRAP positive multinucleated cells with LPS was observed. Interestingly, the PDL cells and gingival fibroblasts seem to have opposite responses to LPS regarding osteoclastogenesis, as Nagasawa et al. (50) have shown increased expression of OPG (and not RANKL) mRNA. Cell culture supernatants of LPS-stimulated gingival fibroblasts also inhibited differentiation of precursor hematopoietic cells in response to RANKL stimulation. It is important to note that the biochemical composition of purified LPS may influence the results and explain some of the conflicting results in the literature. Even though we evaluated our A. actinomycetemcomitans Y4 LPS preparation to assure that it was not contaminated by nucleic acid or protein, the lower limits of detection of the procedures cannot absolutely rule out minor protein contamination.
Recent in vivo data from our lab group supports the role of p38 signaling which is required for periodontal bone loss in an experimental rat model (3). In this model, A. actinomycetemcomitans LPS induced alveolar bone loss was blocked by an orally-active p38 inhibitor. Reduced numbers of osteoclasts were found as well adjacent to the areas of active bone resorption including the periodontal ligament area. Other recent studies have suggested that T- and B-cell derived RANKL are the predominant sources of RANKL in human periodontal diseased tissue compared with healthy sites (22). However, this study utilized only biopsied soft tissue which did not include the periodontal ligament or associated boney tissue from diseased sites. Recently, Taubman et al. (20) discussed the relevance of immune cells, specifically B and T cells to bone resorption in periodontal disease. Although the role of B and T cells on bone resorption is compelling, it is not possible to rule out the role of resident cells, such as endothelial cells of PDL fibroblasts to bone resorption, since they are also capable of expressing functional RANKL. In the complexity of the in vivo host response to the antigens from the subgingival biofilm, it is more than likely that cytokines, chemokines and growth factors produced by resident cells will play a role in the network of events that ultimately can modulate the activity of immune cells. As evidence supporting this assumption, A. actinomycetemcomitans infection was shown to induce bone loss in SCID mice. Even though this bone loss was approximately half of that observed in SCID mice transplanted with T cells from humans with aggressive periodontitis, it was significantly greater than sham-infected control SCID mice (19). Thus, the PDL may still play a prominent source of local RANKL production within the periodontal microenvironment, especially when one considers the dynamics of the homeostasis of the periodontal ligament and the role of the cells in this tissue to regulate alveolar bone turnover. These findings suggest that p38 signaling plays a major role in controlling cytokine expression in the periodontal ligament which can contribute towards inflammatory bone loss associated with periodontal diseases.
Acknowledgments
The authors want to express their gratitude to Janice E. Berry and Dr. Martha J. Somerman for providing the mPDL cells used in this study. Grant support was provided by CAPES-Brazilian Ministry of Education #0192/03-1 (CR), FAPESP #2006/04602-1 (CR), DE14460 (KLK) and DE18290 (KLK).
References
- 1.Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276(23):20659–20672. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- 2.Jiang Y, Mehta CK, Hsu TY, Alsulaimani FF. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect Immun. 2002;70(6):3143–3148. doi: 10.1128/IAI.70.6.3143-3148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossa C, Ehmann K, Liu M, Patil C, Kirkwood KL. MKK3/6-p38 MAPK signaling is required for IL-1beta and TNF-alpha-induced RANKL expression in bone marrow stromal cells. J Interferon Cytokine Res. 2006;26(10):719–729. doi: 10.1089/jir.2006.26.719. [DOI] [PubMed] [Google Scholar]
