

Abstract
Great insight has been gained into the structure and function of the Inositol 1,4,5 trisphosphate receptor (InsP3R) by studies employing mutagenesis of the cDNA encoding the receptor. Notably, early studies using this approach defined the key constituents required for InsP3 binding in the N-terminus and the membrane spanning regions in the C-terminal domain responsible for channel formation, targeting and function. In this article we evaluate recent studies which have used a similar approach to investigate key residues underlying the in vivo modulation by select regulatory factors. In addition, we review studies defining the structural requirements in the channel domain which comprise the conduction pathway and are suggested to be involved in the gating of the channel.
In the 20 years following the cloning of the type-1 inositol 1,4,5-trisphosphate receptor (InsP3R-1) [1] analysis of the predicted amino acid sequence has proceeded with studies of mutant InsP3R proteins in an continuing effort to link structure to function. Indeed, the initial sequence analysis of the InsP3R-1 noted several salient features of the protein; a highly positively charged region in the N-terminus, potentially constituting requirements for the putative InsP3 binding region; two Protein Kinase A (PKA) consensus phosphorylation sites in a predicted central cytoplasmic domain and a hydrophobic region in the C-terminus, indicating a putative transmembrane domain. Strong evidence for the latter prediction was that a deletion mutant lacking the C-terminal 534 amino acid residues was, in contrast to the membrane associated native protein, largely soluble [1]. Additional truncation mutations proved invaluable in assigning function to the N- and C- terminal regions of the protein. In particular, deletion of predicted transmembrane domains in the C-terminus resulted in a soluble, monomeric protein confirming that the transmembrane domains were involved in tetrameric assembly and endoplasmic reticulum localization [2]. This study also demonstrated that expression of the N-terminal 788 residues of the protein retained InsP3 binding while further truncation to residue 519 eliminated binding, indicating the boundary for the InsP3 binding domain in the N-terminus of the protein [2].
These initial findings, establishing the location of the binding site and channel have subsequently been greatly extended. Notably, using an extensive collection of glutathione S-transferase (GST) N-terminal receptor constructs and exhaustive mutagenesis, Mikoshiba and colleagues defined the binding “core” of the receptor between residues 223-608. Within this domain, the key positively charged amino acids, which coordinate the negative phosphate groups of InsP3 to mediate high-affinity specific binding were also elucidated [3, 4]. Notably, 9 of the 12 Arginine or Lysine residues identified in these studies were later confirmed to coordinate the binding of InsP3 in a crystal structure of the InsP3R binding core with InsP3 [5]. Cloning of two further family members revealed that the proteins shared high sequence similarity at the N- and C-termini coupled by a substantial intervening segment with far greater sequence diversity. This region, since termed the regulatory and coupling domain, harbors putative consensus motifs for regulation by a dizzying array of potential modulatory input, including phosphorylation, by cytoplasmic factors such as ATP and by protein binding partners (For review see [6-8]). By virtue of harboring differing subtype specific motifs, the regulatory and coupling domain has been suggested to contribute to the selective regulation of Ca2+ release through individual InsP3R family members.
Despite a wealth of information regarding some aspects of the molecular requirements of InsP3R function, as noted for example InsP3 binding, there is a relative lack of data addressing the determinants of other features of InsP3R structure and function. In particular, the residues in the C-terminal channel domain contributing to the conduction pathway, to the gating of the channel and those responsible for coupling binding in the N-terminus to opening of the pore are not established. Also neglected are studies unequivocally establishing that putative structural motifs are indeed directly responsible for the in vivo modulation of InsP3R activity. This latter issue, when investigated, has provided some surprising results. In this article, we review studies which have primarily employed InsP3R mutagenesis to gain insight into some of these unresolved issues.
The InsP3R modulatory/coupling domain-phosphorylation of InsP3R-1 by PKA
The presence of PKA consensus phosphorylation sites noted in the original reports, were consistent with the observation that the InsP3 binding protein expressed in cerebellar purkinje neurons was a known prominent substrate for PKA [9]. Indeed, in retrospect, the InsP3R-1 was known to be a phosphoprotein prior to its function as Ca2+ release channel being established having been identified in an early proteomic screen of brain phosphoproteins [10, 11]. In general, confirmation that a putative regulatory site has relevance for modulating the activity of the protein in vivo is a multi step process. In the case of a phosphorylation event, the protein must be shown to be phosphorylated and this observation correlated with an effect on protein function. Ideally, these two events should then be conclusively linked by abrogating the effect on activity by mutation of the phosphor-acceptor amino acid. In terms of PKA phosphorylation, early experiments physically demonstrated that InsP3R-1 was stoichiometrically phosphorylated at two putative phosphorylation sites (S1588 and S1755 in the mouse protein). These residues are present in the canonical PKA substrate motif (RRXS/T; R=basic residue) [12, 13] (Figure 1A/B). Moreover, it was suggested that the major peripherally expressed splice variant of the InsP3R-1, the so-called short form (S2- InsP3R-1), in which 40 amino acids (Figure 1B; 1692-1732) are excised from the protein between the two phosphorylation sites, was more readily phosphorylated in vitro at S1588 than the predominately neuronal S2+ InsP3R harboring the 40 amino acid insert which was phosphorylated at both sites [12].
Figure 1. Consensus PKA phosphorylation substrate motifs and “Walker type A” ATP binding motifs in the InsP3R isoforms.
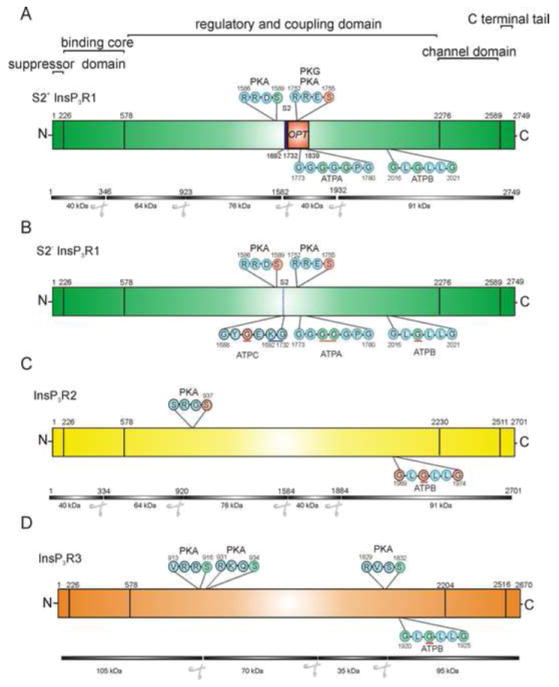
The general domain structure of InsP3R family members is shown for S2+ InsP3R1 in A; S2- InsP3R-1 in B; InsP3R-2 in C and InsP3R-3 in D. In the schematic for each receptor PKA phosphorylation sites are shown above and putative ATP binding domains are shown below. Residues colored red indicate amino acids which when mutated have functional consequences. Residues colored in green indicate amino acids which when mutated did not result in effects on InsP3R activity. Residues within the putative ATP binding domains underlined in red indicate residues which abrogate binding of ATP to GST-fusion proteins containing the particular site. In InsP3R-1 (A and B) the PKA phosphorylation sites Serine 1589 and Serine 1755 are separated by the S2 splice region shown in blue (amino acids 1692-1732) in the top panel. In B the blue underlined residues indicate the N and C terminal boundaries of the S2 splice region. The site of the in frame deletion present in the Opthisthotonos mouse (OPT; amino acids 1732-1839) which results in deletion of a phosphorylation site and the ATPA site is indicated. Below the schematic is a diagram of the fragments of InsP3R-1 produced by limited digestion with trypsin [46, 126, 127]. ATP binding was reported in the C-terminal 40 kDa and 91 kDa fragments [46]. In B, PKA sites and the complement of ATP binding motifs are shown in the S2- InsP3R-1. The amino acid numbering is based on the rat S2+ InsP3R-1. In C, a functionally important PKA phosphorylation site at Serine 937 is shown. The ATPB Walker motif is also functionally relevant in InsP3R-2. Below the schematic a diagram is shown which illustrates a subcloning strategy used to determine phosphorylation sites in InsP3R-2. The fragments of InsP3R2 were based on tryptic sites in InsP3R-1. PKA phosphorylation of the 76 kDa fragment was demonstrated [45]. In D, phosphorylation sites Serine 916, Serine 934 and Serine 1832 are shown, together with the ATPB Walker motif. Mutation of each of these sites appears to be without functional consequence. Below the schematic is a diagram depicting fragments produced by limited chymotryptic digestion of InsP3R-3 [43, 46]. ATP binding was demonstrated in the C-terminal 95 kDa fragment [46]. PKA dependent phosphorylation was shown in the N terminal 105 kDa fragment; the 70 kDa and 35 kDa fragments [43].
Numerous subsequent studies addressed the functional effects of PKA phosphorylation and resulted in contradictory findings; both potentiated and inhibitory effects on InsP3R-1 activity where reported for the S2+ InsP3R-1, while mostly inhibitory effects were reported in experiments were the S2- InsP3R-1 was presumed to be expressed [14-18]. These studies highlight the general problem of linking a regulatory event to the functional consequence; the activity of the InsP3R was measured indirectly as a net Ca2+ flux and secondly, mutagenesis of the putative site was not performed to unequivocally prove that modification was linked to the change in activity. Obviously both these issues were not trivial given the contemporary tools available, in particular, the lack of a suitable InsP3R null expression system to functionally test the effect of mutations in the receptor. A major advance to the field in this respect has undoubtedly been the creation of the DT40 3KO cell line [19-21], a unique, genetically tractable tool which has allowed unambiguous measurement of InsP3R function on a null background.
