

Abstract
Palladium-catalyzed C–H activation/C–C bond–forming reactions have emerged as a promising class of synthetic tools in organic chemistry. Among the many different means of forging C–C bonds using Pd-mediated C–H activation, a new horizon in this field is Pd(II)-catalyzed cross-coupling of C–H bonds with organometallic reagents via a Pd(II)/Pd(0) catalytic cycle. While this type of reaction has proven to be effective for the selective functionalization of aryl C(sp2)–H bonds, the focus of this review is on Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling, a topic of particular importance because reactions of this type enable fundamentally new methods for bond construction. Since our laboratory’s initial report on cross-coupling of C–H bonds in 2006, this area has expanded rapidly, and the unique ability of Pd(II) catalysts to cleave and functionalize alkyl C(sp3)–H bonds has been exploited to develop protocols for forming an array of C(sp3)–C(sp2) and C(sp3)–C(sp3) bonds. Furthermore, enantioselective C(sp3)–H activation/C–C cross-coupling has been achieved through the use of chiral amino acid-derived ligands, offering a novel technique for producing enantioenriched molecules. Although this nascent field remains at an early stage of development, further investigations hold the potential to revolutionalize the way in which chiral molecules are synthesized in industrial and academic laboratories.
Keywords: C–H activation, palladium, C–C coupling, organometallic reagent
1. Introduction
A frontier in modern organic synthesis is the discovery and development of novel reactions to convert unactivated carbon–hydrogen (C–H) bonds to carbon–carbon (C–C) bonds in a selective manner.[1] Many transition metals are capable of mediating such transformations, yet among them, palladium is unique in that the overall catalytic process can be tailored to mimic that of traditional Pd(0)-catalyzed cross-coupling reactions, which are known for their remarkable utility, practicality, and reliability.
In the past several decades, tremendous progress has been made in developing new Pd-catalyzed C–H activation/C–C bond–forming reactions.[1] Research in this field has largely focused on the discovery of new modes of catalysis, and on the expansion of substrate scope to include a broader array of starting materials and coupling partners. Successful examples of Pd-catalyzed C–H activation/C–C bond–forming processes include the direct coupling of olefins and aryl C(sp2)–H bonds (first reported by Fujiwara in 1967),[2,3] C–H arylation reactions using Pd(0)/PR3 catalysts and aryl halides,[4] the coupling of C–H bonds with alkyl- and aryl halides via Pd(II)/Pd(IV) redox chemistry,[5] C–H carbonylation reactions using carbon monoxide,[6] and the cross-coupling of two different arenes through double C–H activation.[7] Despite these efforts, one reaction that had remained elusive prior to our group’s initial report in 2005 was the catalytic coupling of C–H bonds with organometallic nucleophiles along a Pd(II)/Pd(0) cycle.[8]
The advent of cross-coupling reactions of organohalides and organometallic reagents has had a lasting impact in synthetic chemistry, as well as in scientific research more broadly.[9] Since the milestone discovery of the cross-coupling reaction between organomagnesium reagents and organic halides reported independently by Kumada and Corriu in 1972,[10, 11] the scope and practicality of these transformations have advanced dramatically. These improvements have been facilitated by several factors: 1) the use of other organometallic nucleophiles, including organotin (Stille–Migita), -boron (Suzuki–Miyaura), -zinc (Negishi) and -silicon (Hiyama) reagents;[9] 2) a refined understanding of the operative mechanism(s); and 3) the development of novel ligands.[12] These cross-coupling reactions are compatible with a vast array of organometallic coupling partners, including aryl-, vinyl-, alkyl-, allyl- and alkynyl-metal reagents. Recent advancements have broadened the substrate scope such that both alkyl electrophiles and alkyl nucleophiles can be used, leading to the formation of C(sp3)–C(sp2) and C(sp3)–C(sp3) bonds.[13, 14]
An enduring objective in the field of chemical synthesis is to design reaction systems that proceed under mild conditions without producing toxic byproducts and that employ cheap, readily available starting materials.[15] Given that traditional cross-coupling reactions require that both reactants have a reactive functional group, a logical improvement would be to develop methodology in which installation of one of the two functional groups would be obviated. In particular, this could be accomplished by utilizing Pd(II)-mediated C–H activation (rather than oxidative addition of the aryl or alkyl halide to Pd(0)) as a means of entering the catalytic cycle. This strategy would potentially offer several advantages relative to traditional cross-coupling reactions. Firstly, C–H bonds are ubiquitous in organic molecules; thus one could utilize the inherent chemical functionality of the substrate, rather than relying on prefunctionalization. Secondly, the preparation of the organohalide or pseudohalide starting materials would no longer be required, which would greatly reduce waste generation. Thirdly, this reaction could offer tactical advantages when the position-selective introduction of halides is not straightforward in a given synthetic plan.
There exist virtually unlimited opportunities for devising new transformations that use unactivated C–H bonds as reaction partners. C–H bonds can readily be cleaved by transition metal catalysts. For instance, using Pd, cyclopalladation has been extensively documented,[16-19] and a wide variety of heteroatom–containing directing groups have been utilized to achieve catalytic C–H functionalization of both aryl C(sp2)–H and alkyl C(sp3)–H bonds, the latter of which are markedly more inert.[20,21] In the initial stages of our studies to develop C–H activation/C–C cross-coupling reactions, these early studies served as the intellectual foundation for our research. At the same time, it was important for us to recognize that the reaction conditions needed for Pd(II)-catalyzed C–H functionalization would be incompatible with those typically used in Pd(0)-catalyzed cross-coupling. This is due to the fact that modern cross-coupling reactions rely upon bulky, electron-rich phosphine and N-heterocyclic carbine (NHC) ligands to promote oxidative addition of the organohalides and subsequent reductive elimination; however, there ligands are known to be detrimental in Pd(II)-catalyzed C–H activation reactions. Thus, our work drew inspiration from the research areas of both traditional cross-coupling and C–H activation. Our investigations ultimately led to the design and execution of the first controlled sequence of C–H activation followed by transmetallation with organometallic reagents via Pd(II)/Pd(0) redox chemistry.[8]
Catalytic C(sp2)–H activation methods have been widely developed for the functionalization of arenes and heteroarenes in the past two decades.[21] In comparison, much less research has been devoted to the catalytic activation of non-acidic alkyl C(sp3)–H bonds. Despite the remarkable progress made in C(sp3)–C(sp3) cross-coupling protocols using alkyl halides during the past several years,[13] efforts to couple C(sp3)–H bonds with organometallic reagents have generally been far less successful. The main purpose of this short review is to highlight the early endeavors, which mainly took place in our laboratory, that lead to the discovery of Pd(II)-catalyzed cross-coupling of C(sp3)–H bonds with organometallic reagents to form new C(sp3)–C(sp2) and C(sp3)–C(sp3) bonds. The proof of concept for this class of reactions was initially established using aryl C(sp2)–H activation, which is better understood and more predictable. Therefore, the first part of the review will provide a detailed discussion of factors concerning catalysis and reactivity in this class of C–H activation reactions. The early examples of Rh- and Ru-catalyzed C(sp2)–H activation/C–C cross-coupling reactions reported by Oi[22] and Murai,[23] as well as Pd(II)-catalyzed C(sp2)–H activation/C–C cross-coupling reactions reported subsequent to our initial reports have previously been reviewed,[1f, 24] and as such, they are beyond the scope of this review. Key problems in the field and potential solutions are also discussed.