- 4.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 5.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 6.Karring T, Nyman S, Gottlow J, Laurell L. Development of the biological concept of guided tissue regeneration--animal and human studies. Periodontol 2000. 1993;1:26–35. [PubMed] [Google Scholar]
- 7.Oshiro T, Shiotani A, Shibasaki Y, Sasaki T. Osteoclast induction in periodontal tissue during experimental movement of incisors in osteoprotegerin-deficient mice. Anat Rec. 2002;266(4):218–225. doi: 10.1002/ar.10061. [DOI] [PubMed] [Google Scholar]
- 8.Wada N, Maeda H, Tanabe K, Tsuda E, Yano K, Nakamuta H, et al. Periodontal ligament cells secrete the factor that inhibits osteoclastic differentiation and function: the factor is osteoprotegerin/osteoclastogenesis inhibitory factor. J Periodontal Res. 2001;36(1):56–63. doi: 10.1034/j.1600-0765.2001.00604.x. [DOI] [PubMed] [Google Scholar]
- 9.Fukushima H, Kajiya H, Takada K, Okamoto F, Okabe K. Expression and role of RANKL in periodontal ligament cells during physiological root-resorption in human deciduous teeth. Eur J Oral Sci. 2003;111(4):346–352. doi: 10.1034/j.1600-0722.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa T, Kikuiri T, Takeyama S, Yoshimura Y, Mitome M, Oguchi H, et al. Human periodontal ligament cells derived from deciduous teeth induce osteoclastogenesis in vitro. Tissue Cell. 2002;34(1):44–51. doi: 10.1054/tice.2002.0223. [DOI] [PubMed] [Google Scholar]
- 11.Lossdorfer S, Gotz W, Jager A. Immunohistochemical localization of receptor activator of nuclear factor kappaB (RANK) and its ligand (RANKL) in human deciduous teeth. Calcif Tissue Int. 2002;71(1):45–52. doi: 10.1007/s00223-001-2086-7. [DOI] [PubMed] [Google Scholar]
- 12.Beertsen W, van den Bos T. Alkaline phosphatase induces the mineralization of sheets of collagen implanted subcutaneously in the rat. J Clin Invest. 1992;89(6):1974–1980. doi: 10.1172/JCI115805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groeneveld MC, Everts V, Beertsen W. Alkaline phosphatase activity in the periodontal ligament and gingiva of the rat molar: its relation to cementum formation. J Dent Res. 1995;74(7):1374–1381. doi: 10.1177/00220345950740070901. [DOI] [PubMed] [Google Scholar]
- 14.Kanzaki H, Chiba M, Shimizu Y, Mitani H. Dual regulation of osteoclast differentiation by periodontal ligament cells through RANKL stimulation and OPG inhibition. J Dent Res. 2001;80(3):887–891. doi: 10.1177/00220345010800030801. [DOI] [PubMed] [Google Scholar]
- 15.Fukushima H, Jimi E, Okamoto F, Motokawa W, Okabe K. IL-1-induced receptor activator of NF-kappa B ligand in human periodontal ligament cells involves ERK-dependent PGE2 production. Bone. 2005;36(2):267–275. doi: 10.1016/j.bone.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Nukaga J, Kobayashi M, Shinki T, Song H, Takada T, Takiguchi T, et al. Regulatory effects of interleukin-1beta and prostaglandin E2 on expression of receptor activator of nuclear factor-kappaB ligand in human periodontal ligament cells. J Periodontol. 2004;75(2):249–259. doi: 10.1902/jop.2004.75.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Kanzaki H, Chiba M, Shimizu Y, Mitani H. Periodontal ligament cells under mechanical stress induce osteoclastogenesis by receptor activator of nuclear factor kappaB ligand up-regulation via prostaglandin E2 synthesis. J Bone Miner Res. 2002;17(2):210–220. doi: 10.1359/jbmr.2002.17.2.210. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D, Yang YQ, Li XT, Fu MK. The expression of osteoprotegerin and the receptor activator of nuclear factor kappa B ligand in human periodontal ligament cells cultured with and without 1alpha,25-dihydroxyvitamin D3. Arch Oral Biol. 2004;49(1):71–76. doi: 10.1016/s0003-9969(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 19.Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106(6):R59–67. doi: 10.1172/jci10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taubman MA, Valverde P, Han X, Kawai T. Immune response: the key to bone resorption in periodontal disease. J Periodontol. 2005;76(11 Suppl):2033–2041. doi: 10.1902/jop.2005.76.11-S.2033. [DOI] [PubMed] [Google Scholar]
