

Summary
Human cytidine deaminase APOBEC3C (A3C) acts as a potent inhibitor of SIVagm and can be regulated by both HIV-1 and SIVagm Vif. The mechanism by which Vif suppresses A3C is unknown. In the present study, we demonstrate that both HIV-1 and SIVagm Vif can act in a proteasome-dependent manner to overcome A3C. SIVagm Vif requires the Cullin5-ElonginB-ElonginC E3 ubiquitin ligase for the degradation of A3C as well as the suppression of its antiviral activity. Mutation of a residue critical for the species-specific recognition of human or monkey A3G by HIV-1 Vif or SIVagm Vif in A3C had little effect on HIV-1 or SIVagm Vif-mediated degradation of A3C. Although the amino-terminal region of A3G was not important for Vif-mediated degradation, the corresponding region in A3C was critical. A3C mutants that were competent for Vif binding but resistant to Vif-mediated degradation were identified. These data suggest that primate lentiviral Vif molecules have evolved to recognize multiple host APOBEC3 proteins through distinct mechanisms. However, Cul5-E3 ubiquitin ligase appears to be a common pathway hijacked by HIV-1 and SIV Vif to defeat APOBEC3 proteins. Furthermore, Vif and APOBEC3 binding is not sufficient for target protein degradation indicating an important but uncharacterized Vif function.
Introduction
In recent years, members of the apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3 (APOBEC3) protein family have come to be seen as representing a novel arm of innate immunity (Sheehy et al., 2002; Bieniasz, 2004; Goff, 2004; Harris and Liddament, 2004; Navarro and Landau, 2004; Rose et al., 2004; Turelli and Trono, 2005; Cullen, 2006; Chiu and Greene, 2006; Malim, 2006; Yu, 2006). Various APOBEC3 proteins exhibit broad antiviral activity against a wide range of retroviruses (Bieniasz, 2004; Goff, 2004; Harris and Liddament, 2004; Navarro and Landau, 2004; Rose et al., 2004; Turelli and Trono, 2005; Chiu and Greene, 2006; Cullen, 2006; Malim, 2006; Yu, 2006), endogenous retroviruses (Esnault et al., 2005), LINE-1 and Alu retrotransposons (Turelli et al., 2004a; Bogerd et al., 2006; Chen et al., 2006; Chiu et al., 2006; Muckenfuss et al., 2006; Stenglein and Harris, 2006; Hulme et al., 2007; Niewiadomska et al., 2007), hepatitis B virus (Xu et al., 2007; Turelli et al., 2004b), adeno-associated virus (Chen et al., 2006) and CMV/SV40 viral promoters (Zhang et al., 2007).
Studies have indicated that APOBEC3 can be packaged into diverse retroviruses and that they mediate potent antiviral functions in newly infected target cells. Encapsidation of APOBEC3G (A3G) into HIV-1 particles is mediated by Gag molecules (Alce and Popik, 2004; Cen et al., 2004; Douaisi et al., 2004; Luo et al., 2004; Schafer et al., 2004; Zennou et al., 2004; Navarro et al., 2005), and the nucleocapsid domain of Gag is required for efficient A3G packaging through RNA interaction (Alce and Popik, 2004; Cen et al., 2004; Douaisi et al., 2004; Luo et al., 2004; Schafer et al., 2004; Zennou et al., 2004; Navarro et al., 2005). Viral genomic RNA(Svarovskaia et al., 2004; Khan et al., 2005) and 7SL RNA (Wang et al., 2007) are thought to mediate A3G packaging. This virion-packaged A3G induces cytidine deamination of viral DNA during reverse transcription in newly infected target cells (Harris et al., 2003; Lecossier et al., 2003; Mangeat et al., 2003; Mariani et al., 2003; Zhang et al., 2003; Suspene et al., 2004; Yu et al., 2004a). A3G and another APOBEC3 protein, A3F, also reduce the accumulation of viral reverse transcription products (Bishop et al., 2006; Guo et al., 2006; Kaiser and Emerman, 2006; Iwatani et al., 2007; Li et al., 2007; Luo et al., 2007; Mbisa et al., 2007; Yang et al., 2007) and inhibit proviral DNA formation (Luo et al., 2007; Mbisa et al., 2007).
To overcome the restriction imposed by APOBEC3 factors, HIV-1 encodes a regulatory protein, Vif, which triggers the degradation of APOBEC3 proteins through poly-ubiquitination and proteasomal degradation (Conticello et al., 2003; Marin et al., 2003; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003; Mehle et al., 2004; Liu et al., 2004; 2005). Vif recruits Cul5, ElonginB/C E3 ubiquitin ligase (Yu et al., 2003; 2004b; Mehle et al., 2004; Kobayashi et al., 2005; Liu et al., 2005; Luo et al., 2005) through an HCCH motif (Luo et al., 2005) and virus-specific BC-box (Mehle et al., 2004; Yu et al., 2004b). The amino-terminal region of HIV-1 Vif interacts with the A3G or A3F through distinct interfaces (Marin et al., 2003; Simon et al., 2005; Schrofelbauer et al., 2006; Tian et al., 2006; Russell and Pathak, 2007), and three functional domains in the carboxy-terminal region of this Vif protein interact with Cul5 and ElonginB/C. The HCCH motif (Luo et al., 2005), which binds zinc (Mehle et al., 2006; Xiao et al., 2006; 2007a) and contains a consensus sequence Hx2YFxCFx4Φx2AΦx7-8Cx5H (Xiao et al., 2007b), is required for Cul5 binding. The SLQXLA motif, which is conserved between HIV-1 and other lentiviral Vif molecules, is a virus-specific BC-box (Mehle et al., 2004; Yu et al., 2004b) that mediates the interaction with ElonginB/C. There is a downstream LPxL Cul5 box that is also important for Cul5 interaction. In the case of SIVagmTan Vif, the HCCH motif and viral BC-box are present, but the LPxL Cul5 box is lacking (Luo et al., 2005). Vif has also been hypothesized to reduce the translation of A3G and to suppress APOBEC3 through a ubiquitin/proteasome-independent mechanism (Stopak et al., 2003; Opi et al., 2007). In the case of SIVagm, TanVif has been shown to suppress human A3G via a degradation-independent fashion (Takeuchi et al., 2005).