2. The First Example of Pd(II)-Catalyzed Cross-Coupling of C(sp2)–H Bonds and Organotin Reagents
In the early stages of our investigations, we benefited from understanding the reaction mechanisms at play in the Pd(0)-catalyzed Stille–Migita and Suzuki–Miyaura reactions, which allowed us to identify potential problems that we could encounter (Scheme 1).[9] These reactions proceed through a sequence of oxidative addition of the organohalide to Pd(0), transmetallation of the organometallic reagent, and finally reductive elimination to form the desired C–C bond in the product. Based on this knowledge, we aimed to design a novel catalytic cycle in which Pd(II)-catalyzed C–H activation would generate an analogous [Pd(II)–R] species. Following transmetallation and reductive elimination, the resulting Pd(0) species would be reoxidized by a terminal oxidant to close the catalytic cycle (Scheme 2). Comparing the two catalytic cycles, we realized that there were three major obstacles for establishing this new mode of catalysis. Firstly, it was evident that the commonly used phosphine and NHC ligands, which play such a critical role in Pd(0) catalysis, would be incompatible with the Pd(II)-catalyzed C–H cleavage step. Secondly, a reoxidation system would be required to regenerate catalytically active Pd(II) from Pd(0), and this reoxidant would need to be compatible with all other steps in the cycle. Thirdly, Pd(II) could react competitively (or predominantly) with the organometallic reagents, rather than engaging with the directing group and inserting into the proximal C–H bond, which would result in formation of the homocoupling product. We approached each of these challenges in turn during the course of our efforts to merge C–H activation technology with cross-coupling chemistry.
Scheme 1.
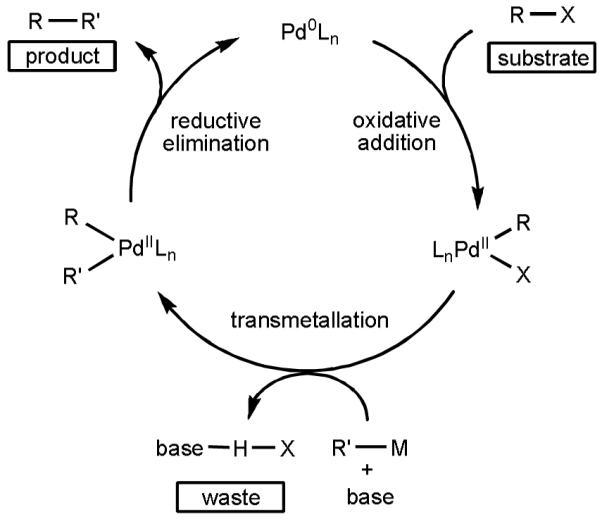
A Simplified Catalytic Cycle for Pd(0)-catalyzed Cross-Coupling Reactions
Scheme 2.
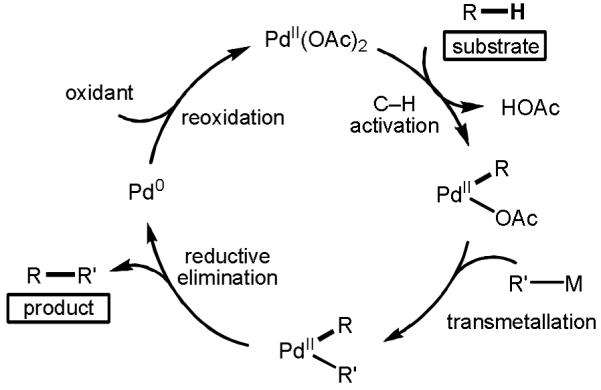
Proposed Catalytic Cycle for Pd(II) -catalyzed C–H Activation/C–C Cross-Coupling Reactions
Based on our proposed catalytic cycle, we attempted to overcome the three aforementioned obstacles by making logical choices of substrate (i.e., directing group), organometallic reagent, and terminal oxidant. When we initiated our investigation in 2004, we had already established that oxazoline gave excellent reactivity as a directing group in the diastereoselective iodination and acetoxylation of C–H bonds via Pd(II)/Pd(IV) catalysis (Scheme 3).[25] Thus, we chose to take advantage of the facile and robust reactivity of the oxazoline directing group in our efforts to establish the proof of concept for C–H activation/C–C cross-coupling via Pd(II)/Pd(0) catalysis. Selecting an appropriate coupling partner from the wide variety of organometallic reagents that have been utilized to date was cumbersome. The choice of organotin reagents was inspired by Hartwig’s early observation of transmetallation between a cyclopalladated complex and Me3SnPh (Scheme 4).[26] In that report, the complex needed to be prepared separately before it could undergo transmetallation with the organotin reagent, and the reductive elimination was facilitated by the intramolecular phosphine ligand. Therefore, our initial aim was to establish reaction conditions in which oxazoline-directed Pd(II)-catalyzed C–H activation would take place preferentially over organotin homocoupling. Subsequently, transmetallation and reductive elimination would fashion the desired product. Each step would need to take place without the aid of the phosphine ligand.
Scheme 3.
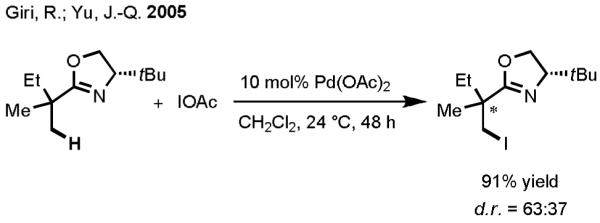
Diastereoselective Iodination of sp3 C–H Bonds via Pd(II)/Pd(IV) catalysis.
Scheme 4.
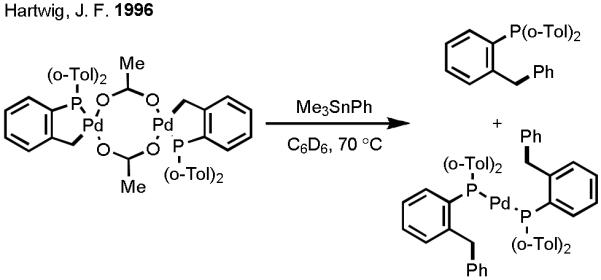
Transmetallation of an Organotin Reagent with a Cyclopalladated Complex.