- 21.Mogi M, Otogoto J, Ota N, Togari A. Differential expression of RANKL and osteoprotegerin in gingival crevicular fluid of patients with periodontitis. J Dent Res. 2004;83(2):166–169. doi: 10.1177/154405910408300216. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169(3):987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D, Xu JK, Figliomeni L, Huang L, Pavlos NJ, Rogers M, et al. Expression of RANKL and OPG mRNA in periodontal disease: possible involvement in bone destruction. Int J Mol Med. 2003;11(1):17–21. doi: 10.3892/ijmm.11.1.17. [DOI] [PubMed] [Google Scholar]
- 24.Iino Y, Hopps RM. The bone-resorbing activities in tissue culture of lipopolysaccharides from the bacteria Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Capnocytophaga ochracea isolated from human mouths. Arch Oral Biol. 1984;29(1):59–63. doi: 10.1016/0003-9969(84)90043-8. [DOI] [PubMed] [Google Scholar]
- 25.Kirby AC, Meghji S, Nair SP, White P, Reddi K, Nishihara T, et al. The potent bone-resorbing mediator of Actinobacillus actinomycetemcomitans is homologous to the molecular chaperone GroEL. J Clin Invest. 1995;96(3):1185–1194. doi: 10.1172/JCI118150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishida E, Hara Y, Kaneko T, Ikeda Y, Ukai T, Kato I. Bone resorption and local interleukin-1alpha and interleukin-1beta synthesis induced by Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis lipopolysaccharide. J Periodontal Res. 2001;36(1):1–8. doi: 10.1034/j.1600-0765.2001.00637.x. [DOI] [PubMed] [Google Scholar]
- 27.Nishihara T, Ueda N, Amano K, Ishihara Y, Hayakawa H, Kuroyanagi T, et al. Actinobacillus actinomycetemcomitans Y4 capsular-polysaccharide-like polysaccharide promotes osteoclast-like cell formation by interleukin-1 alpha production in mouse marrow cultures. Infect Immun. 1995;63(5):1893–1898. doi: 10.1128/iai.63.5.1893-1898.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oikawa AKM, Okamatsu Y, Shinki T, Kamijo R, Yamamoto M, Hasegawa K. Mitogen-activated protein kinases mediate interleukin-1beta-induced receptor activator of nuclear factor-kB ligand expression in human periodontal ligament cells. Journal of Periodontal Research. 2007 doi: 10.1111/j.1600-0765.2006.00959.x. in press. [DOI] [PubMed] [Google Scholar]
- 29.Okahashi N, Inaba H, Nakagawa I, Yamamura T, Kuboniwa M, Nakayama K, et al. Porphyromonas gingivalis induces receptor activator of NF-kappaB ligand expression in osteoblasts through the activator protein 1 pathway. Infect Immun. 2004;72(3):1706–1714. doi: 10.1128/IAI.72.3.1706-1714.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Errico JA, Ouyang H, Berry JE, MacNeil RL, Strayhorn C, Imperiale MJ, et al. Immortalized cementoblasts and periodontal ligament cells in culture. Bone. 1999;25(1):39–47. doi: 10.1016/s8756-3282(99)00096-4. [DOI] [PubMed] [Google Scholar]
- 31.Wilson ME, Hamilton RG. Immunoglobulin G subclass response of localized juvenile periodontitis patients to Actinobacillus actinomycetemcomitans Y4 lipopolysaccharide. Infect Immun. 1992;60(5):1806–1812. doi: 10.1128/iai.60.5.1806-1812.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittrant Y, Theoleyre S, Couillaud S, Dunstan C, Heymann D, Redini F. Relevance of an in vitro osteoclastogenesis system to study receptor activator of NF-kB ligand and osteoprotegerin biological activities. Exp Cell Res. 2004;293(2):292–301. doi: 10.1016/j.yexcr.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 33.Ekuni D, Yamamoto T, Yamanaka R, Tachibana K, Watanabe T. Proteases augment the effects of lipopolysaccharide in rat gingiva. J Periodontal Res. 2003;38(6):591–596. doi: 10.1034/j.1600-0765.2003.00694.x. [DOI] [PubMed] [Google Scholar]