In the present study, we have explored the viral defence of SIVagmTan Vif against A3C, and have obtained clear evidence that SIVagmTan Vif can suppress the antiviral activity of A3C by inducing its proteasome-mediated degradation. Functional Cul5-E3 ubiquitin ligase activity was found to be critical for this defensive function of SIVagmTan Vif. We also found that a distinct region in A3C that is required for its Vif-mediated degradation was dispensable for Vif-mediated degradation of A3G. In human A3G, altering position 128 to the equivalent residue in AGM A3G changes Vif sensitivity from HIV-1 to SIVagmTan Vif and vice versa. We found that altering the 128 position in A3C had little influence on sensitivity to HIV-1 or SIVagmTan Vif. These data suggest that specific residues and functional domains are playing different roles in A3C than they are in A3G.
Results
SIVagmTan Vif induces the degradation of human A3C which is dependent on proteasome activity
We initially screened a variety of human APOBEC3 proteins for their ability to inhibit SIVagm, by testing the infectivity of SIVagmTan and SIVagmTanΔVif viruses produced in the presence of human A3A, A3C, A3F or A3G using a single-round infectivity assay. For this purpose, human 293T cells were co-transfected with either SIVagmTan or SIVagmTanΔVif and one of the haemagglutinin (HA)-tagged human APOBEC3 expression vectors. MAGI-CCR5 reporter cells were then infected using the supernatant from the transfected 293T cells. Viral infectivity in the absence of APOBEC3 was set to 100%. We found that single-domain protein A3A was unable to restrict SIVagmTan infection either with or without Vif (Fig. 1A). In contrast, the double-domain proteins A3G and A3F were able to inhibit the infectivity of both wild-type SIVagmTan or SIVagmTanΔVif (Fig. 1A). Among the human APOBEC3 members assayed, only A3C was able to inhibit the infectivity of the Vif-deficient SIVagmTanΔVif virus but not the wild-type SIVagmTan virus (Fig. 1A), suggesting that SIVagm Vif is able to overcome human A3C antiviral activity. As expected, AGM A3G inhibited the infectivity of the Vif-deficient SIVagmTanΔVif virus but not the wild-type SIVagmTan virus (Fig. 1A).
Fig. 1.

SIVagmTan Vif can selectively overcome the antiviral activity of human A3C.
A. 293T cells were co-transfected with an infectious clone of SIVagmTan or SIVagmTanΔVif plus a control vector or an expression vector encoding an HA-tagged APOBEC3 protein at a 3:1 ratio. The infectivity of the resulting virus was assayed by infecting MAGI cells, which were then fixed and stained for β-galactosidase activity. Virus infectivity in the absence of APOBEC3 was set to 100%.
B. 293T cells were co-transfected with an expression vector for A3A-HA, A3C-HA, A3F-HA, A3G-HA or AGMA3G-HA plus a control vector or an expression vector for SIVagmTan Vif at a 1:3 ratio. Cell lysates were analysed 48 h after transfection by immunoblotting using an anti-HA antibody to detect HA-tagged APOBEC3 proteins, anti-myc antibody to detect myc-tagged SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
C. SIVagmTan Vif and HIV-1 Vif induce the degradation of human A3C in a proteasome-dependent manner. Plasmids expressing HA-tagged A3C were co-transfected with a control plasmid, an expression plasmid for HIV-1 Vif-myc, or an expression plasmid for SIVagmTan Vif-myc into 293T cells at a 1:3 ratio. Transfected cells were treated with the proteasome inhibitor MG132 at 5 mM (lanes 2, 4 and 6) or DMSO (lanes 1, 3 and 5) at 36 h after transfection. Cells were harvested 12 h later and analysed by immunoblotting using anti-HA antibody to detect HA-tagged A3C, anti-myc antibody to detect myc-tagged HIV-1 Vif or SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
D. 293T cells were co-transfected with an infectious clone of SIVagmTanΔVif plus a control vector or an expression vector encoding an HA-tagged APOBEC3 protein in the absence or the presence of SIVagmTanVif-myc. Cell lysates and viral lysates were harvested from each sample and analysed by immunoblotting at 48 h post transfection using the anti-HA antibody to detect HA-tagged APOBEC proteins, the anti-p27 antibody to detect SIVagmTan CA protein, and an antibody against ribosomal P19 to assess protein loading.
We then evaluated the ability of SIVagmTan Vif to degrade the human APOBEC3 proteins. For this purpose, 293T cells were co-transfected with one of the HA-tagged APOBEC3 expression vectors and pSIVagmTan Vif or a control plasmid. We found that A3C expression in the cell lysates was reduced by coexpression with SIVagmTan Vif (Fig. 1B, top panel, compare lanes 3 and 4) but that expression of human A3A, A3F and A3G was unchanged when each of these proteins was individually coexpressed with SIVagmTan Vif (Fig. 1B). As expected, the AGM A3G expression in the cell lysates was reduced by coexpression with SIVagmTan Vif (Fig. 1B, top panel, compare lanes 9 and 10). We have also observed that expression of human A3B, A3DE and A3H was not affected by SIVagmTan Vif (data not shown).
To begin to analyse the mechanism by which Vif suppresses A3C, we asked whether the degradation of A3C by Vif was affected by the presence of the proteasome inhibitor MG132. 293T cells were co-transfected with A3C plus the HIV-1 Vif or SIVagmTanVif expression vectors, MG132 was added at 36 h after transfection, and the cells were harvested 12 h later. We found that SIVagmTan Vif induced the degradation of A3C (Fig. 1C, top panel, compare lanes 1 and 5), and this degradation could be blocked by the proteasome inhibitor (Fig. 1C, top panel, compare lanes 5 and 6). SIVagmTan Vif proteins were also stabilized by the addition of the proteasome inhibitor (Fig. 1C, middle panel, lanes 5 and 6), suggesting that Vif turnover is also dependent on proteasome machinery. The HIV-1 Vif-induced degradation of A3C was also inhibited by the proteasome inhibitor (Fig. 1C, lanes 3 and 4).
We also examined the virion incorporation of APOBEC3 proteins. Our data demonstrate that the packaging of A3A into SIVagm virions was inefficient compared with other APOBEC3 proteins (Fig. 1D). Human A3G and A3F can be packaged into the virions in the absence or the presence the SIVagmTan Vif. Human A3C and AGMA3G packaging was reduced by SIVagmTan Vif (Fig. 1D, lanes 4 and 10).