Even given this substantial biochemical and functional data, mutational analyses have provided unexpected insights into the effects of phosphorylation on InsP3R-1. A common approach has been to express and compare WT and mutant InsP3R in the DT40-3KO background. In particular these studies have unambiguously assigned the effects of phosphorylation to particular amino acids [22]. Analyses of alanine substitutions at S1589 and S1755 in the rat receptor established that, despite both sites being phosphorylated in vitro, phosphorylation of S1755 was necessary and sufficient for markedly increasing the activity of the neuronal S2+ splice variant. In contrast, phosphorylation of either S1589 or S1755 could augment the activity of the peripheral S2- isoform. Further insights into the mechanisms by which PKA enhances InsP3R-1 activity were obtained by using “phosphomimetic” glutamate substitutions at S1589 and S1755 [23]. The utility of these mutations is that any effects on activity reflect the inherent properties of the channel and are therefore independent of any cell type specific events such as targeting of PKA through anchoring proteins [24]. The glutamate substitutions exhibited increased sensitivity following agonist stimulation and direct activation by InsP3 uncaging. Once again, the S1755E mutation was sufficient for this effect in the S2+ isoform (S1589E retained activity similar to WT) and both S1589E and S1755E were required for the maximal increase in sensitivity in the context of the S2- splice variant.
Another unique attribute of the DT40-3KO cell is that direct electrophysiological measurements of InsP3R activity following expression of defined homotetrameric channels can be made in native membranes. These measurements can be performed in the “on nucleus” configuration pioneered by Foskett and colleagues [25-29] or by whole-cell measurements of channels expressed in the plasma membrane as detailed by Taylor and co-workers [30-32]. Mechanistic information regarding the enhancing effects of phosphorylation was provided by single channel measurements of plasma membrane InsP3R-1 in stable DT40 cell lines. This technique, allows for direct channel measurements from a small numbers of channels in the plasma membrane (typically less than 4) while in the whole-cell patch clamp configuration. Similar to Ca2+ imaging experiments and lipid bilayer channels measurements [16], the primary effect of the phosphomimetic mutations, like actual phosphorylation, was to cause an increase in Po at sub-saturating [InsP3]. Interestingly, there was no apparent effect on InsP3 binding or the biphasic effects of Ca2+ on the receptor. There were however, clear effects on the kinetics of channel opening and closing such that the phosphorylated channel predominately gated in a “bursting mode” characterized by extended periods of rapid channel openings and closings [33].
The InsP3R modulatory/coupling domain-phosphorylation of InsP3R-1 by PKG
The identical PKA-phosphorylation sites are also potential targets of cGMP-dependent Protein Kinase (PKG) [34-36]. Despite phosphorylation of S1755 being theoretically a preferred site of PKG due to the presence of an upstream aromatic residue (F1759), S1588 has been reported to be the predominant target of PKG in S2+ InsP3R-1 [36]. Again, there is little consensus in the literature as to the site responsible for the functional effects of PKG phosphorylation. Interestingly a similar mutational analysis indicated that the splicing of the InsP3R-1 also appeared to determine the susceptibility of phosphorylation by PKG. Activating PKG resulted in augmented Ca2+ signals in S2+ InsP3R-1 expressing cells, an effect attributed to phosphorylation of S1755. Surprisingly, PKG had no effect in cells expressing S2- InsP3R-1 [23]. The parsimonious explanation for these data, given that phosphate incorporation at either site in the S2- InsP3R-1 (following PKA activation) results in enhanced activity, is that the particular conformation of the S2- InsP3R renders the protein a poor substrate for PKG. These data also indicate that the reported functional effects of PKG activation in tissues such as smooth muscle and megakaryocytes [37] which express S2- InsP3R-1 are likely to be the consequence of phosphorylation of other InsP3R family members co-expressed. Alternatively, these effects could be mediated indirectly via an accessory protein such as InsP3R-associated cGMP kinase substrate (IRAG) [38].
The InsP3R modulatory/coupling domain- PKA phosphorylation of InsP3R-2 and InsP3R-3
Functional effects of PKA phosphorylation have also been attributed to phosphorylation of InsP3R-2 and InsP3R-3 [18, 39-42]. It should be noted however that neither receptor is stoichiometrically phosphorylated following PKA activation [18, 43]. For example, in AR4-2J pancreatoma cells, parotid acinar cells and hepatocytes expressing a predominance of InsP3R-2, an increase in receptor activity has been demonstrated following PKA stimulation [18, 44]. In contrast, in cells expressing a majority of InsP3R-3, such as pancreatic acinar cells and Rin M5F insulinoma cells, both an inhibitory and enhancing effect have been reported [18, 41, 42]. It should be noted that without exception, all these studies have again relied on indirect measurements of InsP3R activity, further confounded by the fact that all the cells studied express multiple InsP3R family members to varying extents. Moreover, although PKA consensus motifs are conserved in InsP3R-1 of all species with genomes encoding multiple InsP3R proteins, these consensus sites are absent in InsP3R-2 and InsP3R-3 protein (Figure 1C and D). Here the problem of conclusive linkage between the site responsible for cause and effect has lagged behind the determination of actual sites phosphorylated in the individual receptors.
The InsP3R modulatory/coupling domain- PKA phosphorylation of InsP3R-2
Scores of serine and threonine residues are present in minimal PKA consensus sequences (RXXS/T or RRXS/T) within the cytosolic domains of InsP3R-2 making a strictly mutagenesis approach somewhat unattractive. To circumvent this issue, a subcloning approach was taken to identify phosphorylation sites in InsP3R-2 [45]. This strategy was based on the fact that limited trypsin digestion of the InsP3R-1 or InsP3R-3 attacks solvent exposed sites and results in 5 (InsP3R-1) or 4 (InsP3R-3) globular domains [3, 43, 46]. In a similar manner to maintain tertiary structure, the sub-clones were designed around 5 predicted tryptic digest sites in InsP3R-2. Only fragments 3 and 5, corresponding to residues 920-1583 and 1884-2701 were phosphorylated in vitro. Mutation of S937A (among 5 serine residues) eliminated phosphorylation in fragment 3 while mutation of S2633A abrogated the effect in fragment 5, identifying these residues as potential PKA phosphorylation sites in InsP3R-2. S2633 is conserved in all InsP3R subtypes and has in fact been demonstrated to be phosphorylated by Akt kinase in vivo [47, 48]. Because mutation of established PKA sites in both InsP3R-1 and InsP3R-3 abolished all the effects of PKA activation, S2633 is unlikely to represent a PKA phosphorylation site in the intact protein. In contrast, S937 is unique to the InsP3R-2 (Figure 1C) and is conserved in all InsP3R-2 genes in the NCBI database. Of interest, S937 was also independently identified as a phosphorylated residue in a proteomic screen of liver phosphoproteins [49]. Subsequent experiments demonstrated that an antibody raised against phosphorylated S937 recognized a protein product of appropriate size in forskolin treated COS-7 cells expressing InsP3R-2 but not S937A InsP3R-2 and importantly, native receptor in forskolin treated parotid acinar cells. Finally, PKA activation resulted in enhanced Ca2+ signals and single InsP3R-2 channel activity in DT40-3KO cells expressing InsP3R-2 but the effect was absent in S937A InsP3R-2 expressing cells [45]. These experiments, taking a deliberate methodological approach establish that S937 is a bone fide, functionally relevant phosphorylation site in InsP3R-2. Further, work is obviously necessary to establish the physiological role of this regulation in cells which express predominately InsP3R-2.
The InsP3R modulatory/coupling domain- PKA phosphorylation of InsP3R-3
Wojcikiewicz and colleagues have reported that of 33 serines and 18 threonine residues present in minimal PKA consensus motifs of InsP3R-3, only S–A substitutions in three sites (S916, S934, S1832) reduced phosphorylation of InsP3R-3 in vivo (Figure 1D) [43]. These sites appear to represent the sole sites in the intact InsP3R-3 as a construct harboring S-A mutations in each of the three serine residues is not phosphorylated following PKA activation. Interestingly, while S934 in particular was phosphorylated prominently in DT40-3KO cells stably expressing InsP3R-3, no effect on Ca2+ release stimulated by activation of endogenous DT40 cell phospholipase C-coupled receptors was observed in populations of cells [50]. These data again highlight that the physical demonstration of a phosphorylation event does not necessarily correlate with effects on Ca2+ release. Further experiments directly measuring channel activity are necessary to rule out that PKA-phosphorylation of InsP3R-3 results in subtle effects on channel activity not evident in population [Ca2+] measurements. An alternative is that phosphorylation of InsP3R-3 has consequences other than directly effecting Ca2+ flux for example, by influencing the complement of interacting proteins/factors and thus altering function as a signaling integrator and adaptor protein.