In order to establish suitable conditions in a systematic manner, we initiated our endeavors by carrying out a detailed analysis of each individual step.[8] First, it was necessary to ascertain whether or not the cyclopalladated complex prepared by reacting the oxazoline substrate with 1 equiv of Pd(OAc)2 could undergo transmetallation with Me4Sn. Gratifyingly, exposing the preformed complex to Me4Sn led to formation of the expected methylated complex, albeit in a mere 20% yield (Scheme 5). Next, the reductive elimination step was investigated. In the case of the previously reported C–H iodination reaction using Pd(II)/Pd(IV) catalysis, the reductive elimination step is facilitated by the high oxidation state and steric bulk of the Pd(IV)-intermediate. An added benefit there is that catalytically active Pd(II) is regenerated upon reductive elimination from Pd(IV). However, in our Pd(II)/Pd(0) reaction system, the proposed reductive elimination of the desired cross-coupling product would need to take place from a relatively stable Pd(II) intermediate to give Pd(0), a process that is not nearly as straightforward. In the Pd(0)-catalyzed Stille–Migita coupling reaction, the reductive elimination step is accelerated by the presence of phosphine or NHC ligands. Thus, despite the fact that these specific types of ligands are incompatible with our reaction system, we nevertheless screened a wide array of available ligands in an effort to identify one that could promote reductive elimination. Gratifyingly, we made a crucial discovery that reductive elimination could be promoted by the addition of 1 equiv of 1,4-benzoquinone (BQ) to furnish the desired cross-coupling product. It is important to note that BQ had previously been used to promote C–C bond formation in an arene C–H activation/olefination reaction, a finding that inspired our attempts.[27] With several steps in the putative catalytic cycle established, we proceeded to identify suitable conditions to carry out this stepwise transformation catalytically in a single pot.
Scheme 5.
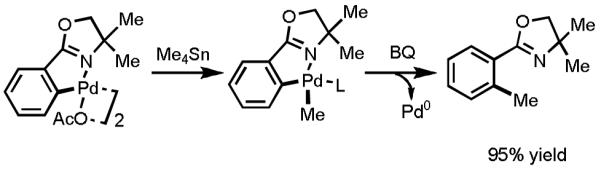
Stepwise Analysis of the Transmetallation and Reductive Elimination Steps.
As predicted, our efforts to devise a one-pot Pd(II)-catalyzed C–H activation/C–C cross-coupling reaction with organotins were met with tremendous challenges. At the outset, we feared that the Pd(II) species would react preferentially with the organotin reagents, rather than engaging the C–H bonds. Indeed, to our dismay, our initial attempts to treat oxazoline substrates with Pd(OAc)2 and organotin reagents under various conditions consistently resulted in full recovery of Pd(0) precipitates (Scheme 6), despite the fact that each individual step in the putative catalytic cycle had been firmly established previously. For the purposes of intelligence gathering in our screening studies, we decided to add the organotin reagents batchwise in order to reduce the rate of Pd(II)-mediated organotin homocoupling. This procedural maneuver ended up being vital, as it allowed us to observe meaningful quantities of the desired coupling products and to develop a reliable assay. To indentify a suitable reoxidation system to regenerate Pd(II) at the end of the catalytic cycle, common oxidants were surveyed. Reactions were carried out by stirring the oxazoline substrates with 10 mol% Pd(OAc)2 and BQ in the presence of various oxidants, and one batch of 0.037 equiv Me4Sn was added every 3 hours for 60 hours (20 batches total). From these studies, we determined that the combination of Cu(OAc)2 and BQ in CH3CN gave the highest yields (Scheme 7). This protocol represents the first example of Pd(II)-catalyzed cross-coupling between aryl C(sp2)–H bonds and organotin reagents.
Scheme 6.
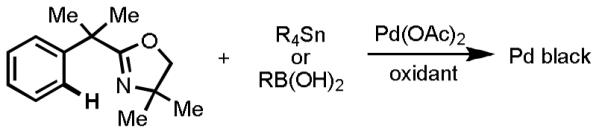
Failed Experiments. Precipitation of Pd Black.
Scheme 7.
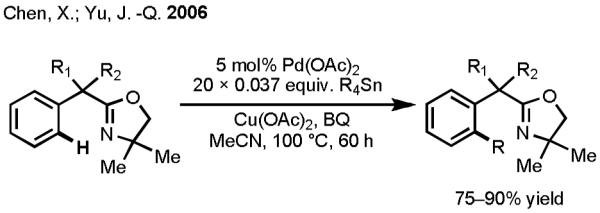
First Example of Pd(II)-catalyzed C(sp2)–H Activation/C–C Cross-Coupling with Organotin Reagents.
3. Pyridine-directed Pd(II)-Catalyzed Cross-Coupling of C(sp2)–H and C(sp3)–H Bonds and Organoboron Reagents
The Pd(II)-catalyzed C–H activation/C–C cross-coupling reaction with alkyltin reagents described in the preceding section established a proof of concept, demonstrating that organometallic reagents could be catalytically coupled with C–H bonds.[8] However, several unsolved problems remained to be addressed, including using more environmentally benign organometallic reagents and improving the substrate scope. Given these objectives, the development of Pd-catalyzed C–H activation/C–C cross-coupling using organoboron reagents (in analogy to the Suzuki–Miyaura reaction) seemed highly desirable. In particular, we viewed the use of organoboron reagents as advantageous because they are generally safe, stable, and because a large number are commercially available. We also hypothesized that the transmetalation rate (hence, the rate of undesired homocoupling) would be slower than in the case of organotin reagents, which we thought would be beneficial, potentially allowing us to avoid the cumbersome batchwise addition protocol that we resorted to earlier.
Moreover, one of our group’s major research interests is the development of new methods for C(sp3)–H functionalization.[1f] Pd(II) is capable of activating and functionalizing alkyl C(sp3)–H bonds; however, Pd(II)-mediated C(sp3)–H cleavage is far less facile than C(sp2)–H cleavage. Consequently, devising reaction conditions in which C(sp3)–H activation occurs faster than competitive homocoupling of the organometallic reagent is a tremendous challenge. Here, we postulated that it would be possible to address the homocoupling problem by using the highly efficient C(sp3)–H activation systems established in previous reports[1f, 25] in conjunction with organoboron reagents, which are known to undergo homocoupling more slowly.