SIVagmTan Vif interacts specifically with human A3C
To determine whether the interaction of APOBEC3 with SIVagmTan Vif was correlated with APOBEC3 protein degradation, we attempted to co-immunoprecipitate SIVagmTan Vif and the various APOBEC3 proteins. For this purpose, 293T cells were co-transfected with pSIVagmTanVif and an HA-tagged A3A, A3C, A3F or A3G expression plasmid. In order to block proteasome-mediated degradation and increase protein stability, the cells were treated with 10 μM MG132 12 h prior to harvesting. Proteins bound to APOBEC3-HA-tagged proteins were immobilized by incubating the cell lysates with anti-HA agarose beads, and bound proteins were eluted from the beads under acidic conditions. Immunoblot analysis revealed that although SIVagmTan Vif was coexpressed with all the tested human APOBEC3 proteins in the cell lysates (Fig. 2A, left), SIVagmTan Vif bound only to HA-tagged A3C (Fig. 2A, lane 6) and not to A3A, A3F or A3G (Fig. 2A, lanes 5, 7 and 8 respectively).
Fig. 2.

Interaction of SIVagm Vif with A3C.
A. 293T cells were co-transfected with an expression vector for SIVagmTan Vif plus an expression vector for HA-tagged A3A, A3C, A3F or A3G at a 2:1 ratio. Cells were harvested 48 h later and subjected to immunoprecipitation analysis using the anti-HA antibody conjugated to agarose beads. Cell lysates or coprecipitated proteins were analysed by immunoblotting using an anti-HA antibody to detect HA-tagged APOBEC3 proteins and an anti-myc antibody to detect myc-tagged SIVagmTan Vif.
B. Co-precipitation of HIV-1 Vif-myc or SIVagmVif-myc with A3C-HA, as determined by co-precipitation analysis.
We then asked whether HIV-1 and SIVagm Vif were both capable of interaction with A3C. 293T cells were co-transfected with vectors expressing HA-tagged A3C and HIV-1 Vif, SIVagm Vif, or the GFP control vector. MG132 was added at 36 h after transfection in order to increase protein stability. The cell lysates were incubated with anti-HA agarose beads, and the bound proteins were eluted. Immunoblot analysis indicated that both HIV-1 Vif and SIVagm Vif co-immunoprecipitated with A3C (Fig. 2B, top panel, lanes 5 and 6).
SIVagm Vif-mediated suppression of A3C requires the BC-box and HCCH motif
It is currently understood that HIV-1 Vif overcomes A3G and A3F activity by inducing their degradation through the Cul5-Elongin B/C E3 ubiquitin ligase complex (Yu et al., 2003; 2004b; Mehle et al., 2004; Kobayashi et al., 2005; Liu et al., 2005; Luo et al., 2005). Our model for SIVagmTan Vif-induced degradation of A3C protein predicts that SIVagmTan Vif also acts as a substrate receptor to bring its target to a Cul5-E3 ubiquitin ligase, where the target protein is poly-ubiquitinated and degraded via the proteasome machinery (Fig. 3). The SLQYLA motif in SIVagmTan Vif is part of a viral BC box that interacts with Elongin C (Luo et al., 2005). Mutation of the SLQ motif ablates its interaction with Elongin C and inhibits the recruitment of E3 ligase by the substrate receptor, thereby preventing the degradation of the substrate (Fig. 3C). The HCCH zinc-binding motif of SIVagmTan Vif interacts with Cul5 (Luo et al., 2005). Mutations in the HCCH zinc-binding motif prevent the formation of the complete E3 ubiquitin ligase (Fig. 3D). Indeed, both the BC-box and HCCH mutants of SIVagmTan Vif could not induce the degradation of AGM A3G (Fig. 3E, lanes 3-7) compared with the wild-type SIVagmTan Vif (Fig. 3E, lane 2).
Fig. 3.
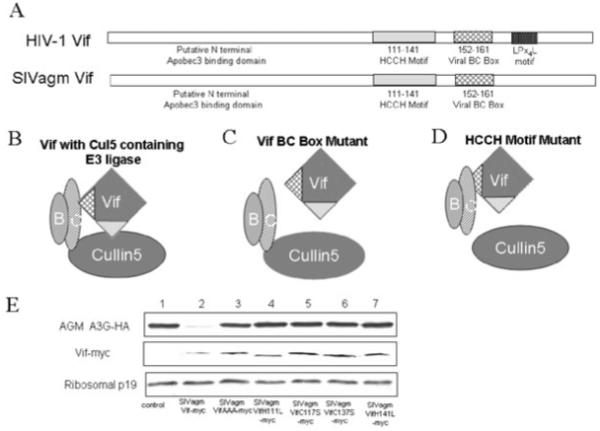
Model for SIVagmTan Vif-induced proteasome degradation.
A. Like HIV-1 Vif, SIVagmTan Vif contains an HCCH motif for interacting with Cul5 and a BC-box for binding to ElonginB-ElonginC. The amino-terminal region is the putative APOBEC3 interaction domain.
B. A functional Cul5-ElonginB/C-SIVagmTan Vif complex.
C. Mutation of the BC-box (SLQ motif) ablates the interaction with ElonginC as well as Cul5.
D. Mutations in the HCCH zinc-binding motif prevent Cul5 recruitment.
E. Degradation of AGM A3G-HA was blocked by mutations in the SIVagm Vif BC-box (SIVagmVifAAA) or HCCH motif (H111L, C117S, C137S and H141L). The ratio is 3:1 of Vif and AGM A3G-HA.
To investigate the contribution of the viral BC-box to the SIVagmTan Vif-induced degradation of A3C, we co-transfected pSIVagmTanVif or the BC box mutant pSIVagmTanVif-AAA along with A3C-HA into 293T cells. Immunoblot analysis of the cell lysates showed that the degradation of A3C induced by SIVagmTan Vif (Fig. 4A, lane 2) was not seen with SIVagmTanVif-AAA (Fig. 4A, lane 3). The incorporation of A3C into SIVagmTanΔVif virions in the presence of the SIVagm VifAAA mutant was increased compared with that in the presence of the wild-type SIVagmTan Vif (Fig. 4B, compares lanes 3 and 4). The functionality of the SIVagmTan Vif viral BC box mutant was tested in a single-round tissue culture infectivity assay. 293T cells were co-transfected with plasmids expressing A3C and an infectious clone, SIVagmTanΔVif, plus pSIVagmTanVif or pSIVagmTanVif-AAA. Virus infectivity was tested in the MAGI-CCR5 reporter cell line. Virus infectivity in the absence of A3C was set to 100%. SIVagmTan Vif suppressed the antiviral activity of A3C (Fig. 4C), while expression of the SIVagmTanVif-AAA mutant protein did not (Fig. 4C). These results suggest that the mechanism by which SIVagmTanVif overcomes A3C antiviral activity requires the viral BC-box that interacts with Elongin C (Luo et al., 2005).