The InsP3R modulatory/coupling domain-regulation by adenine nucleotides
A further example where mutagenesis of “established” regulatory sites has yielded unexpected results is the modulation of receptor activity by adenine nucleotides. This form of modulation may be important in a variety of physiological and pathological situations by providing direct coupling between the metabolic status of the cell and Ca2+ release. ATP was initially demonstrated to increase Ca2+ flux [51] and the InsP3-gated activity of channels from smooth muscle [52]. Subsequently, it was established that 8-azido-α32P-labelled ATP bound stoichiometrically to the purified cerebellar InsP3R at a single binding site [53] and was a potent modulator of InsP3-induced calcium release and channel activity via the reconstituted receptor [53, 54]. A number of studies documented that sub-millimolar concentrations of ATP (and other adenine nucleotides) increase the activity of the channel, while higher concentrations appear to inhibit [55, 56]. ATP modulates all InsP3R family members, albeit with InsP3R specific characteristics [57-59]. For example, in Ca2+ release assays, where Ca2+ is clamped to ~ basal cytosolic levels, ATP appears to modulate Ca2+ release through InsP3R-1 and InsP3R-3 at all [InsP3] while InsP3R-2 is only subject to modulation at sub-maximal [InsP3] [59, 60]. In isolation, the individual receptors also differ in their affinities for ATP modulation, with InsP3R-2 having a ~10 fold higher sensitivity than InsP3R-3 and InsP3R-1 exhibiting intermediate affinity [55, 57-60]. Consistent with these data, native cells expressing a majority of InsP3R-2 are more sensitive to ATP modulation than cells expressing predominately InsP3R-3 [61]. Mechanistically, the inhibitory effect is thought to occur as high [ATP] inhibit InsP3 binding to the receptor [55, 56] while the increase in activity occurs as ATP allosterically modulates the Ca2+ co-agonist sensitivity of the InsP3R [27, 28]. The effects of ATP do not require hydrolysis [54-56], but are thought to be mediated by ATP binding to specific sites in the regulatory domains of the receptors [6, 25, 62].
It has been proposed, and widely accepted that ATP binds to glycine rich regions containing the sequence G-X-G-X-X-G (Figure 1). This sequence is reminiscent of Walker type A motifs common in many ATPase and GTPases [6, 25, 62, 63]. The primary sequence of InsP3R family members contains a number of such motifs (Figure 1A-D). A region termed the ATPB site is conserved in all three family members (aa 2016-2021 in InsP3R-1), the so-called ATPA site is unique to the InsP3R-1 (aa 1773-1780; Figure 1 A-D) and a further site termed ATPC is introduced by excision of the S2 splice site forming S2- InsP3R-1 (aa 1574-1765; Figure 1B). It should be noted that it has been demonstrated quite convincingly that these sites in isolation bind ATP. For example, GST fusion proteins harboring the particular sites have been shown to bind fluorescent TNP-ATP or α32P-ATP specifically [59, 64, 65]. Furthermore, mutation of the binding sites by a G-A substitution in the putative ATP binding pocket eliminates binding in this context [59, 65]. Perhaps the most compelling evidence for the relevance of these sites are data which show that 8-azido α32P-ATP can be cross-linked to recombinant InsP3R which when subjected to controlled proteolysis is found in fragments which are predicted to contain the glycine rich motifs [46]. Specifically, InsP3R-1 was shown to have two binding sites in fragments containing ATPA and ATPB (Figure 1A), while InsP3R-3 bound one ATP molecule in a fragment predicted to harbor ATPB (Figure 1D) [46]. No mutagenesis was carried out in these studies and thus it is formally possible that sites other than GXGXXG motifs are present in these fragments. The presence of two putative binding sites in InsP3R-1 has also been used to explain the higher functional sensitivity of the InsP3R-1 to ATP when compared with InsP3R-3. This explanation is consistent with the reduced functional sensitivity to ATP modulation of the Opisthotonos (opt) mutation in InsP3R-1 which results in a deletion of 107 amino acids including the ATPA site [66]. This mutation does however result in deletion of an extended stretch of amino acids (removal of G1732-Q1839) and thus a possibility exists that disruption of overall InsP3R structure in opt alters ATP binding outside the immediate deleted region.
Role of the ATPC site in S2- InsP3R-1
Despite the weight of persuasive evidence that these domains bind ATP and that the presence of these sites correlates with regulation of function, a firm causal link is missing in the absence of mutagenesis in the full length receptors. Efforts to address this issue have been made by mutating all recognized ATP binding motifs in each of the InsP3R family members. The primary motivation for this work was to unequivocally establish that the effects of ATP binding at particular sites indeed leads to regulation of InsP3R function. Since the creation of the ATPC site in S2- InsP3R-1 is the only obvious structural difference between the S2+ and S2- splice variants this alteration could potentially contribute to the previously noted splice specific regulation of these receptors [22, 66, 67]. To confirm nucleotide binding to this putative site, TNP-ATP was shown to specifically interact with GST fusion proteins containing the ATPC site. Importantly, this binding was eliminated by substitution of the central glycine residue for alanine in the binding motif (G1690A). In cells expressing S2- InsP3R-1, Ca2+ release was shown to be markedly increased by ATP, however the mutation of ATPC in the full length S2- InsP3R-1 (ΔATPC S2- InsP3R-1) did not alter this modulation. Instead, the mutation eliminated PKA phosphorylation and the resultant potentiated Ca2+ release [65]. The conclusion from these data was two-fold; that ATP binding to ATPC appears to be necessary for a conformation of the S2- InsP3R-1 which is susceptible to PKA phosphorylation and secondly that ATP regulation of Ca2+ release per se occurs at an alternative site, presumably the ATPA or ATPB also present in InsP3R-1.
Role of the ATPB site in InsP3R-2 and InsP3R-3
Experiments addressed whether the single conserved ATPB site in InsP3R-2 and InsP3R-3 is responsible for ATP regulation of Ca2+ release in these receptors [59]. To ensure disruption of the ATP binding pocket each glycine residue in InsP3R-2 ATPB was replaced with alanine (Figure 1C; G1969A, G1971A and G1974A; termed ΔATPB InsP3R-2). This construct eliminated ATP regulation of InsP3-induced Ca2+ release. In addition, both the oscillation frequency and amplitude of Ca2+ signals following B cell receptor (BcR) activation were reduced in cells expressing ΔATPB InsP3R-2 when compared to WT InsP3R-2. These data confirm that the ATPB site in the context of InsP3R-2 is solely responsible for mediating the effects of ATP binding and that nucleotide binding has functional consequences after stimulation of an endogenous signaling pathway. In contrast, in the InsP3R-3 a similar mutation in ATPB (Figure 1D; G1920A, G1922A, G1925A; termed ΔATPB InsP3R-3) regulation of InsP3-induced Ca2+ release and BcR-induced Ca2+ signaling were unchanged [59]. These data indicate that despite evidence of ATP binding in a region that contains the ATPB site that the site itself is functionally irrelevant for regulation of Ca2+ release via InsP3R-3. This finding although surprising, nevertheless appears in hindsight, consistent with InsP3R-2 and InsP3R-3 having markedly different characteristics of ATP modulation [58, 59].
Role of the ATPA and ATPB site in S2+ InsP3R-1
In a similar fashion to InsP3R-3, mutation of the InsP3R-1 to create ΔATPB InsP3R-1 (G2016A, G2018A, G2021A InsP3R-1) resulted in no obvious alteration in the sensitivity of Ca2+ release to ATP [60]. Given the presumed higher affinity of the ATPA site potentially masking any role of ATPB these data were not unexpected. Remarkably, however when S2+ InsP3R-1 was constructed devoid of any known ATP binding sites (mutation of G1773, G1775A,G1777A, G1780A in ATPA and G2016A, G2018A, G2021A in ATPB; S2+ InsP3R-1 ΔATPA/B) the modulation of Ca2+ release by ATP was unaltered. Indeed, at the single channel level, monitored in the “on-nucleus patch” configuration, modulation of Po by ATP, the single channel conductance and the predominant mode of gating of the mutant channel were identical to native S2+ InsP3R-1 [60]. The inescapable conclusion from these data are that the known ATP binding sites in InsP3R-1, similar to InsP3R-3, are not responsible for mediating the functional effects of ATP binding on Ca2+ release. Given the absence of any further known predicted nucleotide binding sites in the primary sequence, a novel ATP binding sequence must mediated these effects. A possibility is that the interaction is dependent on the tertiary structure of the InsP3R. Alternatively, a cryptic ATP binding site may be exposed following the large conformational changes the receptor undergoes when binding Ca2+ [68-70]. Conceivably, the functional effects of ATP could also be mediated through a tightly bound accessory protein.
Mutagenesis of The InsP3R channel domain
The original prediction of six transmembrane (TM) domains in the C-terminal segment of the receptor based on hydropathy analysis has been confirmed experimentally [71-73]. Mutagenesis within the C-terminal channel domain (extending from ~amino-acid 2270-2749 of InsP3R-1) has primarily been carried out to investigate the permeation/gating mechanisms of the channel [74-79], to study the role of the transmembrane domains in oligomerization [72, 80, 81], and to explore potential regulatory protein-protein interactions of the cytosol-exposed C-terminal tail [16, 82-84]. In general, studies on the conduction mechanism of InsP3R channels have been hampered by the lack of a detailed knowledge of the three dimensional structure of the channel domain, as well as the absence of convenient screening methods to analyze the behavior of large numbers of mutant channels. Hence, studies using mutagenesis approaches on the channel domain are quite limited. Figure 2A indicates the location of point mutations that have been engineered in the InsP3R-1 pore domain together with their functional consequences. For comparison, the corresponding mutations in ryanodine receptors (RyR) are also shown. The information obtained from these mutations are summarized and discussed below.
Figure 2. The pore and C-terminal tail of InsP3 receptors.