As we examined the literature, we took note of the inspiring report by Buchwald and coworkers, in which Pd(0)/PR3-catalyzed oxidative addition/intramolecular C–H activation sequence was elegantly combined with a Suzuki–Miyaura coupling reaction to effect the arylation of C(sp3)–H bonds using external phenylboronic acids (Scheme 8).[28] Moreover, similar to Hartwig’s observation of a transmetallation event with organotin reagents, Sames observed transmetallation between a cyclopalladated complex and a vinylboronic acid.[29] Encouraged by our success in developing Pd(II)-catalyzed C–H activation/C–C cross-coupling using organotin reagents and inspired by the insights gleaned from these related studies, we moved forward to establish a Pd(II)-catalyzed cross-coupling of C–H bonds and organoboron reagents. To this end, we began by investigating suitable reaction conditions, first focusing our attention on systems proceeding via aryl C(sp2)–H activation.
Scheme 8.
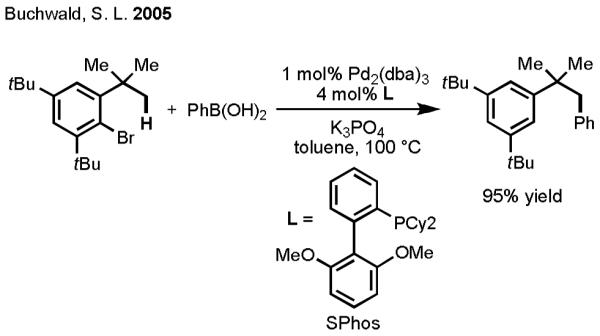
Pd(0)/PR3-catalyzed Cross-Coupling of C(sp3)–H Bonds with Phenylboronic Acid.
We first attempted to perform Pd(II)-catalyzed oxazoline-directed coupling of C(sp2)–H bonds with organoboronic acids but found that the reaction gave a mere 10% yield. As we explored other directing groups, we were pleased to find that reactions between substrates containing a pyridine directing group and alkylboronic acids (as well as methylboroxine) gave improved results (40–93% yield) (Scheme 9).[30] The use of Ag(I) as the reoxidant was critical for catalytic turnover and also for promoting transmetallation.[31] Intriguingly, arylboronic acids gave poorer results in these experiments (20–30% yield). Suzuki–Miyaura coupling reactions typically work better with arylboronic acids than with alkylboronic acids because the former undergo faster transmetallation than the latter and also because the resulting [Pd(II)–aryl] intermediate cannot participate in undesired β-hydride elimination, which can take place in the case of [Pd(II)–alkyl] intermediates.[9c] A plausible explanation for our observation is that arylboronic acids are more prone to Pd(II)-mediated homocoupling and are hence consumed more rapidly during the course of the reaction. In subsequent projects, we employed more stable arylboronic esters or trifuloroborates as our aryl nucleophile sources. After establishing this reaction with aryl C(sp2)–H activation, we then proceeded to apply this novel transformation to C(sp3)–H bonds.
Scheme 9.
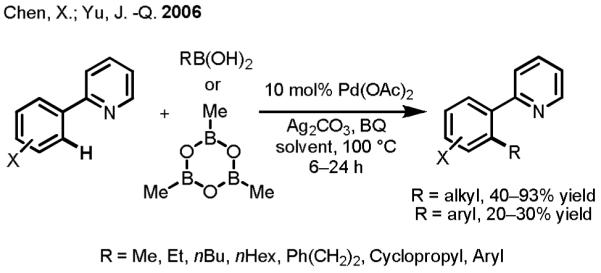
First Example of Pd(II)-catalyzed C(sp2)–H Activation/C–C Cross-Coupling with Organoboron Reagents.
Compared to many other transition metal catalysts, Pd(II) is superior in its ability to cleave alkyl C(sp3)–H bonds under relatively mild conditions.[1f, 20, 21] Having established that organoboron reagents could be coupled with aryl C(sp2)–H bonds, we were enticed to pursue the unprecedented alkyl C(sp3)–H activation variant of this transformation. To our delight, we were able to demonstrate the feasibility of such a process using the conditions developed for the C(sp2)–H activation reaction, leading to the discovery of the first Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling sequence (Scheme 10). Although generally, the primary alkyl C(sp3)–H bonds were found to give the best results, secondary methylene C(sp3)–H bonds were also reactive under these conditions, though they gave much lower yields.
Scheme 10.
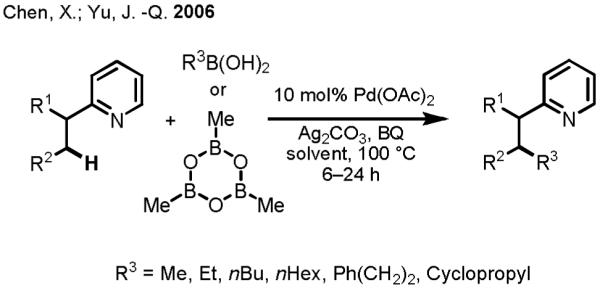
An Early Example of C(sp3)–H Activation/C–C Coupling
With the conceptual underpinnings of this new class of transformation firmly in place, we shifted our attention to expanding the synthetic utility by pursuing substrate classes with more synthetic relevance. At the same time, we were also aware of the need to find less expensive and more environmentally benign oxidants, in order to replace the copper and silver salts, which we used in superstoichiometric amounts in the studies described above.
4. Pd(II)-Catalyzed Cross-Coupling of C(sp2)–H and C(sp3)–H Bonds and Organoboron Reagents Directed by Carboxylic Acids and Hydroxamic Acids
As discussed earlier, a major problem in these coupling reactions is the undesired Pd(II)-mediated homocoupling of the organometallic reagents. This side reaction becomes predominant if C–H activation of the substrate is not sufficiently fast. In the two reports described above, the presence of strongly coordinating nitrogen-containing directing groups (i.e., oxazoline and pyridine) ensured rapid binding of the substrates with Pd(II) in solution. Owing to this strong coordination, C–H insertion in these systems is facile. However, these types of directing groups are disadvantageous because they are synthetically restrictive. Thus, we viewed that expanding the scope of the directing groups to include synthetically versatile and convertible functional groups, such as carboxylic acids and simple amides, as a major prerequisite for widespread use and general applicability.
Organic molecules that contain carboxylic acid moieties are versatile, ubiquitous, and inexpensive. Thus, using carboxylic acids to direct C–H functionalization seemed appealing to us in charting a path forward. Despite the attractiveness of this direction, our preliminary forays to establish carboxylate-directed Pd(II)-mediated insertion into inert aryl C(sp2)–H and alkyl C(sp3)–H bonds were largely unsuccessful. The key discovery that propelled our efforts in a productive direction was the observation that a wide range of cationic counter ions, including Na+, promoted carboxylate-directed Pd(II)-mediated insertion into C–H bonds.[32] In our working model, the sodium cation coordinates with the carboxylate group in a κ2 fashion, which forces Pd(II) to coordinate with the unhindered oxygen lone pair electrons. This finding enabled us for the first time to apply our C–H activation/C–C cross-coupling protocol to substrates that did not contain strong directing groups.