Fig. 4.

The BC-box motif is required for SIVagmTan Vif activity against A3C.
A. The plasmid expressing HA-tagged A3C was co-transfected with a control plasmid, SIVagmTan Vif-myc or SIVagmTan Vif-AAA-myc expression plasmid into 293T cells at a 1:3 ratio. Cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA, anti-myc antibody to detect myc-tagged SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
B. 293T cells were co-transfected with an infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc, or SIVagmTan Vif-AAA-myc plus A3C-HA. The ratio is 3:3:1 (SIVagmTanΔVif : SIVagmTanVif-myc or SIVagmTan Vif-AAA-myc : A3C-HA). Viral lysates were harvested from each sample and analysed by immunoblotting at 48 h post transfection using the anti-HA antibody to detect HA-tagged A3C protein, the anti-p27 antibody to detect SIVagmTan CA protein.
C. 293T cells were co-transfected with an infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc, or SIVagmTan Vif-AAA-myc plus A3C-HA [the ratio is same as (B)]. The infectivity of the resulting virus was assayed by infecting MAGI cells. Cells were fixed and stained for β-galactosidase activity. Virus infectivity in the absence of APOBEC3 was set to 100%.
To further assess SIVagmTan Vif function, we looked at the ability of SIVagmTanVif HCCH mutants to suppress A3C. Expression vectors for SIVagmTanVif and HCCH mutants were co-transfected with the A3C-HA expression vector into 293T cells. We used SIVagmTanVif mutants that had single residues in the HCCH motif mutated and were unable to bind Cul5 (Luo et al., 2005). Mutations in the HCCH motif destroyed the ability of mutant SIVagmTan Vif to induce the degradation of A3C (Fig. 5A, lanes 3, 4, 5, 6). Mutations in the HCCH motif of SIVagmTan Vif also reduced their ability to exclude A3C into SIVagmTanΔVif virions (Fig. 5B). In a single-round infectivity assay, we tested the ability of the HCCH mutants to rescue viral infectivity in the presence of A3C. The same HCCH mutations that disrupted the ability of mutant SIVagmTan Vif to induce the degradation of A3C (Fig. 5A) also disrupted the ability of the mutant Vif protein to rescue virus infectivity (Fig. 5C).
Fig. 5.

The HCCH motif is required for SIVagmTan Vif activity against A3C.
A. The plasmid expressing HA-tagged A3C was co-transfected with a control plasmid, SIVagmTan Vif-myc or one of the SIVagmTan Vif HCCH mutant expression plasmids into 293T cells at a 1:3 ratio of A3C and SIVagmTanVif. Cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA, anti-myc antibody to detect myc-tagged SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
B. 293T cells were co-transfected with an infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc, or SIVagmTan Vif HCCH mutant plus A3C. The ratio is 3:3:1 (SIVagmTanΔVif : SIVagmTanVif-myc or SIVagmTan Vif HCCH mutant : A3C). Viral lysates were harvested from each sample and analysed by immunoblotting at 48 h post transfection using the anti-HA antibody to detect HA-tagged A3C protein, the anti-p27 antibody to detect SIVagmTan CA protein.
C. 293T cells were co-transfected with an infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc, or SIVagmTan Vif HCCH mutant plus A3C [the ratio is same as (B)]. Infectivity of the produced virus was assayed by infecting MAGI cells. Cells were fixed and stained for β-galactosidase activity. Virus infectivity in the presence of parental SIVagmTan Vif was set to 100%.
A dominant-negative Cul5 mutant disrupts SIVagmTanVif activity against A3C
To study the role of Cul5 SIVagmTan Vif-induced A3C degradation, we used a dominant-negative Cul5ΔNedd8 mutant to inhibit Cul5 function. Neddylation of cullins is critical for E3 ubiquitin ligase activity. Cul5ΔNedd8 is able to interact with Vif but is defective in E3 ligase activity, resulting in inhibition of Vif activity. When 293T cells were co-transfected with A3C and pSIVagmTan Vif plus pCul5ΔNedd8 or a control vector, we found that the presence of a dominant-negative Cul5 mutant inhibited SIVagmTan Vif-induced A3C degradation (Fig. 6A, lane 3), suggesting that Cul5 plays a role in the SIVagmTan Vif-induced turnover of A3C. Cul5ΔNedd8 also inhibited SIVagmTan Vif-induced AGM A3G degradation (Fig. 6A, lane 6). The ability of SIVagmTan Vif to exclude A3C virion packaging was inhibited by Cul5ΔNedd8 (Fig. 6B). We then asked whether a disruption of Cul5 function could influence the ability of SIVagmTan Vif to suppress A3C antiviral activity. For this purpose, 293T cells were co-transfected with the infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc and A3C in the absence or the presence of pCul5ΔNedd8. The supernatants were collected from transfected cells and used to infect the MAGI-CCR5 reporter cell line. The results showed that infectivity of virus produced in the presence of the mutant Cul5ΔNedd8 was markedly reduced (Fig. 6C) when compared with that of virus produced in the absence of Cul5ΔNedd8 (Fig. 6C), indicating that Cul5 activity is required for SIVagmTan Vif-mediated suppression of A3C.
Fig. 6.

SIVagmTan Vif activity against A3C antiviral activity requires a functional Cul5.
A. 293T cells were co-transfected with the plasmid expressing HA-tagged A3C or AGMA3G and SIVagmTan Vif-myc plus a control vector or a vector expressing Cul5ΔNedd8 into 293T cells. The ratio is 3:1:1 of SIVagmTanVif-myc and Cul5ΔNedd8 and A3C or AGMA3G. Cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA or AGMA3G, anti-myc antibody to detect myc-tagged SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
B. 293T cells were co-transfected with the infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc plus A3C in the absence or the presence of Cul5ΔNedd8. Viral lysates were harvested from each sample and analysed by immunoblotting at 48 h post transfection using the anti-HA antibody to detect HA-tagged A3C protein, the anti-p27 antibody to detect SIVagmTan CA protein.