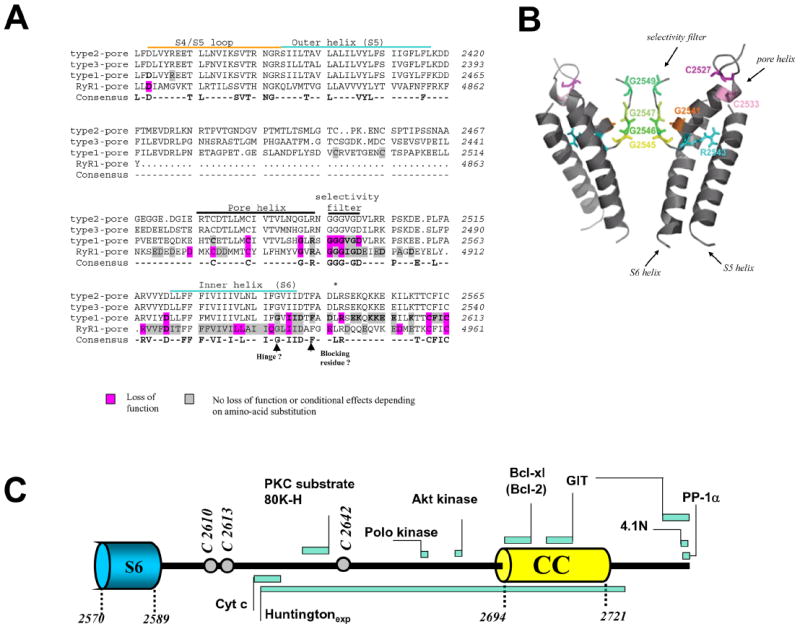
Panel A shows the sequence of a portion of the pore region of InsP3Rs. The consensus residues shown in bold are identical in all 3 InsP3R isoforms and all 3 RyR isoforms although only the sequence of RyR1 is shown in the Figure. Residues which have been mutated in InsP3R1 are shown together with their functional consequences. The analogous mutations in RyRs are shown. For additional information see text and Ref. (9). The R2596 position is indicated by an asterisk. Panel B A homology model of the InsP3R based on a KirBac 1.1 template is shown. The image is taken from Ref. (9) and is used with permission. Only the location of selected mutations from Panel A are shown. The linkers between S5 and the pore helix, and between the selectivity filter and S6, were not included in the model. Panel C shows a cartoon of the C-terminal tail with endogenous cysteines indicated by open circles and bars to indicate the approximate location of interacting proteins. CC = coiled coil domain. GIT; G-protein coupled receptor kinase-interacting protein, PP1-a; protein phosphates 1-a, 4.1N; neuron specific isoform of erythrocyte protein band 4.1. For additional details see text.
Permeation and selectivity of InsP3R channels
A basic feature of InsP3R and related ryanodine receptor (RyR) channels are high divalent cation conductances (~50pS with 50 mM Ca2+), even higher monovalent cation conductances (200-350pS with 140mM K+), and only modest discrimination between divalent and monovalent ions (PCa2+/K+ ~6-8) (reviewed in [25]). It has recently been suggested that the high K+ conductance of RyR and InsP3R may yield an unexpected benefit in permitting these channels to provide their own countercurrent to compensate for the charge movement due to Ca2+ release from intracellular stores [85]. In contrast to K+ channels [86] and voltage-gated Ca2+ channels [87], there is no compelling evidence for multi-ion occupancy of the channel, suggesting that electrostatic repulsion between ions residing in the pore is probably not the mechanism that accounts for the prodigious ion flux in InsP3R or RyR channels (but cf.[88]). This, and other biophysical properties, have led to the prediction that these channels would have short/wide pores with rings of negatively charged residues at either end to facilitate the rapid delivery and exit of ions through the pore [25, 89].
Our knowledge of InsP3R pore structure is primarily based on comparisons to the several K+ channels for which crystal structures are available. The retention of channel activity in an InsP3R mutant from which TM 1-4 have been deleted strongly suggests that the pore of InsP3R is distal to TM 5 [75]. Within the primary sequence of this region a number of structural features common to many other channels can be recognized. These include sequentially: an outer helix (TM 5), a pore helix, a selectivity filter and an inner helix (TM 6). A homology model of the closed InsP3R pore has been constructed by da Fonseca and Morris using the KirBac1.1 K+ channel as template [76] and is shown in Figure 2B. Several of the residues that are completely conserved in all three InsP3R and RyR isoforms within the pore-helix/ selectivity filter/ TM 6 segment have been mutated to alanine [76]. Not unexpectedly many of these mutations entirely eliminate channel function – presumably, as a result of disrupting critical structures such as the pore-helix or the selectivity filter. In particular, the removal of a negative charge at the luminal mouth of the receptor (D2550A) inactivates the channel. This mutant has been widely used by investigators as a “pore-dead” version of InsP3R [33, 90-94]. It is of interest that mutation of the analogous aspartate in RyR1 (D4899A) or RyR2 (D4831A) does not completely inactivate the channel [95-97], possibly because there is an adjacent glutamate residue that may fulfill a similar role. In addition, RyR possess a second pair of negative charges one residue away from the first pair, which in InsP3R are both positively charged residues (Figure 2A). The distribution of negative charges at the luminal entrance is therefore very different between the two channels and the implications of this for ion permeation and selectivity remain to be investigated. Surprisingly, the mutation of positively and negatively charged residues to alanine at the cytosolic exit of the channel has little impact on channel function with the exception of R2596 which is the only charged residue that is conserved in all InsP3R and RyR isoforms (asterisk in Figure 2A).
The selectivity filter of the InsP3R channel has the “signature sequence” GGGVG with the valine replaced by an isoleucine in RyR channels. Mutation of the first three glycines to alanine inactivates the InsP3R channel whereas mutation of the fourth glycine does not [76]. Substitution of the bulky valine for the smaller isoleucine in InsP3R increased K+ conductance to that seen in wild type RyR without changing selectivity [30, 33, 74]. It has been argued, based on modeling and molecular dynamics simulations, that the rather wide pore diameter of RyR (~7Å; [89]) and InsP3R (6.5Å; [98]) means that the selectivity filter actually exerts little effect on channel selectivity [99]. Instead, the density of negative charges at the luminal entrance has been proposed to play the major role in discriminating monovalent from divalent cations [89, 99, 100]. Some support for this hypothesis is the finding that the conservative substitution D2550E eliminated selectivity without altering K+ conductance [74].
Some of the caveats of mutagenesis studies in InsP3R can be illustrated with specific examples. The positioning of a highly conserved positively charged residue (R2543) at the C-terminal end of the pore helix would not seem conducive to the rapid movement of positively charged ions through the channel. However, mutation of the residue to alanine has little effect in 45Ca2+ flux assays, suggesting that the side-chain projects away from the permeation path as predicted by the homology model [74, 76]. Surprisingly, the R2543A mutation alters Ca2+ regulation of the receptor [74, 76]. That mutations within the pore of the channel can alter Ca2+ regulation is not without precedent in RyR studies [97] and could indicate that some point mutations can induce global conformational changes in the protein that have secondary regulatory effects. Another concern relates to the methods used to assay the function of InsP3R channels. Two glycine mutants in the selectivity filter were shown to be functional (G2549A) or non-functional (G2546A) in 45Ca2+ flux assays and when anti-IgM was used to stimulate Ca2+ release in DT40 cell lines stably expressing the mutants [97]. However, both mutants were equally active when assayed by measuring K+ conductance in patch clamp studies of the receptors expressed in plasma membranes of DT40 cells [97]. Thus the effects of mutagenesis on the conduction properties of the channel may differ if Ca2+ or K+ is monitored as the permeant ion.
Gating mechanism of the channel
The binding of InsP3 at the N-terminal ligand binding domain (LBD) induces a conformational change in the receptor [101, 102] but how this change is transmitted to the opening of the channel in the C-terminal segment of the protein is unknown. Early studies based on cross-linking and co-immunoprecipitation assays suggested that a 40 kDa N-terminal tryptic fragment on one monomeric subunit can be directly cross-linked to a C-terminal 95 kDa tryptic fragment of an adjacent subunit [103]. This supports the idea of a direct interaction between LBD and channel domains. Based on experiments with fusion proteins and mutagenesis studies of full-length receptors, the site of interaction in the channel domain has been narrowed to the cytosol-exposed loop between TM 4 and TM 5. This conclusion is supported by experiments in which the deletion of two 10 amino-acid segments, which together constitute the majority of the TM4,5-loop, disrupted channel function and the interaction between N and C-terminal tryptic fragments [77]. Point mutations indicate that the C-N interactions may involve polar residues particularly in the proximal portion of the TM4,5 loop [77]. By analogy with other channels [104-106] it has been proposed that the TM4,5 loop is an amphipathic helix lying parallel to the membrane that constricts the cytosolic face of the TM 6 pore-lining helix and therefore maintains the channel in its closed state [77]. The conformational change resulting from binding of InsP3 leads to the mechanical movement of the TM4,5 loop that allows the TM 6 domains to separate and permits ion flux. A mutant InsP3R from which the TM 1-4 helices have been removed generates a constitutively open channel [75]. Although the TM 4,5 loop is retained in this mutant, it is unlikely to be oriented correctly to perform its normal function of compressing the TM 6 helices and this probably accounts for the constitutive activity of the mutant.