In 2007, our laboratory reported an example of carboxylic acid–directed Pd(II)-catalyzed C–H activation/C–C cross-coupling using both alkyl and aryl boronic acids (Scheme 11).[33] Again, our initial efforts to establish reactivity focused on aryl C(sp2)–H activation. The yields were generally poor, and the substrate scope was limited to only a few benzoic acids. On the boronic acid side, the only alkyl boronic acid that was found to work was MeB(OH)2. Upon transmetallation, other alkylboronic acids that contain β-hydrogen atoms from a [Pd(II)–alkyl] species, which is capable of undergoing rapid β-hydride elimination, rather than the desired reductive elimination to give the cross-coupling product. In Fu’s pioneering work on Pd(0)-catalyzed cross-coupling between alkyl halides and alkylboron reagents, β-hydride elimination is suppressed by the presence of sterically bulky, electron-donating trialkylphosphine ligands.[13,14] In our case, devising a strategy to suppress β-hydride elimination seemed daunting, and it remained a major unsolved problem until the work discussed at the end of this section. The use of Ag2CO3 as a stoichiometric oxidant was another significant practical drawback. Nevertheless, despite these limitations, we were encouraged that our goal to use simple functional groups to promote Pd(II)-mediated C–H insertion for the purposes catalytic coupling could be achieved.
Scheme 11.
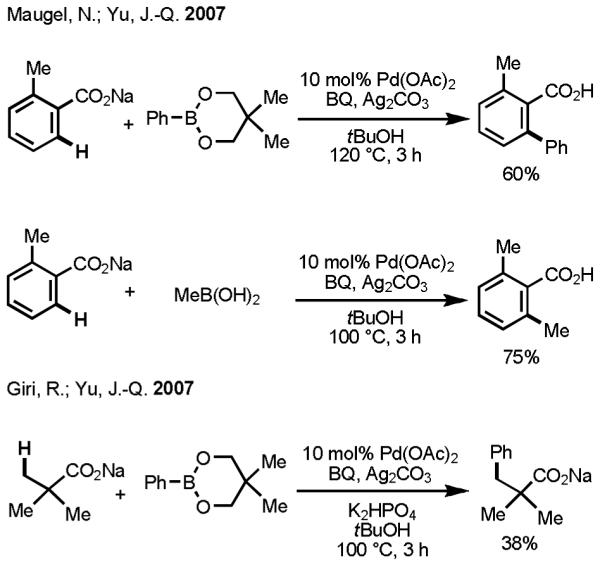
Carboxylate-Directed Coupling of C–H Bonds with Organoboron Reagents
Extensive screening was carried out in an effort to find suitable reaction conditions for carboxylate-directed Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling with organoboron reagents; however success was limited. Even under the best conditions, Pd was observed to precipitate out of solution in the form of Pd black or other aggregates after only three hours. At this point, no further C–H functionalization was found to take place. We elected to pursue two approaches to improve this rather challenging but potentially powerful transformation. Firstly, we aimed to improve drastically the rate of C(sp3)–H activation, which would ideally allow us to reduce the reaction temperature. To accomplish this, we sought to modify the directing group in such a way that reactivity could be improved while maintaining the overall synthetic versatility. Second, we aimed to develop a better reoxidation system to improve both the catalytic turnover number and reduce the cost. To this end, we examined the possibility of converting the carboxylic acid group into a simple hydroxamic acid (CONHOMe) moiety. Due to its structural similarity to a carboxylic acid, we hypothesized that this amide could mimic the coordination mode of an acid while also providing improved reactivity by offering stronger coordination. In addition, we thought that the methoxy group could provide steric hindrance, which is believed to be crucial for preventing β-hydride elimination in C(sp3)–C(sp3) cross-coupling reactions.
To our delight, after an extensive survey of possible reaction conditions, we established an improved protocol for β-C–C bond formation with aliphatic acid derivatives, which gave substantially higher yields at lower reaction temperatures (70 °C) (Scheme 12).[34] Furthermore, using sterically hindered 2,2,5,5-tetramethyltetrahydrofuran as solvent, we found success in coupling alkylboronic acids possessing β-hydrogen atoms with C(sp3)–H bonds in good yields. We believe that this solvent behaves as a bulky spectator ligand which inhibits β-hydride elimination from the [Pd(II)–alkyl] species. Moreover, we found that this transformation could be carried out in the absence of costly Ag(I) salts by using high pressure (20 atm) O2 or air as the terminal oxidant (Scheme 13). Although the need for a high pressure vessel severely detracts from the operational simplicity of this protocol (particularly on larger scale), the ability to use air for reoxidation is a significant step forward towards truly economical and green C–C bond–forming reactions via Pd(II)-catalyzed C–H activation.
Scheme 12.
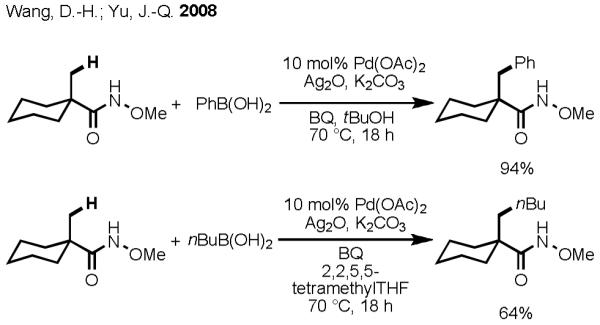
Hydroxamic Acid–Directed C(sp3)–H Activation/C–C Cross-Coupling with Aryl and Alkyl Boronic Acids
Scheme 13.