C. 293T cells were co-transfected with the infectious clone of SIVagmTanΔVif plus a control vector or an expression vector for SIVagmTan Vif-myc plus A3C in the absence or the presence of Cul5ΔNedd8. Infectivity of the produced virus was assayed by infecting MAGI cells. Virus infectivity in the absence of A3C was set to 100%.
The N-terminal region upstream from the active site of A3C is necessary for Vif-induced degradation
To identify the regions of A3C that are determinants for Vif-induced degradation, we designed chimeric proteins involving two single-domain proteins (Fig. 7A): A3C, which is recognized by SIVagmTanVif, and A3A, which is not (Fig. 1). These chimeras were then co-transfected with a control plasmid or pSIVagmTanVif into 293T cells. We found that A3A/C1 and A3A/C2 were resistant to SIVagmTanVif-induced degradation (Fig. 7B, compares lanes 3 and 4 as well as lanes 5 and 6). On the other hand, A3C/A1 was still sensitive to SIVagmTanVif-induced degradation (Fig. 7B, compare lanes 7 and 8). A3A/C1 contains N-terminal region, active site and linker sequences from A3A, suggesting these regions in A3C are critical for SIVagmTanVif-induced degradation. A3A/C2 contains only the amino-terminal region of A3A and is resistant to SIVagmTanVif-induced degradation. This is surprising, as amino-terminal region of A3G is not required for Vif interaction or Vif-induced degradation. Deletion of the amino-terminal region (amino acids 2-47) of A3C also blocked SIVagmTanVif-induced degradation (Fig. 7C).
Fig. 7.

Regions of A3C that are necessary for Vif-induced degradation.
A. The chimeras produced between A3A and A3C were generated in regions between the predicted secondary structures of A3A and A3C. A3CΔN contains an in-frame deletion of amino acids 2-47.
B. Various expression vectors for parental A3C-HA or chimeras were co-transfected with SIVagmTan Vif-myc or a control vector into 293T cells at a 1:3 ratio of APOBEC and SIVagmTan Vif-myc. Transfected cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA, anti-myc antibody to detect myc-tagged SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
C. Expression vectors for A3C-HA or A3CΔN-HA (in-frame deletion of amino acids 2-47) were co-transfected with SIVagmTanVif-myc or SIVagmTanVifC117S-myc expression vector into 293T cells. The ratio of APOBEC and Vif is 1:3. Cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA or A3CΔN-HA, anti-myc antibody to detect myc-tagged SIVagmTanVif-myc or SIVagmTanVifC117S-myc and an antibody against ribosomal P19 to assess protein loading.
D. 293T cells were co-transfected with an expression vector for SIVagmTan Vif plus various expression vectors for parental A3C-HA or chimeras at a 2:1 ratio. Cells were harvested 48 h later and subjected to immunoprecipitation analysis using the anti-HA antibody conjugated to agarose beads. Cell lysates or coprecipitated proteins were analysed by immunoblotting using an anti-HA antibody to detect HA-tagged A3C or chemeras proteins and an anti-myc antibody to detect myc-tagged SIVagmTan Vif.
Interestingly, A3A/C1, A3A/C2 and A3C/A1 all maintained the ability to interact with SIVagmTanVif (Fig. 7D) despite their divergent abilities to be degraded by SIVagmTanVif (Fig. 7B). These data demonstrate that interaction with Vif is not sufficient to trigger Vif-mediated APOBEC3 degradation.
Although A3C and A3G may adapt a similar fold (Fig. 8A) based on the model structure of APOBEC2 (Prochnow et al., 2007), the regions in A3C and A3G that are important for Vif mediated degradation are different. The amino-terminal region of A3C (amino acids 2-47) was required for HIV-1 Vif mediated degradation (Fig. 8B). In contrast, the amino-terminal region of A3G (amino acids 2-47) was not required for HIV-1 Vif-mediated degradation (Fig. 8C). These findings suggest that the epitope recognition by Vif and/or the site of poly-ubiquitination may be located in different regions in A3C and A3G. In the case of A3G, the linker #1 (amino acids 105-245), but not the amino-terminal region, are important for Vif-induced degradation (Zhang et al., 2008). In A3C, the amino-terminal region was required (Fig. 8B).
Fig. 8.

The regions in A3C and A3G that are important for Vif mediated degradation.
A. The dimer of A3C and the full-length A3G were modelled on the structure of APOBEC2. Residues 1-47 of A3C are shown in black. Residues 105-245 of A3G are also shown in black.
B. Expression vectors for A3C-HA or A3CΔN-HA (in-frame deletion of amino acids 2-47) were co-transfected with HIV-1 Vif-myc or HIV-1 VifL145A-myc expression vector into 293T cells. Transfected cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3C-HA or A3CΔN-HA, anti-myc antibody to detect HIV-1 Vif-myc or HIV-1 VifL145A-myc and an antibody against ribosomal P19 to assess protein loading.
C. Expression vectors for A3G-HA or A3GΔN-HA (in-frame deletion of amino acids 2-47) were co-transfected with HIV-1 Vif-myc or HIV-1 VifL145A-myc expression vector into 293T cells. Transfected cells were harvested 48 h after transfection and analysed by immunoblotting using anti-HA antibody to detect A3G-HA or A3GΔN-HA, anti-myc antibody to detect HIV-1 Vif-myc or HIV-1 VifL145A-myc and an antibody against ribosomal P19 to assess protein loading.