The N-terminal segments of the receptor that interact with the TM 4,5 loop have not been identified but a possible candidate is the suppressor domain (aa1-223). Deletion of this domain enhances InsP3 binding to the receptor and eliminates channel function [79]. Interestingly, the TM4,5 loop deletion mutants also display enhanced InsP3 binding, supporting the view that the loop and the LBD can influence each other [77]. Binding measurements of synthetic InsP3 ligands to InsP3R constructs retaining or lacking the suppressor domain have led Taylor & colleagues [107] to propose that the energy from InsP3 binding is used to re-arrange an intramolecular interaction between the suppressor domain and the inner ligand-binding core. Furthermore, they conclude that coupling to the channel occurs exclusively through the suppressor domain. This is consistent with the finding that mutations in the suppressor domain that diminish the ability of the suppressor domain to inhibit InsP3 binding also decrease the open probability of the channel [107]. Despite these studies there is no direct experimental evidence that the suppressor domain is the N-terminal interaction site of the TM 4,5 loop. Alternative candidates could include other portions of the LBD, such as the α-helical domain (aa 427-605), which has the ability to gate endogenous InsP3R when targeted to ER membranes [108]. Uchida et al., reported [79] that deletion of a segment from the regulatory domain (aa 651-1130) interferes with channel function but this mutation also showed a large decrease in InsP3 binding affinity which could be indicative of gross structural defects in the protein. Many of the mutations that have been used to study gating have involved large deletions of the full-length protein. A point mutation in Drosophila InsP3R (corresponding to S217F in the rat InsP3R-1 sequence) shows an enhanced affinity for InsP3 in Ca2+ flux assays without significant changes in InsP3 binding affinity [78]. This mutation has therefore been characterized as a gain-of-function “gating” mutation. Additional point mutations in the suppressor or regulatory domains that specifically interfere with gating, without affecting InsP3 binding, remain to be identified.
In common with other voltage-gated channels a major barrier to ion-flux is expected to be located at the constriction formed at the cytosolic end of the pore-lining TM 6 helices (Figure 2B). Identifying the residue(s) at the channel gate is problematic since the exact boundaries of the pore-lining helix have not been established. Many channels have bulky, hydrophobic residues at the gate and mutation of these residues to alanine disrupts channel function [109, 110]. There is a conserved phenylalanine residue at the end of the TM 6 helix that could potentially play such a role. However, the mutation F2592A does not inactivate the channel, suggesting that the gate of InsP3R may be located elsewhere [76]. Crystal structures of open K+ channels suggest that bending of the inner pore-lining helix at a conserved glycine is part of the gating mechanism [109, 111]. A conserved glycine that could potentially function as a “gating hinge” is present in TM 6 but its mutation (G2586A) also does not inactivate the channel [76]. This suggests that if InsP3R possess a gating hinge it is probably not located at G2586. Nevertheless, the deliberate introduction of a bend in the helix with a G2586P substitution does produce a constitutively open channel [76]. It is possible that the TM 6 helix is inherently flexible and movement at a hinge may not be involved in the gating process, as suggested for the hERG K+ channel [112].
Properties of the C-terminal tail
The C-terminal tail consists of ~160 amino-acids projecting into the cytosol from the putative boundary of the pore-lining TM 6 helix. Interestingly, the C-terminal tail of RyR are significantly shorter (~100 amino-acids) suggesting that this portion of the protein may have distinct functions in InsP3R and RyR. Initial studies indicated that deletion of the terminal 13 amino-acids from the InsP3R had no effect but deletion of 139 amino-acids eliminated channel function [16, 79]. A study using progressive deletion mutants found that channel function was lost somewhere between removal of 43 and 60 amino-acids. This study also showed that the C-tail contained a coiled-coil domain in the region being deleted and that a fusion protein of the coiled-coil could form tetramers in vitro [77]. Whether this occurs in full-length InsP3R in vivo is not known but there is evidence that the C-tail does contribute to the stability of the InsP3R tetramer [80]. Two highly conserved cysteine residues in the tail (C2610, C2613) have been identified whose mutation to serine or alanine inactivates the channel [76, 79]. The proximity of these residues to the terminus of the TM 6 helix has led to the suggestion that these residues may function as “gate keepers” [79]. The exact role played by the cysteines or the coiled-coil domain in channel function has not been established but it is likely that they are critical to maintaining the architecture of the C-terminal tail and providing a normal exit pathway for the conducting ion. In other channels [96, 113], including RyR [114], there is evidence for salt-bridge interactions between charged residues in the TM 4,5 loop and the C-tail. Additional approaches are required to assess whether this occurs in InsP3R and to determine how such interactions may contribute to the mechanics of the gating process.
The C-tail is also the site of interaction of a number of regulatory proteins (Figure 2C) and the phosphorylation site of Akt kinase (S2618) [47, 48] and Polo kinase (T2656) [115]. The interacting proteins cover a range of sizes and include: cytochrome c (12kDa, [116]), BclXl (26kDa, [84]), PP1α (37kDa, [16], Huntington-associated protein 1A (70kDa,[83]), 80K-H protein kinase C substrate (80kDa, [117]), G-protein coupled receptor kinase-interacting protein (95kDa; [118]) and 4.1N (98kDa; [119]). The localization of the binding sites to specific regions of the C-tail has been carried out using C-tail fusion proteins or yeast 2-hybrid assays. It is generally assumed that these relatively large proteins have free access to the entire length of the C-tail in the intact receptor. However, reaction of cysteine substitution mutants introduced into the C-tail of full length receptors with 5kDa PEG-maleimide suggest that freely accessible residues are limited to the terminal ~35 amino-acids. Using this assay, Ca2+-induced conformational changes in the receptor were observed to enhance accessibility of the C-tail[68]. Structural information on the C-tail, particularly its orientation with respect to the membrane and the rest of the receptor, is needed to determine its role in InsP3R function and regulation.
Properties of the intraluminal loop
The intraluminal loop between TM 5 and the start of the pore-helix is ~62 amino-acids long in InsP3R compared to only ~14 amino-acids in RyR. Although the sequences show considerable variability between the three InsP3R isoforms, all three receptors contain 2 highly conserved cysteines in the loop (C2596, C2504 in InsP3R-1). Mutation of neither cysteine affects channel function [120]. The loop has been proposed to be the binding site for ERp44, an ER luminal protein of the thioredoxin family that inhibits InsP3R channel function [120]. The binding appears to require both cysteines to be in their reduced state, although other determinants for binding must be involved since association with ERp44 is specific to the InsP3R-1 isoform [120]. Recent studies have shown that the cysteines can form an intramolecular disulphide bond, at least in fusion proteins encoding the loop [121]. The disulphide bond status of the cysteines in full-length receptors and their role in redox regulation of InsP3R in intact cells remain to be evaluated.
Concluding Remarks
Clearly, analysis of mutant InsP3R family members using a combination of Ca2+ flux biochemical and electrophysiological techniques has yielded valuable information regarding InsP3R structure and function. Nevertheless, future molecular modeling based on more detailed knowledge of the 3 dimensional structure coupled with functional analysis of mutant receptors will undoubtedly be pivotal in addressing important remaining questions. Again, a fruitful approach may be to use related ion channels and shared protein binding partners as a framework for design of constructs. Obvious subjects to address include the sites of regulation of InsP3R by Ca2+. Mutation of E2100 in InsP3R and corresponding conserved residues in RyR, significantly alters Ca2+ sensitivity of Ca2+ release [122, 123]. However, this single residue is unlikely to represent the sole determinant of the complex effects of Ca2+ on InsP3R. Because of the critical importance of Ca2+ as a co-agonist at the InsP3R, further defining regions of the protein responsible for this action is vital for an understanding of receptor function. By analogy with other Ca2+ binding motifs this will probably entail defining a pocket formed by coordinating residues within the 3 dimensional structure of the protein. Similarly, further work is also needed to gain a thorough understanding of the gating mechanism of the receptor, in particular an expanded characterization of the residues necessary for coupling InsP3 binding to channel opening.