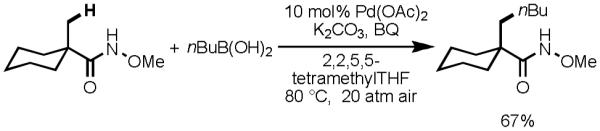
C(sp3)–H Activation/C–C Cross-Coupling of Alkyl Boronic Acids Reoxidized by High-Pressure Air
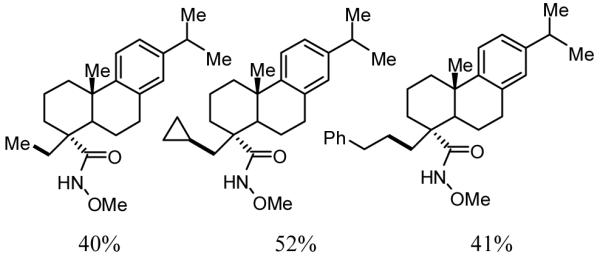
One of the considerations that motivated our efforts to pursue new classes of directing groups (particularly those commonly found in organic molecules) was our desire to enable novel synthetic applications. To demonstrate how Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling could be used drug diversification, we procured a sample of dehydroabietic acid, a natural product identified as an efficient BK channel opener used in the treatment of diseases such as acute stroke, epilepsy and asthma.[35] After conversion to the corresponding hydroxamic acid, the β-methyl group of this compound could be smoothly alkylated with a range of different alkyl boronic acids (Table 1). This technique holds tremendous promise in medicinal chemistry for the rapid synthesis of analog libraries, particularly given the fact that diversification of core structures of this type is generally prohibitively difficult since the molecule does not contain many reactive functional groups.
Table 1.
Dehydroabietic acid derivatives synthesized using Pd(II)-catalyzed C(sp3)–H Activation/C–C Cross-coupling
|
5. Current Challenges in C(sp3)–H Activation/C–C Cross-Coupling Reactions
In the three reports discussed above, we disclosed several notable discoveries that we made as part of our broader effort to develop novel C–H activation/C–C bond–forming reactions for organic synthesis. Despite these achievements, the field of C–H activation/C–C cross-coupling is still at an early stage, and these reactions have not yet reached high levels of practicality, reliability, and versatility, especially in comparison to Pd(0)-catalyzed cross-coupling reactions with aryl and alkyl halide electrophililes. To bridge this divide, several major hurdles must be surmounted in order for these reactions to find widespread application in synthetic settings. At the present time, our research laboratory is particularly intrigued and excited by three frontiers in this field.
Firstly, the substrate scope needs to be dramatically expanded. One severe limitation for carboxylic acid– and hydroxamic acid–directed C(sp3)–H activation/C–C cross-coupling reactions is that they are incompatible with substrates containing α-hydrogen atoms, which restricts the range of starting materials to those possessing quaternary centers at the α-position (Scheme 14).[33,34] The lack of reactivity can be explained by the loss of a favorable Thope–Ingold effect (comparing substrates with quaternary centers at the α-position to those containing α-hydrogen atoms).[36] In addition, for α-hydrogen-containing substrates, following Pd(II)-mediated C–H activation, the resulting palladacycle can undergo β-hydride elimination with the α-hydrogen atom.
Scheme 14.
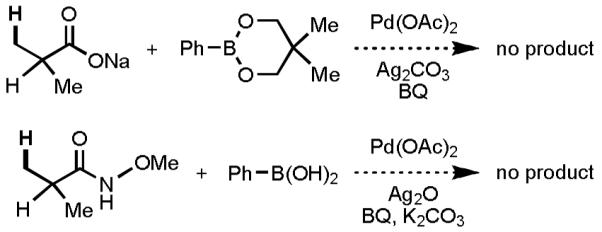
Incompatibility of Substrates Containing α-Hydrogen Atoms
Secondly, just as increasing the range of compatible substrate is a major challenge, so too is improving the scope of organometallic nucleophiles that can be used as coupling partners. One decisive advantage of modern cross-coupling chemistry in comparison to C–H activation/C–C cross-coupling is the fact that a massive library of organometallic reagents is available from commercial sources, nearly all of which perform reliably in cross-coupling. C–H activation/C–C cross-coupling, on the other hand, suffers from much narrower scope in this respect. For example, C–H activation/C–C cross-coupling with heteroaryl (pyridine, indole, thiazole, etc.)- and vinylboron reagents has heretofore been challenging. Thus, the coupling partner scope has been limited to only the simplest aryl and alkyl reagents. The root cause of this limitation is the fact that Pd(II) catalysts competitively coordinate to heteroatoms or olefins when they are present in the coupling partners. This coordination hampers directed Pd(II)-mediated C–H insertion. In addition, similar to the issues described above, Pd(II)-mediated homocoupling of these reagents outpaces C–H insertion.
Thirdly, we aim to develop a broadly applicable asymmetric C(sp3)–H activation/C–C cross-coupling protocol, with the hope of delivering an unprecedented disconnection for asymmetric C–C bond construction. Reactions of this type hold the potential to expedite campaigns in target-oriented synthesis and would enable new strategies that take advantage of simpler and more abundant starting materials. It is also anticipated that the chiral ligands discovered may be the key to expanding scope of the reactions as mentioned above.
In 2008, we disclosed the first example of Pd(II)-catalyzed enantioselective C–H activation/C–C cross-coupling using monoprotected amino acids as chiral ligands to effect stereoinduction with substrates containing pyridine directing groups.[37] In that report, we focused on desymmetrization of the prochiral C–H bonds on geminal diaryl or dimethyl groups. Notable among the mechanistic insights gleaned from this investigation was the finding that mono-N-protection of the amino acid ligand was crucial for chiral recognition. Using Boc-Leu-OH as the ligand, we successfully obtained the enantiomerically enriched C(sp2)–H activation/C–C cross-coupling product in 63% yield and 90% ee (Scheme 17). In an effort to expand the scope of this reaction, we also attempted to perform enantioselective C(sp3)–H activation of prochiral methyl groups. To our disappointment, despite extensively screening ligands and reaction conditions, we were able to obtain only 38% yield of the desired product with a maximum ee of just 37%.
Scheme 17.
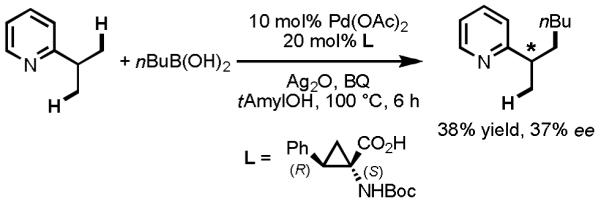
Desymmetrization of Prochiral C(sp3)–H bonds through Enantioselective C–H Activation
One problem that we faced during this investigation was that the high reaction temperatures required for C(sp3)–H activation proved to be deleterious to chiral induction. Thus, for future studies, it is clear that improved reactivity is needed, such that high yields can be obtained even at low temperatures. Only when this is achieved will this type of asymmetric C–C bond–forming reaction gain traction in practical applications.
These three formidable challenges are among the main unsolved problems in the field of Pd(II)-catalyzed C–H activation. Ultimately, throughout the above discussion, a pervasive issue that has often hampered reaction development is the slow rate of C–H insertion by Pd(II), which often leads to low product yield, substrate decomposition, and side product formation. Thus, as is evident from the research presented in this review, a principal consideration in the reaction discovery process in our laboratory is the design and/or selection of directing groups that are capable of positively influencing the rate of C–H insertion. Given that C(sp3)–H cleavage with Pd(II) is slower than C(sp2)–H cleavage, directing group development becomes even more important when probing new C(sp3)–H fuctionalization methods. With these factors in mind, a major investment has been made in our laboratory to invent truly practical and powerful directing groups that can simultaneously address the three aforementioned challenges.