Residue 128 in A3C does not affect the species specificity of Vif-induced degradation
A negatively charged residue at position 128 of A3G has been shown to be required for HIV-1 Vif-induced degradation, while a positively charged residue at position 128 of A3G is required for SIVagm Vif-induced degradation (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). However, an uncharged residue (alanine) at this position in A3G allowed degradation by both HIV-1 and SIVagm Vif (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). Interestingly, position 128 in human A3C is an uncharged residue, tyrosine, and human A3C could be degraded by both HIV-1 and SIVagm Vif. To test the possibility that a charged residue at position 128 of A3C might influence its degradation by Vif, we generated A3C point mutants in which residue 128 was changed from a tyrosine to either an aspartic acid (the equivalent residue in human A3G) or lysine (the equivalent residue in AGM A3G) (Fig. 9A). We co-transfected A3C or A3C mutants with a control vector, an expression vector for HIV-1 Vif, or SIVagmTan Vif into 293T cells. Cell lysates were analysed by immunoblotting using anti-HA antibody to detect A3C, anti-myc antibody to detect myc-tagged HIV-1 or SIVagmTan Vif, or antiribosome p19 antibody to assess protein loading (Fig. 9B). We found that the parental A3C, A3CY128D and A3CY128K proteins were all sensitive to both HIV-1 and SIVagmTan Vif-induced degradation. These results suggest that the ability of A3C to be degraded by both HIV-1 and SIVagmTan Vif is not determined by residue 128, a situation that is different from that for A3G.
Fig. 9.

Residue 128 in human A3C does not affect the species specificity of SIVagmTan or HIV-1 Vif-induced degradation.
A. It has been shown that altering residue 128 of human or AGM A3G affects HIV-1 or SIVagm Vif-induced degradation. A3C mutants at residue 128 were generated by changing a tyrosine to either an aspartic acid (equivalent residue in human A3G) or lysine (equivalent residue in AGM A3G).
B. 293T cells were co-transfected with expression vectors for A3C-HA or A3C mutants plus a control vector, an expression vector for HIV-1 Vif, or SIVagmTan Vif at a 1:3 ratio of APOBEC and Vif. Cell lysates were analysed 48 h after transfection by immunoblotting using anti-HA antibody to detect HA-taged A3C or A3C mutants, anti-myc antibody to detect myc-tagged HIV-1 Vif or SIVagmTan Vif and an antibody against ribosomal P19 to assess protein loading.
Discussion
In the present study, we have demonstrated for the first time that Cul5-E3 ubiquitin ligase function is required for SIVagmTanVif-mediated suppression of the antiviral activity of A3C protein (Figs 3-6), despite the fact that SIVagmTan lacks the LPxL Cul5 box that is present in HIV-1 Vif. The degradation of AGM-A3G by SIVagmTan Vif also requires functional interaction with Cul5-ElonginB/C complex (Fig. 3). This conservation of function suggests that, despite a significant difference in protein structure HIV-1 and SIVagmTan Vif molecules have evolved to use an evolutionarily conserved Cul5-containing E3 ubiquitin ligase to degrade APOBEC3 proteins.
We also found that, like HIV-1 Vif (Liu et al., 2004), SIVagmTan Vif could be stabilized by the use of a proteasome inhibitor (Fig. 1). Blocking Cul5 E3 ubiquitin ligase function also stabilized SIVagmTan Vif (Fig. 6). Studies have previously indicated that HIV-1 Vif may be regulated by the Cul5-E3 ubiquitin ligase (Mehle et al., 2004; Liu et al., 2005). Our findings suggest that SIVagmTan Vif may also be autoregulated by the Cul5-ElonginB/C-SIVagmTan Vif E3 ubiquitin ligase complex. Degradation of Vif molecules by the Cul5-E3 ubiquitin ligase may also limit the virion packaging of Vif. Our previous study indicated that the proteasome inhibitor MG132 could significantly increase Vif packaging into HIV-1 virions (Liu et al., 2004).
Interestingly, studying of the interaction of Vif with various A3C and A3A chimera mutants demonstrated that interaction with Vif is not sufficient to trigger Vif-mediated APOBEC3 degradation. A3A/C1, A3A/C2 and A3C/A1 all maintained the ability to interact with SIVagmTanVif (Fig. 7D). However, A3A/C1 and A3A/C2 were resistant to Vif mediated degradation (Fig. 7B). These data demonstrate that Vif is not simply a linker between the E3 ubiquitin ligase complex and the substrate. Vif plays an important yet uncharacterized role in substrate ubiquitination and/or degradation.
In examining the ability of SIVagmTan Vif to overcome APOBEC3 antiviral activity across species, we found that of the human APOBEC3 proteins tested including A3A, A3B, A3C, A3DE, A3F, A3G and A3H, only human A3C could be efficiently degraded by SIVagmTan Vif. SIVagm Vif has been shown to suppress AGM A3G and A3F. The APOBEC3 genes in AGM have not been fully characterized. However, the ability of SIVagm Vif to overcome human A3C suggests that primate lentiviral Vif proteins may also act on antiviral proteins that the virus may not even encounter in its natural host. One possible interpretation is that SIVagmTan Vif recognizes human A3C in a degenerate fashion through its homology with AGM A3G or A3F. Alternatively, AGM may also encode A3C, which shares a similar structure with human A3C, and both molecules are recognized by SIVagmTan Vif.
The results of our comparative study of parental A3A, A3C and their chimeras have indicated that the amino-terminal region of A3C upstream from the active site of A3C determines the molecule’s sensitivity to Vif. This argument is further supported by the observation that amino-terminal deletion of amino acids 2-47 made mutant A3C resistant to Vif-induced degradation. Interestingly, amino acids 2-47 of A3G was dispensable for Vif-induced degradation (Fig. 8C). Our results are consistent with the previous observation that amino acids 54-124 of A3G is sufficient for interaction with HIV-1 Vif (Conticello et al., 2003) and residues 104-245 in A3G are sufficient for HIV-1 Vif-induced degradation (Zhang et al., 2008). Further studies are therefore needed to understand why the amino-terminal region of A3C but not A3G is required for Vif-mediated degradation.
Mutation of A3C at position 128 had no significant effect on the sensitivity of the protein to either HIV-1 or SIVagmTan Vif. This result is distinctly different from the situation observed with regard to human and AGM A3G (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). These observations further support the idea that different regions in A3G and A3C are critical for Vif-induced degradation. This result is also comparable to those obtained with A3F mutants, in which the equivalent mutations in A3F did not affect Vif sensitivity (Liu et al., 2005). Given the inability to generate the same species-specificity mutation in A3F and A3C, the charge alone at that position may not be the species determinant.