An additional approach is to exploit InsP3R isoform specific differences in structure and function by the generation of chimerical receptors. For example, this paradigm has been used to investigate the particular molecular motifs responsible for the distinct InsP3 binding affinities and Ca2+ sensitivities exhibited by individual InsP3R isoforms [58, 124, 125]. An obvious further use for this approach is in elucidating legitimate nucleotide binding sites in InsP3R-1 and InsP3R-3. To this end an approach using the template of InsP3R-2 ΔATPB, which is not regulated by ATP, [59] and serial substitution of portions of InsP3R-1/-3 may be valuable in identifying the sites of nucleotide regulation of these isoforms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–8. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 2.Mignery GA, Sudhof TC. The ligand binding site and transduction mechanism in the inositol-1,4,5-triphosphate receptor. Embo J. 1990;9:3893–8. doi: 10.1002/j.1460-2075.1990.tb07609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshikawa F, Iwasaki H, Michikawa T, Furuichi T, Mikoshiba K. Cooperative formation of the ligand-binding site of the inositol 1,4, 5-trisphosphate receptor by two separable domains. The Journal of biological chemistry. 1999;274:328–34. doi: 10.1074/jbc.274.1.328. [DOI] [PubMed] [Google Scholar]
- 4.Yoshikawa F, Morita M, Monkawa T, Michikawa T, Furuichi T, Mikoshiba K. Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. The Journal of biological chemistry. 1996;271:18277–84. doi: 10.1074/jbc.271.30.18277. [DOI] [PubMed] [Google Scholar]
- 5.Bosanac I, Alattia JR, Mal TK, Chan J, Talarico S, Tong FK, Tong KI, Yoshikawa F, Furuichi T, Iwai M, Michikawa T, Mikoshiba K, Ikura M. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 6.Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 1999;25:247–64. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- 7.Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem. 2004;73:437–65. doi: 10.1146/annurev.biochem.73.071403.161303. [DOI] [PubMed] [Google Scholar]
- 8.Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005;38:261–72. doi: 10.1016/j.ceca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto H, Maeda N, Niinobe M, Miyamoto E, Mikoshiba K. Phosphorylation of P400 protein by cyclic AMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase II. J Neurochem. 1989;53:917–23. doi: 10.1111/j.1471-4159.1989.tb11792.x. [DOI] [PubMed] [Google Scholar]
- 10.Walaas SI, Nairn AC, Greengard P. Regional distribution of calcium- and cyclic adenosine 3’:5’-monophosphate-regulated protein phosphorylation systems in mammalian brain. I. Particulate systems. J Neurosci. 1983;3:291–301. doi: 10.1523/JNEUROSCI.03-02-00291.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walaas SI, Nairn AC, Greengard P. PCPP-260, a Purkinje cell-specific cyclic AMP-regulated membrane phosphoprotein of Mr 260,000. J Neurosci. 1986;6:954–61. doi: 10.1523/JNEUROSCI.06-04-00954.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danoff SK, Ferris CD, Donath C, Fischer GA, Munemitsu S, Ullrich A, Snyder SH, Ross CA. Inositol 1,4,5-trisphosphate receptors: distinct neuronal and nonneuronal forms derived by alternative splicing differ in phosphorylation. Proc Natl Acad Sci U S A. 1991;88:2951–5. doi: 10.1073/pnas.88.7.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun. 1991;175:192–8. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- 14.Nakade S, Rhee SK, Hamanaka H, Mikoshiba K. Cyclic AMP-dependent phosphorylation of an immunoaffinity-purified homotetrameric inositol 1,4,5-trisphosphate receptor (type I) increases Ca2+ flux in reconstituted lipid vesicles. The Journal of biological chemistry. 1994;269:6735–42. [PubMed] [Google Scholar]
- 15.Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proc Natl Acad Sci U S A. 1988;85:8747–50. doi: 10.1073/pnas.85.22.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase a and protein phosphatase 1alpha. J Neurosci. 2003;23:403–15. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tertyshnikova S, Fein A. Inhibition of inositol 1,4,5-trisphosphate-induced Ca2+ release by cAMP-dependent protein kinase in a living cell. Proc Natl Acad Sci U S A. 1998;95:1613–7. doi: 10.1073/pnas.95.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wojcikiewicz RJ, Luo SG. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. The Journal of biological chemistry. 1998;273:5670–7. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- 19.Kubista H, Hawkins T, Moss SE. Characterisation of calcium signalling in DT40 chicken B-cells. Biochim Biophys Acta. 1998;1448:299–310. doi: 10.1016/s0167-4889(98)00132-3. [DOI] [PubMed] [Google Scholar]
- 20.Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. Embo J. 1999;18:1303–8. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugawara H, Kurosaki M, Takata M, Kurosaki T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. Embo J. 1997;16:3078–88. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner LE, 2nd, Li WH, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2- splice variants. The Journal of biological chemistry. 2003;278:45811–7. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- 23.Wagner LE, 2nd, Li WH, Joseph SK, Yule DI. Functional consequences of phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. The Journal of biological chemistry. 2004;279:46242–52. doi: 10.1074/jbc.M405849200. [DOI] [PubMed] [Google Scholar]
- 24.Tu H, Tang TS, Wang Z, Bezprozvanny I. Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. The Journal of biological chemistry. 2004;279:19375–82. doi: 10.1074/jbc.M313476200. [DOI] [PubMed] [Google Scholar]
- 25.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mak DO, Foskett JK. Single-channel inositol 1,4,5-trisphosphate receptor currents revealed by patch clamp of isolated Xenopus oocyte nuclei. The Journal of biological chemistry. 1994;269:29375–8. [PubMed] [Google Scholar]
- 27.Mak DO, McBride S, Foskett JK. ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca(2+) activation. The Journal of biological chemistry. 1999;274:22231–7. doi: 10.1074/jbc.274.32.22231. [DOI] [PubMed] [Google Scholar]
- 28.Mak DO, McBride S, Foskett JK. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J Gen Physiol. 2001;117:435–46. doi: 10.1085/jgp.117.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mak DO, McBride S, Raghuram V, Yue Y, Joseph SK, Foskett JK. Single-channel properties in endoplasmic reticulum membrane of recombinant type 3 inositol trisphosphate receptor. J Gen Physiol. 2000;115:241–56. doi: 10.1085/jgp.115.3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dellis O, Dedos SG, Tovey SC, Taufiq UR, Dubel SJ, Taylor CW. Ca2+ entry through plasma membrane IP3 receptors. Science. 2006;313:229–233. doi: 10.1126/science.1125203. [DOI] [PubMed] [Google Scholar]
- 31.Dellis O, Rossi AM, Dedos SG, Taylor CW. Counting functional inositol 1,4,5-trisphosphate receptors into the plasma membrane. The Journal of biological chemistry. 2008;283:751–5. doi: 10.1074/jbc.M706960200. [DOI] [PubMed] [Google Scholar]
- 32.Taylor CW, Dellis O. Plasma membrane IP3 receptors. Biochem Soc Trans. 2006;34:910–2. doi: 10.1042/BST0340910. [DOI] [PubMed] [Google Scholar]
- 33.Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–96. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. The Journal of biological chemistry. 1994;269:8701–7. [PubMed] [Google Scholar]
- 35.Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor. Cyclic GMP-dependent protein kinase mediates cAMP and cGMP dependent phosphorylation in the intact rat aorta. The Journal of biological chemistry. 1996;271:21933–8. doi: 10.1074/jbc.271.36.21933. [DOI] [PubMed] [Google Scholar]
- 36.Soulsby MD, Alzayady K, Xu Q, Wojcikiewicz RJ. The contribution of serine residues 1588 and 1755 to phosphorylation of the type I inositol 1,4,5-trisphosphate receptor by PKA and PKG. FEBS Lett. 2004;557:181–4. doi: 10.1016/s0014-5793(03)01487-x. [DOI] [PubMed] [Google Scholar]
- 37.Tertyshnikova S, Yan X, Fein A. cGMP inhibits IP3-induced Ca2+ release in intact rat megakaryocytes via cGMP- and cAMP-dependent protein kinases. J Physiol. 1998;512(Pt 1):89–96. doi: 10.1111/j.1469-7793.1998.089bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, Wilm M, Hofmann F, Ruth P. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 2000;404:197–201. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- 39.Bruce JI, Shuttleworth TJ, Giovannucci DR, Yule DI. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. The Journal of biological chemistry. 2002;277:1340–8. doi: 10.1074/jbc.M106609200. [DOI] [PubMed] [Google Scholar]
- 40.Arguin G, Regimbald-Dumas Y, Fregeau MO, Caron AZ, Guillemette G. Protein kinase C phosphorylates the inositol 1,4,5-trisphosphate receptor type 2 and decreases the mobilization of Ca2+in pancreatoma AR4-2J cells. J Endocrinol. 2007;192:659–68. doi: 10.1677/JOE-06-0179. [DOI] [PubMed] [Google Scholar]
- 41.Giovannucci DR, Groblewski GE, Sneyd J, Yule DI. Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. The Journal of biological chemistry. 2000;275:33704–11. doi: 10.1074/jbc.M004278200. [DOI] [PubMed] [Google Scholar]
- 42.Straub SV, Giovannucci DR, Bruce JI, Yule DI. A role for phosphorylation of inositol 1,4,5-trisphosphate receptors in defining calcium signals induced by Peptide agonists in pancreatic acinar cells. The Journal of biological chemistry. 2002;277:31949–56. doi: 10.1074/jbc.M204318200. [DOI] [PubMed] [Google Scholar]