In light of our observation that hydroxamic acid directing groups were capable of promoting facile C(sp3)–H activation, we conjectured that by systematically modifying the steric and electronic properties of an amide directing group, we could find a solution to our reactivity problems (Scheme 18). In early 2010, we reported the first example of Pd(II)-catalyzed olefination of C(sp3)–H bonds using a novel N-arylamide (CONHAr) directing group.[38] A wide array of N-arylamide substrates were prepared by merging carboxylic acids and commercially available anilines. Extensive fine tuning of the directing group was carried out in order to study the influence of steric and electronic properties on the observed reactivity. Utlimately we found that N-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl)amide directing groups, which are both highly electron-deficient and relatively sterically unencumbered, gave the highest reactivity. Remarkably, using this directing group, C(sp3)–H activation could efficiently take place, even in the presence of competitively coordinating olefins. In addition, substrates containing α-hydrogen atoms could now be used in C(sp3)–H functionalization reactions, thus substantially expanding the substrate scope. Further studies have revealed that the C(sp3)–H insertion occurs at reaction temperatures as low as 25 °C, albeit in slower rates.
Scheme 18.
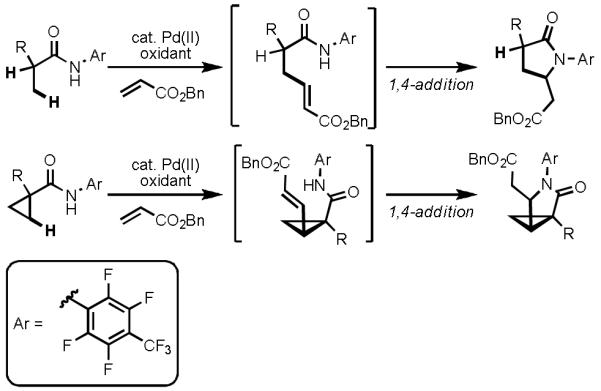
First Example of Pd(II)-catalyzed Olefination of C(sp3)–H Bonds Enabled by a Novel N-Arylamide Directing Group
The discovery of this novel directing group has since allowed us to perform a wide variety of C(sp3)–H functionalization reactions under mild conditions with substrates containing α-hydrogen atoms. We have already disclosed a novel Pd(0)/phosphine-catalyzed intermolecular C(sp3)–H arylation reaction,[4i] as well as a C(sp3)–H arylation reaction using Pd(II)/Pd(IV) catalysis.[5h] Additional manuscripts describing Pd(II)-catalyzed intermolecular carbonynation, oxygenation, and intramolecular amination are currently in preparation. In summary, this directing group offers an unprecedented levels of compatibility with three types of Pd-catalyzed C–H activation, namely Pd(0)/Pd(II), Pd(II)/(0), and Pd(II)/(IV) catalysis. This versatility has allowed us to convert C(sp3)–H bonds into a wide array of C–C and C–heteroatom bonds. However, the ultimate goal of our work is to apply this newly improved reactivity to address problems in enantioselective C(sp3)–H activation/C–C cross-coupling. Our preliminary results in this direction are extremely promising, and the results will be disclosed in the very near future.
6. Summary and Outlook
The myriad recent advances in the field of Pd-catalyzed C–H activation/C–C bond formation are highly encouraging, suggesting that this class of transformation could not only serve as a complement to cross-coupling reactions with organohalides, but could eventually replace this traditional chemistry in some contexts. Moreover, Pd(II)-catalyzed C(sp3)–H activation/C–C cross-coupling, the main subject of this review, holds the promise to generate carbogenic complexity in an expedient and stereocontrolled fashion. Our group developed the first protocol for successful C–H activation/C–C cross-coupling with organometallic reagents along a Pd(II)/Pd(0) catalytic cycle. Since this initial discovery, this class of reactions has been applied to increasingly diverse classes of substrates and coupling partners. After establishing aryl C(sp2)–H activation/C–C cross-coupling, we have gone on to explore alkyl C(sp3)–H activation/C–C cross-coupling, a tremendously challenging transformation. As part of our group’s focus on developing simple, practical reactions, we have pursued new strategies for C(sp3)–H activation directed by simple, ubiquitous functional groups. In the future, expanding the range of compatible substrates and coupling partners will continue to usher this catalytic transformation into the mainstream. Simultaneously, careful investigation into enantioselective C(sp3)–H activation/C–C cross-coupling has the potential to change the ways in which carbogenic stereocenters are synthesized in academic and industrial laboratories. Success on this front will likely hinge on the design and synthesis of new chiral ligands, as well as improved directing groups
Scheme 16.
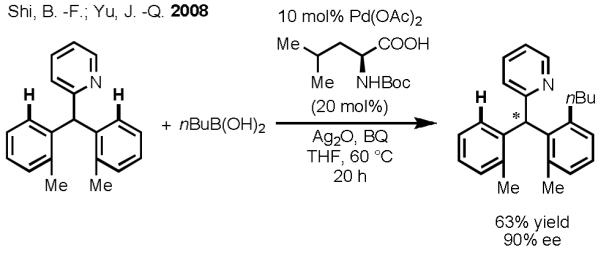
Desymmetrization of Prochiral C(sp2)–H Bonds through Enantioselective C–H Activation
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS, 1 R01 GM084019-01A1) for financial support, Bristol Myers Squibb for a predoctoral fellowship (M. W.), and the National Science Foundation, the Department of Defense, and the Skaggs Oxford Scholarship program for predoctoral fellowships (K.M.E.).
References
- [1].For reviews on C–H activation chemistry, see: Crabtree RH. Chem. Rev. 1985;85:245. Shilov AE, Shul’pin GB. Chem. Rev. 1997;97:2879. doi: 10.1021/cr9411886. Stahl SS, Labinger JA, Bercaw JE. Angew. Chem. Int. Ed. 1998;37:2181. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2180::AID-ANIE2180>3.0.CO;2-A. Bergman RG. Nature. 2007;446:391. doi: 10.1038/446391a. Brookhart M, Green MLH, Parkin G. Proc. Natl. Acad. Sci. 2007;104:6908. doi: 10.1073/pnas.0610747104. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273.