The homology between A3G and A3C suggests that these two factors may be similarly sensitive to Vif-induced degradation involving their respective active sites and linker regions. However, the chimera and deletion mutant data seem to indicate that the two proteins are recognized and poly-ubiquitinated in different ways. The contribution of the amino-terminus to the Vif-induced degradation of A3C is still unclear. Further investigation into the interaction of Vif with A3C and A3G should shed light on the different epitopes involved in Vif recognition. These interactions between Vif and APOBEC3 proteins are all potential targets for drugs that are designed to shift the balance between viral and host defences in the host’s favor.
Experimental procedures
Plasmids
A3A and A3C HA-tagged expression plasmids were obtained as a kind gift from Dr Michael Malim in the Department of Infectious Diseases, Guy’s, King’s and St Thomas’ School of Medicine, King’s College, London. The AGM A3G-HA expression vector was a gift from Nathaniel R. Landau (The Salk Institute for Biological Studies, La Jolla, CA). The A3F HA-tagged plasmid has been described previously (Liu et al., 2005). Plasmids A3G-HA and Cul5ΔNedd8 (pCul5ΔNedd8) have been described (Yu et al., 2003). All experiments were normalized for transfected DNA with control plasmid pcDNA3.1 (+) (Invitrogen) or pcDNA 3.1-GFP. The SIVagmTan Vif mutant clone, SIVagmTanΔVif, was constructed by inserting four nucleotides at the Age site in the Vif coding region of pSIVagmTan-1. The expression vectors pSIVagmTanVif-myc, pSIVagmTanVif AAA-myc, pSIVagmTanVifH111L-myc, pSIVagmTanVifC117S-myc, pSIVagmTanVifC135S-myc and pSIVagmTanVifH141L-myc have been described previously (Luo et al., 2005). HIV-1 Vif-myc and HIV-1 VifL145A have been described previously (Yu et al., 2004b). The A3G deletion construct used has been described previously (Luo et al., 2004). The A3CΔN-HA was amplified with the following primers: forward 5′-CTCGAGACC ATGGTCTCCTGGAAGACG-3′, reverse 5′-GAATCCCTACGCG TAATCTGGGAC-3′ containing XhoI and EcoRI site, respectively, and C-terminal HA-tag using A3C-HA as template. The PCR product was cloned into pcDNA3.1 to generate A3CΔN-HA plasmid. The A3CY128D and A3CY128K mutant constructs derived from the A3C-HA expression vector were constructed by the PCR-based mutagenesis method.
We constructed A3C and A3A chimeras by using overlapping template PCR to amplify the individual amino- and carboxy-terminals from pCMV4A3A or pCMV4A3C. We then used these PCR products as a template for a second PCR reaction, with primers specific for each specific amino- or carboxy-terminus, then cloned the products into the pcrBluntII TOPO vector. The resulting chimeras were digested with HindIII and SmaI and cloned into the expression vector pCMV4.
Antibodies and cell culture
The following antibodies were used for this study: anti-myc MAb (Sigma, catalogue No. M5546), anti-HA MAb (Covance; catalogue No. MMS-101R-10000), anti-human ribosomal P antigen (Immunovision; catalogue No. HPO -0100). 293T cells (ATCC; catalogue No. CRL-11268) and MAGI-CCR5 cells (AIDS Research Reagents Program, Division of AIDS, NIAID, NIH; catalogue No. 3522) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 10% fetal bovine serum and penicillin-streptomycin (D-10 medium) and passaged upon confluence.
Transfections and virus purification
DNA transfection was carried out using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. Virus in cell culture supernatants was cleared of cellular debris by centrifugation at 3000 r.p.m. for 15 min in a Sorvall RT 6000B centrifuge and filtration through a 0.22 μm pore size membrane (Millipore). Virus particles were then concentrated by centrifugation through a 20% sucrose cushion by ultracentrifugation at 100 000 g for 2 h at 4°C in a Sorvall Ultra80 ultracentrifuge. Viral pellets were re-suspended in lysis buffer [phosphate-buffered saline (PBS) containing 1% Triton X-100 and complete protease inhibitor cocktail (Roche)]. Viral lysates were analysed by immunoblotting.
Immunoblot analysis
Cells were collected 48 h after transfection, and cell and viral lysates were prepared as previously described (Yu et al., 2003). Cells (105) were lysed in 1× loading buffer [0.08 M Tris (pH 6.8), 2.0% sodium dodecyl sulfate (SDS), 10% glycerol, 0.1 M dithiothreitol and 0.2% bromophenol blue]. The samples were boiled for 10 min, and proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE). The secondary antibodies used were alkaline phosphatase-conjugated anti-human and anti-mouse antibodies (Jackson Immunoresearch), and staining was carried out with 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium solutions prepared from chemicals obtained from Sigma.
Immunoprecipitation
Transfected 293T cells were harvested, washed twice with cold PBS and lysed in lysis buffer [50 mM Tris (pH 7.5), with 150 mM NaCl, 1% Triton X-100 and complete protease inhibitor cocktail tablets] at 4°C for 1 h, then centrifuged at 10 000 g for 30 min. For anti-HA immunoprecipitation, precleared cell lysates were mixed with anti-HA antibody-conjugated agarose beads (Roche) and incubated at 4°C for 3 h, then washed three times with washing buffer [20 mM Tris (pH 7.5), with 100 mM NaCl, 0.1 mM EDTA and 0.05% Tween-20]. The beads were eluted with elution buffer (0.1 M glycine-HCl at pH 3.5 for anti-myc immunoprecipitation or 0.1 M glycine-HCl at pH 2.0 for anti-HA immunoprecipitation). The eluted materials were then analysed by SDS-PAGE and immunoblotting as previously described (Yu et al., 2003).
Single-round infectivity MAGI assays
Viral infection was determined by MAGI assay as follows: MAGI-CCR-5 cells were prepared in six-well plates in D-10 medium 1 day before infection; the cells were at 30-40% confluence on the day of infection. Cells were infected by removing the medium from each well and adding dilutions of virus in a total volume of 500 μl of complete DMEM with 20 μg of DEAE-dextran per well. After a 2 h incubation at 37°C in a 5% CO2 incubator, 2 ml of complete DMEM was added to each well, and the cells were incubated for 48 h under the same conditions. Supernatants were removed, and 800 μl of fixing solution (1% formaldehyde, 0.2% glutaraldehyde in PBS) was added. After a 5 min incubation, the cells were washed twice with PBS. The staining solution [20 μl of 0.2 M potassium ferrocyanide, 20 μl of 0.2 M potassium ferricyanide, 2 μl of 1 M MgCl2 and 10 μl of 40 mg ml-1 5-bromo-4-chloro-3-indolyl-β-d-galactosidaseactopyranoside (Xgal)] was added. Cells were incubated for 2 h at 37°C in a non-CO2 incubator. Staining was stopped by removing the staining solution, and the cells were thoroughly washed twice with PBS. β-Galatosidase activity is under the control of the HIV-1 LTR promoter, which is also transactivated with SIV integration; in this system, positive blue dots indicate the presence of integrated virus. Positive blue dots were counted, and viral infectivity was determined after normalizing the amount of input virus in terms of p27 antigen content. All results represent infections done in triplicate.