- 34.Garlet GP, Martins W, Jr, Ferreira BR, Milanezi CM, Silva JS. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. J Periodontal Res. 2003;38(2):210–217. doi: 10.1034/j.1600-0765.2003.02012.x. [DOI] [PubMed] [Google Scholar]
- 35.Hasegawa T, Yoshimura Y, Kikuiri T, Yawaka Y, Takeyama S, Matsumoto A, et al. Expression of receptor activator of NF-kappa B ligand and osteoprotegerin in culture of human periodontal ligament cells. J Periodontal Res. 2002;37(6):405–411. doi: 10.1034/j.1600-0765.2002.01603.x. [DOI] [PubMed] [Google Scholar]
- 36.Tiranathanagul S, Yongchaitrakul T, Pattamapun K, Pavasant P. Actinobacillus actinomycetemcomitans lipopolysaccharide activates matrix metalloproteinase-2 and increases receptor activator of nuclear factor-kappaB ligand expression in human periodontal ligament cells. J Periodontol. 2004;75(12):1647–1654. doi: 10.1902/jop.2004.75.12.1647. [DOI] [PubMed] [Google Scholar]
- 37.Melcher AH. On the repair potential of periodontal tissues. J Periodontol. 1976;47(5):256–260. doi: 10.1902/jop.1976.47.5.256. [DOI] [PubMed] [Google Scholar]
- 38.Nyman S, Karring T, Lindhe J, Planten S. Healing following implantation of periodontitis-affected roots into gingival connective tissue. J Clin Periodontol. 1980;7(5):394–401. doi: 10.1111/j.1600-051x.1980.tb02012.x. [DOI] [PubMed] [Google Scholar]
- 39.Karring T, Nyman S, Lindhe J. Healing following implantation of periodontitis affected roots into bone tissue. J Clin Periodontol. 1980;7(2):96–105. doi: 10.1111/j.1600-051x.1980.tb01952.x. [DOI] [PubMed] [Google Scholar]
- 40.Gottlow J, Nyman S, Karring T, Lindhe J. New attachment formation as the result of controlled tissue regeneration. J Clin Periodontol. 1984;11(8):494–503. doi: 10.1111/j.1600-051x.1984.tb00901.x. [DOI] [PubMed] [Google Scholar]
- 41.Lee SE, Woo KM, Kim SY, Kim HM, Kwack K, Lee ZH, et al. The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone. 2002;30(1):71–77. doi: 10.1016/s8756-3282(01)00657-3. [DOI] [PubMed] [Google Scholar]
- 42.Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NFkappa B ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem. 2002;277(50):48868–48875. doi: 10.1074/jbc.M208494200. [DOI] [PubMed] [Google Scholar]
- 43.Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002;17(9):1667–1679. doi: 10.1359/jbmr.2002.17.9.1667. [DOI] [PubMed] [Google Scholar]
- 44.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19(4):2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo XH, Guo LJ, Xie H, Yuan LQ, Wu XP, Zhou HD, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res. 2006;21(10):1648–1656. doi: 10.1359/jbmr.060707. [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL) J Biol Chem. 2000;275(40):31155–31161. doi: 10.1074/jbc.M001229200. [DOI] [PubMed] [Google Scholar]
- 47.Mbalaviele G, Anderson G, Jones A, De Ciechi P, Settle S, Mnich S, et al. Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. J Pharmacol Exp Ther. 2006;317(3):1044–1053. doi: 10.1124/jpet.105.100362. [DOI] [PubMed] [Google Scholar]
- 48.Huang H, Ryu J, Ha J, Chang EJ, Kim HJ, Kim HM, et al. Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-kappaB transactivation by RANKL. Cell Death Differ. 2006;13(11):1879–1891. doi: 10.1038/sj.cdd.4401882. [DOI] [PubMed] [Google Scholar]
- 49.Wada N, Maeda H, Yoshimine Y, Akamine A. Lipopolysaccharide stimulates expression of osteoprotegerin and receptor activator of NF-kappa B ligand in periodontal ligament fibroblasts through the induction of interleukin-1 beta and tumor necrosis factor-alpha. Bone. 2004;35(3):629–635. doi: 10.1016/j.bone.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 50.Nagasawa T, Kobayashi H, Kiji M, Aramaki M, Mahanonda R, Kojima T, et al. LPS-stimulated human gingival fibroblasts inhibit the differentiation of monocytes into osteoclasts through the production of osteoprotegerin. Clin Exp Immunol. 2002;130(2):338–344. doi: 10.1046/j.1365-2249.2002.01990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]