- 43.Soulsby MD, Wojcikiewicz RJ. The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. The Biochemical journal. 2005;392:493–7. doi: 10.1042/BJ20051325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Regimbald-Dumas Y, Arguin G, Fregeau MO, Guillemette G. cAMP-dependent protein kinase enhances inositol 1,4,5-trisphosphate-induced Ca2+ release in AR4-2J cells. J Cell Biochem. 2007;101:609–18. doi: 10.1002/jcb.21221. [DOI] [PubMed] [Google Scholar]
- 45.Betzenhauser MJ, Fike JL, Wagner LE, 2nd, Yule DI. Protein kinase A increases type-2 inositol 1,4,5-trisphosphate receptor activity by phosphorylation of serine 937. The Journal of biological chemistry. 2009;284:25116–25. doi: 10.1074/jbc.M109.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maes K, Missiaen L, Parys JB, De Smet P, Sienaert I, Waelkens E, Callewaert G, De Smedt H. Mapping of the ATP-binding sites on inositol 1,4,5-trisphosphate receptor type 1 and type 3 homotetramers by controlled proteolysis and photoaffinity labeling. The Journal of biological chemistry. 2001;276:3492–7. doi: 10.1074/jbc.M006082200. [DOI] [PubMed] [Google Scholar]
- 47.Khan MT, Wagner L, 2nd, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. The Journal of biological chemistry. 2006;281:3731–7. doi: 10.1074/jbc.M509262200. [DOI] [PubMed] [Google Scholar]
- 48.Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, Roderick HL. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A. 2008;105:2427–32. doi: 10.1073/pnas.0711324105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488–93. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soulsby MD, Wojcikiewicz RJ. Calcium mobilization via type III inositol 1,4,5-trisphosphate receptors is not altered by PKA-mediated phosphorylation of serines 916, 934, and 1832. Cell Calcium. 2007;42:261–70. doi: 10.1016/j.ceca.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith JB, Smith L, Higgins BL. Temperature and nucleotide dependence of calcium release by myo-inositol 1,4,5-trisphosphate in cultured vascular smooth muscle cells. The Journal of biological chemistry. 1985;260:14413–6. [PubMed] [Google Scholar]
- 52.Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature. 1988;336:583–6. doi: 10.1038/336583a0. [DOI] [PubMed] [Google Scholar]
- 53.Maeda N, Kawasaki T, Nakade S, Yokota N, Taguchi T, Kasai M, Mikoshiba K. Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J Biol Chem. 1991;266:1109–1116. [PubMed] [Google Scholar]
- 54.Ferris CD, Huganir RL, Snyder SH. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc Natl Acad Sci U S A. 1990;87:2147–51. doi: 10.1073/pnas.87.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bezprozvanny I, Ehrlich BE. ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron. 1993;10:1175–84. doi: 10.1016/0896-6273(93)90065-y. [DOI] [PubMed] [Google Scholar]
- 56.Iino M. Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J Gen Physiol. 1991;98:681–98. doi: 10.1085/jgp.98.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maes K, Missiaen L, De Smet P, Vanlingen S, Callewaert G, Parys JB, De Smedt H. Differential modulation of inositol 1,4,5-trisphosphate receptor type 1 and type 3 by ATP. Cell Calcium. 2000;27:257–67. doi: 10.1054/ceca.2000.0121. [DOI] [PubMed] [Google Scholar]
- 58.Tu H, Wang Z, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophysical journal. 2005;88:1046–55. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Betzenhauser MJ, Wagner LE, 2nd, Iwai M, Michikawa T, Mikoshiba K, Yule DI. ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. The Journal of biological chemistry. 2008;283:21579–87. doi: 10.1074/jbc.M801680200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Betzenhauser MJ, Wagner LE, 2nd, Park HS, Yule DI. ATP regulation of type-1 inositol 1,4,5-trisphosphate receptor activity does not require walker Atype ATP-binding motifs. The Journal of biological chemistry. 2009;284:16156–63. doi: 10.1074/jbc.M109.006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park HS, Betzenhauser MJ, Won JH, Chen J, Yule DI. The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells. The Journal of biological chemistry. 2008;283:26081–8. doi: 10.1074/jbc.M804184200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maeda N, Niinobe M, Mikoshiba K. A cerebellar Purkinje cell marker P400 protein is an inositol 1,4,5-trisphosphate (InsP3) receptor protein. Purification and characterization of InsP3 receptor complex. Embo J. 1990;9:61–7. doi: 10.1002/j.1460-2075.1990.tb08080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wierenga RK, Hol WG. Predicted nucleotide-binding properties of p21 protein and its cancer-associated variant. Nature. 1983;302:842–4. doi: 10.1038/302842a0. [DOI] [PubMed] [Google Scholar]
- 64.Maes K, Missiaen L, Parys JB, Sienaert I, Bultynck G, Zizi M, De Smet P, Casteels R, De Smedt H. Adenine-nucleotide binding sites on the inositol 1,4,5-trisphosphate receptor bind caffeine, but not adenophostin A or cyclic ADP-ribose. Cell Calcium. 1999;25:143–52. doi: 10.1054/ceca.1998.0011. [DOI] [PubMed] [Google Scholar]
- 65.Wagner LE, 2nd, Betzenhauser MJ, Yule DI. ATP binding to a unique site in the type-1 S2- inositol 1,4,5-trisphosphate receptor defines susceptibility to phosphorylation by protein kinase A. The Journal of biological chemistry. 2006;281:17410–9. doi: 10.1074/jbc.M601340200. [DOI] [PubMed] [Google Scholar]
- 66.Tu H, Miyakawa T, Wang Z, Glouchankova L, Iino M, Bezprozvanny I. Functional characterization of the type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/-) splice variants and the Opisthotonos mutant form. Biophysical journal. 2002;82:1995–2004. doi: 10.1016/S0006-3495(02)75548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin C, Widjaja J, Joseph SK. The interaction of calmodulin with alternatively spliced isoforms of the type-I inositol trisphosphate receptor. The Journal of biological chemistry. 2000;275:2305–11. doi: 10.1074/jbc.275.4.2305. [DOI] [PubMed] [Google Scholar]
- 68.Anyatonwu G, Khan MT, Schug ZT, da Fonseca PC, Morris EP, Joseph SK. Calcium dependent conformational changes in Inositol trisphosphate receptors. J Biol Chem. 2009 doi: 10.1074/jbc.M110.123208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamada K, Miyata T, Mayanagi K, Hirota J, Mikoshiba K. Two-state conformational changes in inositol 1,4,5-trisphosphate receptor regulated by calcium. The Journal of biological chemistry. 2002;277:21115–8. doi: 10.1074/jbc.C200244200. [DOI] [PubMed] [Google Scholar]
- 70.Hamada K, Terauchi A, Mikoshiba K. Three-dimensional rearrangements within inositol 1,4,5-trisphosphate receptor by calcium. The Journal of biological chemistry. 2003;278:52881–9. doi: 10.1074/jbc.M309743200. [DOI] [PubMed] [Google Scholar]
- 71.Galvan DL, Borrego-Diaz E, Perez P, Mignery GA. Subunit oligomerization and topology of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1999;274:29483–29492. doi: 10.1074/jbc.274.41.29483. [DOI] [PubMed] [Google Scholar]
- 72.Joseph SK, Boehning D, Pierson S, Nicchitta CV. Membrane insertion, glycosylation and oligomerization of Inositol Trisphosphate receptors in a cell-free translation system. J Biol Chem. 1997;272:1579–1588. doi: 10.1074/jbc.272.3.1579. [DOI] [PubMed] [Google Scholar]
- 73.Michikawa T, Hamanaka H, Otsu H, Yamamoto A, Miyawaki A, Furuichi T, Tashiro Y, Mikoshiba K. Transmembrane topology and sites of N-glycosylation of inositol 1,4,5-trisphosphate receptor. The Journal of biological chemistry. 1994;269:9184–9. [PubMed] [Google Scholar]
- 74.Boehning D, Mak DO, Foskett JK, Joseph SK. Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. The Journal of biological chemistry. 2001;276:13509–12. doi: 10.1074/jbc.C100094200. [DOI] [PubMed] [Google Scholar]
- 75.Ramos-Franco J, Galvan D, Mignery G, Fill M. Location of the permeation pathway in the recombinant type-I Inositol 1,4,5-trisphosphate pathway. J Gen Physiol. 1999;114:243–250. doi: 10.1085/jgp.114.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schug ZT, da Fonseca PC, Bhanumathy CD, Wagner L, Zhang X, Bailey B, Morris EP, Yule DI, Joseph SK. Molecular characterization of the inositol 1,4,5-trisphosphate pore-forming segment. J Biol Chem. 2007;283:2939–2948. doi: 10.1074/jbc.M706645200. [DOI] [PubMed] [Google Scholar]
- 77.Schug ZT, Joseph SK. The Role of the S4-S5 Linker and C-terminal Tail in Inositol 1,4,5-Trisphosphate Receptor Function. J Biol Chem. 2006;281:24431–24440. doi: 10.1074/jbc.M604190200. [DOI] [PubMed] [Google Scholar]
- 78.Srikanth S, Wang Z, Tu H, Nair S, Mathew MK, Hasan G, Bezprozvanny I. Functional properties of the Drosophila melanogaster inositol 1,4,5-trisphosphate receptor mutants. Biophys J. 2004;86:3634–3646. doi: 10.1529/biophysj.104.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uchida K, Miyauchi H, Furuichi T, Michikawa T, Mikoshiba K. Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2003;278:16551–16560. doi: 10.1074/jbc.M300646200. [DOI] [PubMed] [Google Scholar]
- 80.Galvan DL, Mignery GA. Carboxyl-terminal sequences critical for inositol 1,4,5-trisphosphate receptor subunit assembly. J Biol Chem. 2002;277:48248–48260. doi: 10.1074/jbc.M209990200. [DOI] [PubMed] [Google Scholar]
- 81.Parker AK, Gergely FV, Taylor CW. Targeting of inositol 1,4,5-trisphosphate receptors to the endoplasmic reticulum by multiple signals within their transmembrane domains. The Journal of biological chemistry. 2004;279:23797–805. doi: 10.1074/jbc.M402098200. [DOI] [PubMed] [Google Scholar]