Acknowledgements
We thank B. Liu and Z. Xiao for their technical assistance; M. Malim, N. Landau and Y. Zheng for critical reagents; and D. McClellan for editorial assistance. This work was supported by a grant from the NIH (AI062644) and NIH (AI071769) to X.-F. Yu.
References
- Alce TM, Popik W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004;279:34083–34086. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol. 2004;5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci USA. 2004;101:3770–3774. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004;279:33177–33184. doi: 10.1074/jbc.M402062200. [DOI] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, et al. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Greene WC. APOBEC3 cytidine deaminases: distinct antiviral actions along the retroviral life cycle. J Biol Chem. 2006;281:8309–8312. doi: 10.1074/jbc.R500021200. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, et al. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA. 2006;103:15588–15593. doi: 10.1073/pnas.0604524103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80:1067–1076. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaisi M, Dussart S, Courcoul M, Bessou G, Vigne R, Decroly E. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem Biophys Res Commun. 2004;321:566–573. doi: 10.1016/j.bbrc.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Esnault C, Heidmann O, Delebecque F, Dewannieux M, Ribet D, Hance AJ, et al. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature. 2005;433:430–433. doi: 10.1038/nature03238. [DOI] [PubMed] [Google Scholar]
- Goff SP. Retrovirus restriction factors. Mol Cell. 2004;16:849–859. doi: 10.1016/j.molcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006;80:11710–11722. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Hulme AE, Bogerd HP, Cullen BR, Moran JV. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene. 2007;390:199–205. doi: 10.1016/j.gene.2006.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80:875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, et al. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005;280:18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol. 2004;78:11841–11852. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA. 2005;102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007;81:7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH. Natural resistance to HIV infection: The Vif-APOBEC interaction. C R Biol. 2006;329:871–875. doi: 10.1016/j.crvi.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Liao S, Trono D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 2004;279:14481–14483. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dor-weiler IJ, Svarovskaia ES, et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281:17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, et al. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Navarro F, Landau NR. Recent insights into HIV-1 Vif. Curr Opin Immunol. 2004;16:477–482. doi: 10.1016/j.coi.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PT, Yu XF. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol. 2007;81:9577–9583. doi: 10.1128/JVI.02800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol. 2007;81:8236–8246. doi: 10.1128/JVI.02694-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnow C, Bransteitter R, Klein MG, Goodman MF, Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007;445:447–451. doi: 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- Rose KM, Marin M, Kozak SL, Kabat D. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J Biol Chem. 2004;279:41744–41749. doi: 10.1074/jbc.M406760200. [DOI] [PubMed] [Google Scholar]
- Russell RA, Pathak VK. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007;81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–168. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif) Proc Natl Acad Sci USA. 2004;101:3927–3932. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Senger T, Manning G, Landau NR. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006;80:5984–5991. doi: 10.1128/JVI.00388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Suspene R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, et al. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32:2421–2429. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, et al. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem. 2004;279:35822–35828. doi: 10.1074/jbc.M405761200. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Kao S, Miyagi E, Khan MA, Buckler-White A, Plishka R, Strebel K. Production of infectious SIVagm from human cells requires functional inactivation but not viral exclusion of human APOBEC3G. J Biol Chem. 2005;280:375–382. doi: 10.1074/jbc.M408987200. [DOI] [PubMed] [Google Scholar]
- Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol. 2006;80:3112–3115. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P, Trono D. Editing at the crossroad of innate and adaptive immunity. Science. 2005;307:1061–1065. doi: 10.1126/science.1105964. [DOI] [PubMed] [Google Scholar]
- Turelli P, Vianin S, Trono D. The innate antiretroviral factor APOBEC3G does not affect human LINE-1 retrotransposition in a cell culture assay. J Biol Chem. 2004a;279:43371–43373. doi: 10.1074/jbc.C400334200. [DOI] [PubMed] [Google Scholar]
- Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004b;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- Wang T, Tian C, Zhang W, Luo K, Sarkis PT, Yu L, et al. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol. 2007;81:13112–13124. doi: 10.1128/JVI.00892-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Assembly of HIV-1 Vif-Cul5, E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu XF. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. Faseb J. 2007a;21:217–222. doi: 10.1096/fj.06-6773com. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Xiong Y, Zhang W, Tan L, Ehrlich E, Guo D, Yu XF. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J Mol Biol. 2007b;373:541–550. doi: 10.1016/j.jmb.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci USA. 2004;101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology. 2007;46:1810–1820. doi: 10.1002/hep.21893. [DOI] [PubMed] [Google Scholar]
- Yang B, Chen K, Zhang C, Huang S, Zhang H. Virion-associated uracil DNA Glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited Nascent HIV-1 DNA. J Biol Chem. 2007;282:11667–11675. doi: 10.1074/jbc.M606864200. [DOI] [PubMed] [Google Scholar]
- Yu X. Innate cellular defenses of APOBEC3 cytidine deaminases and viral counter-defenses. Current Opinion HIV AIDS. 2006;1:187–193. doi: 10.1097/01.COH.0000221590.03670.32. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004a;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004b;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol. 2004;78:12058–12061. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KL, Mangeat B, Ortiz M, Zoete V, Trono D, Telenti A, Michielin O. Model structure of human APOBEC3G. PLoS ONE. 2007;2:e378. doi: 10.1371/journal.pone.0000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang X, Tian C, Wang T, Sarkis PT, Fang Y, et al. Cytidine deaminase APOBEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell Microbiol. 2008;10:112–121. doi: 10.1111/j.1462-5822.2007.01020.x. [DOI] [PubMed] [Google Scholar]