- 82.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 83.Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–8. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gillespie D, Fill MD. Intracellular calcium release channels mediate their own countercurrent: The ryanodine receptor case study. Biophysical journal. 2008 doi: 10.1529/biophysj.108.131987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 87.Ellinor PT, Yang J, Sather WA, Zhang J, Tsien RW. Ca2+ channel selectivity at a single locus for hih affinity interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 88.Gillespie D, Xu L, Wang Y, Meissner G. (De)constructing the ryanodine receptor: modeling ion permeation and selectivity of the calcium release channel. J Phys Chem B. 2005;109:15598–15610. doi: 10.1021/jp052471j. [DOI] [PubMed] [Google Scholar]
- 89.Williams AJ, West DJ, Sitsapesan R. Light at the end of the Ca(2+)-release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q Rev Biophys. 2001;34:61–104. doi: 10.1017/s0033583501003675. [DOI] [PubMed] [Google Scholar]
- 90.Alzayady KJ, Wojcikiewicz RJ. The role of Ca2+ in triggering inositol 1,4,5-trisphosphate receptor ubiquitination. Biochem J. 2005;392:601–606. doi: 10.1042/BJ20050949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhanumathy CD, Nakao SK, Joseph SK. Mechanism of proteasomal degradation of inositol trisphosphate receptors in CHO-K1 cells. J Biol Chem. 2006;281:3722–3730. doi: 10.1074/jbc.M509966200. [DOI] [PubMed] [Google Scholar]
- 92.Dellis O, Dedos SG, Tovey SC, Taufiq Ur R, Dubel SJ, Taylor CW. Ca2+ entry through plasma membrane IP3 receptors. Science. 2006;313:229–33. doi: 10.1126/science.1125203. [DOI] [PubMed] [Google Scholar]
- 93.Khan MT, Bhanumathy CD, Schug ZT, Joseph SK. Role of Inositol 1,4,5-trisphosphate receptors in apoptosis in DT40 lymphocytes. J Biol Chem. 2007;282:32983–32990. doi: 10.1074/jbc.M705183200. [DOI] [PubMed] [Google Scholar]
- 94.van Rossum DB, Patterson RL, Kiselyov K, Boehning D, Barrow RK, Gill DL, Snyder SH. Agonist-induced Ca2+ entry determined by inositol 1,4,5-trisphosphate recognition. Proc Natl Acad Sci U S A. 2004;101:2323–7. doi: 10.1073/pnas.0308565100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen SR, Li P, Zhao M, Li X, Zhang L. Role of the proposed pore-forming segment of the Ca2+ release channel (ryanodine receptor) in ryanodine interaction. Biophys J. 2002;82:2436–2447. doi: 10.1016/s0006-3495(02)75587-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Du GG, Guo X, Khanna VK, MacLennan DH. Functional characterization of mutants in the predicted pore region of the rabbit cardiac muscle Ca(2+) release channel (ryanodine receptor isoform 2) J Biol Chem. 2001;276:31760–31771. doi: 10.1074/jbc.M102751200. [DOI] [PubMed] [Google Scholar]
- 97.Gao L, Balshaw D, Xu L, Tripathy A, Xin C, Meissner G. Evidence for a role of the lumenal M3-M4 loop in skeletal muscle Ca2+ release channel (Ryanodine receptor) activity and conductance. Biophys J. 2000;79:828–840. doi: 10.1016/S0006-3495(00)76339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marius P, Lea EJ, Dawson AP. Estimation of the pore size of the type-I IP3 receptor reconstituted in single unilamellar vesicles. Biophysical journal. 1980 [Google Scholar]
- 99.Welch W, Rheault S, West DJ, Williams AJ. A model of the putative pore region of the cardiac ryanodine receptor channel. Biophys J. 2004;87:2335–2351. doi: 10.1529/biophysj.104.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu L, Wang Y, Gillespie D, Meissner G. Two rings of negative charges in the cytosolic vestibule of type-I ryanodine receptors modulate ion fluxes. Biophysical journal. 2006;90:443–453. doi: 10.1529/biophysj.105.072538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chan J, Whitten AE, Jeffries CM, Bosanac I, Mal TK, Ito J, Porumb H, Michikawa T, Mikoshiba K, Trewhella J, Ikura M. Ligand-induced conformational changes via flexible linkers in the amino-terminal region of the inositol 1,4,5-trisphosphate receptor. J Mol Biol. 2007;373:1269–1280. doi: 10.1016/j.jmb.2007.08.057. [DOI] [PubMed] [Google Scholar]
- 102.Mignery G, Sudhof TC. Molecular analysis of inositol 1,4,5-trisphosphate receptors. Methods in Neurosciences. 1993;18:247–265. [Google Scholar]
- 103.Boehning D, Joseph SK. Direct association of ligand-binding and pore domains in homo- and heterotetrameric inositol 1,4,5-trisphosphate receptors. EMBO J. 2000;19:5450–5459. doi: 10.1093/emboj/19.20.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Decher N, Chen J, Sanguinetti MC. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers. J Biol Chem. 2004;279:13859–13865. doi: 10.1074/jbc.M313704200. [DOI] [PubMed] [Google Scholar]
- 105.Ferrer T, Rupp J, Piper DR, Tristani-Firouzi M. The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-a’-go-go-related gene (hERG) K+ channel. J Biol Chem. 2006;281:12858–12864. doi: 10.1074/jbc.M513518200. [DOI] [PubMed] [Google Scholar]
- 106.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 107.Rossi AM, Riley AM, Tovey SC, Rahman T, Dellis O, Taylor EJ, Veresov VG, Potter BV, Taylor CW. Synthetic partial agonists reveal key steps in IP3 receptor activation. Nat Chem Biol. 2009;5:631–639. doi: 10.1038/nchembio.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varnai P, Balla A, Hunyady L, Balla T. Targeted expression of the inositol 1,4,5-triphosphate receptor (IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R channels. Proc Natl Acad Sci U S A. 2005;102:7859–7864. doi: 10.1073/pnas.0407535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 110.Rojas A, Wu J, Wang R, Jiang C. Gating of the ATP-sensitive K+ channel by a pore-lining phenylalanine residue. Biochim Biophys Acta. 2007;1768:39–51. doi: 10.1016/j.bbamem.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 111.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 112.Hardman RM, Stansfeld PJ, Dalibalta S, Sutcliffe MJ, Mitcheson JS. Activation gating of hERG potassium channels: S6 glycines are not required as gating hinges. J Biol Chem. 2007;282:31972–31981. doi: 10.1074/jbc.M705835200. [DOI] [PubMed] [Google Scholar]
- 113.Tristani-Firouzi M, Chen J, Sanguinetti MC. Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J Biol Chem. 2002;277:18994–19000. doi: 10.1074/jbc.M200410200. [DOI] [PubMed] [Google Scholar]
- 114.Lee EH, Allen PD. Homo-dimerization of RyR1 C-terminus via charged residues in random coils or in an alpha-helix. Exp Mol Med. 2007;39:594–602. doi: 10.1038/emm.2007.65. [DOI] [PubMed] [Google Scholar]
- 115.Vanderheyden V, Wakai T, Bultynck G, De Smedt H, Parys JB, Fissore RA. Regulation of inositol 1,4,5-trisphosphate receptor type 1 function during oocyte maturation by MPM-2 phosphorylation. Cell Calcium. 2009;46:56–64. doi: 10.1016/j.ceca.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boehning D, van Rossum DB, Patterson RL, Snyder SH. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc Natl Acad Sci U S A. 2005;102:1466–1471. doi: 10.1073/pnas.0409650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kawaai K, Hisatsune C, Kuroda Y, Mizutani A, Tashiro T, Mikoshiba K. 80K-H interacts with inositol 1,4,5-trisphosphate (IP3) receptors and regulates IP3-induced calcium release activity. J Biol Chem. 2009;284:372–380. doi: 10.1074/jbc.M805828200. [DOI] [PubMed] [Google Scholar]
- 118.Zhang S, Hisatsune C, Matsu-ura T, Mikoshiba K. G-protein-coupled receptor kinase-interacting proteins inhibit apoptosis by inositol 1,4,5-triphosphate receptor-mediated Ca2+ signal regulation. J Biol Chem. 2009;284:29158–29169. doi: 10.1074/jbc.M109.041509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fukatsu K, Bannai H, Inoue T, Mikoshiba K. 4.1N binding regions of inositol 1,4,5-trisphosphate receptor type 1. Biochem Biophys Res Commun. 2006;342:573–6. doi: 10.1016/j.bbrc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 120.Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell. 2005;120:85–98. doi: 10.1016/j.cell.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 121.Kang S, Kang J, Kwon H, Frueh D, Yoo SH, Wagner G, Park S. Effects of redox potential and Ca2+ on Inositol 1,4,5-trisphosphate receptor L3-1 loop region: implications for receptor regulation. J Biol Chem. 2008;283:25567–25575. doi: 10.1074/jbc.M803321200. [DOI] [PubMed] [Google Scholar]
- 122.Tu H, Nosyreva E, Miyakawa T, Wang Z, Mizushima A, Iino M, Bezprozvanny I. Functional and biochemical analysis of the type 1 inositol (1,4,5)-trisphosphate receptor calcium sensor. Biophysical journal. 2003;85:290–9. doi: 10.1016/S0006-3495(03)74474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miyakawa T, Mizushima A, Hirose K, Yamazawa T, Bezprozvanny I, Kurosaki T, Iino M. Ca(2+)-sensor region of IP(3) receptor controls intracellular Ca(2+) signaling. Embo J. 2001;20:1674–80. doi: 10.1093/emboj/20.7.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Iwai M, Michikawa T, Bosanac I, Ikura M, Mikoshiba K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. The Journal of biological chemistry. 2007;282:12755–64. doi: 10.1074/jbc.M609833200. [DOI] [PubMed] [Google Scholar]
- 125.Tu H, Wang Z, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophysical journal. 2005;88:1056–69. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Joseph SK, Pierson S, Samanta S. Trypsin digestion of the inositol trisphosphate receptor: implications for the conformation and domain organization of the protein. The Biochemical journal. 1995;307(Pt 3):859–65. doi: 10.1042/bj3070859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yoshikawa F, Iwasaki H, Michikawa T, Furuichi T, Mikoshiba K. Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. The Journal of biological chemistry. 1999;274:316–27. doi: 10.1074/jbc.274.1.316. [DOI] [PubMed] [Google Scholar]