- [2].Moritani I, Fujiwara Y. Tetrahedron Lett. 1967;8:1119. [Google Scholar]
- [3].For selected examples of Pd-catalyzed C–H olefination reactions, see: Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J. Org. Chem. 1998;63:5211. Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992. doi: 10.1126/science.287.5460.1992. Yokota T, Tani M, Sakaguchi S, Ishii Y. J. Am. Chem. Soc. 2003;125:1476. doi: 10.1021/ja028903a. Dams M, De Vos DE, Celen S, Jacobs PA. Angew. Chem. Int. Ed. 2003;42:3512. doi: 10.1002/anie.200351524. Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. 2010;327:315. doi: 10.1126/science.1182512.
- [4].For selected examples of C–H arylation reactions using Pd(0)/PR3 catalysts and aryl halides, see: Nakamura N, Tajima Y, Sakai K. Heterocycles. 1982;17:235. Akita Y, Ohta A. Heterocycles. 1982;19:329. Akita Y, Ohta A. Heterocycles. 1982;19:329. Hennings DD, Iwasa S, Rawal VH. J. Org. Chem. 1997;62:2. doi: 10.1021/jo961876k. Pivsa-Art S, Satoh T, Kawamura Y, Miura M, Nomura M. Bull. Chem. Soc. Jpn. 1998;71:467. Roesch KR, Larock RC. J. Org. Chem. 1998;63:5306. Hennessy EJ, Buchwald SL. J. Am. Chem. Soc. 2003;125:12084. doi: 10.1021/ja037546g. Ren H, Knochel P. Angew. Chem. Int. Ed. 2006;45:3462. doi: 10.1002/anie.200600111. Satoh T, Kawamura Y, Miura M, Nomura M. Angew. Chem. Int. Ed. 1997;36:1740. Campeau L-C, Rousseaux S, Fagnou K. J. Am. Chem. Soc. 2005;127:18020. doi: 10.1021/ja056800x. Chiong HA, Pham Q-N, Daugulis O. J. Am. Chem. Soc. 2007;129:9879. doi: 10.1021/ja071845e. Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p.
- [5].For selected examples of C–C bond forming–reactions via Pd(II)/Pd(IV) catalysis, see: Tremont SJ, Rahman HU. J. Am. Chem. Soc. 1984;106:5759. Xia M, Chen Z-C. Syn. Commun. 2000;30:531. Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. Daugulis O, Zaitsev VG. Angew. Chem. Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. Deprez NR, Kalyani D, Krause A, Sanford MS. J. Am. Chem. Soc. 2006;128:4972. doi: 10.1021/ja060809x. Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. Reddy BVS, Reddy LR, Corey EJ. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j. Wasa M, Yu J-Q. Tetrahedron. 2010;66:4811. doi: 10.1016/j.tet.2010.03.111. Shabashov D, Daugulis O. J. Am. Chem. Soc. 2010;1327:3965. doi: 10.1021/ja910900p.
- [6].For selected examples of C–H carbonylation reactions using carbon monoxide, see: Fujiwara Y, Kawauchi T, Taniguchi H. J. Chem. Soc., Chem. Commun. 1980:220. Itahara T. Chem. Lett. 1982:1151. T. Itahara T. Chem. Lett. 1983:127. Fujiwara Y, Takaki K, Taniguchi Y. Synlett. 1996:591. Orito K, Horibata A, Nakamura T, Ushito H, Nagasaki H, Yuguchi M, Yamashita S, Tokuda M. J. Am. Chem. Soc. 2004;126:14342. doi: 10.1021/ja045342+. Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082. doi: 10.1021/ja8063827.
- [7].For selected examples of cross–coupling reactions between two different arenes through double C–H activation, see: Itahara T. Synthesis. 1979:151. Li R, Jiang L, Lu W. Organometallics. 2006;25:5973. Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. Stuart DR, Villemure E, Fagnou K. J. Am. Chem. Soc. 2007;129:12072. doi: 10.1021/ja0745862. Xia J-B, You S-L. Organometallics. 2007;26:4869. Hull KL, Sanford MS. J. Am. Chem. Soc. 2007;129:11904. doi: 10.1021/ja074395z. Li B-J, Tian S-L, Fang Z, Shi Z-J. Angew. Chem., Int. Ed. 2008;47:1115. doi: 10.1002/anie.200704092. Brasche G, Garcia-Fortanet J, Buchwald SL. Org. Lett. 2008;10:2207. doi: 10.1021/ol800619c. Cho SH, Hwang SJ, Chang S. J. Am. Chem. Soc. 2008;130:9254. doi: 10.1021/ja8026295. Hull KL, Sanford MS. J. Am. Chem. Soc. 2009;131:9651. doi: 10.1021/ja901952h. Zhao X, Yeung CS, Dong VM. J. Am. Chem. Soc. 2010;132:5837. doi: 10.1021/ja100783c.
- [8].Chen X, Li J-J, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:78. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]
- [9].For reviews, see: Stille JK. Angew. Chem. Int. Ed. 1986;25:508. Heck RF. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Pergamon; Oxford: 1991. p. 833. Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 1998. Luh T-Y, Leung M-K, Wong K-T. Chem. Rev. 2000;100:3187. doi: 10.1021/cr990272o. Hiyama T. J. Organomet. Chem. 2002;653:58. Negishi E.-i., Hu Q, Huang Z, Qian M, Wang G. Aldrichimica Acta. 2005;38:71. Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368. Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338. doi: 10.1002/anie.200800497. Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369. Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486. doi: 10.1021/ar800037p. Molander GA, Canturk B. Angew. Chem. Int. Ed. 2009;48:2. doi: 10.1002/anie.200904306.
- [10].a) Tamao K, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:4374. [Google Scholar]; b) Corriu RJP, Massse JP. J. Chem. Soc., Chem. Commun. 1972:144. [Google Scholar]
- [11].For the first example of Pd-catalyzed cross-coupling of organomagnesium reagents and organic halides, see: Yamamura M, Moritani I, Murahashi S. J. Organomet. Chem. 1975;91:C39.
- [12].For advances in ligand developments, see: Old DW, Wolfe JP, Buchwald SL. J. Am. Chem. Soc. 1998;120:9722. Littke AF, Fu GC. Angew. Chem. Int. Ed. 1998;37:3387. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P. Herrmann WA, Reisinger C-P, Spiegler M. J. Organomet. Chem. 1998;557:93. Hamann BC, Hartwig JF. J. Am. Chem. Soc. 1998;120:7369. Zhang C, Huang J, Trudell ML, Nolan SP. J. Org. Chem. 1999;64:3804. Rataboul F, Zapf A, Jackstell R, Harkal S, Riermeier T, Monsees A, Dingerdissen U, Beller M. Chem. Eur. J. 2004;10:2983. doi: 10.1002/chem.200306026.
- [13].For reviews of β-alkyl Suzuki-Miyaura cross–coupling reaction, see: Chemler SR, Trauner D, Danishefsky SJ. Angew. Chem. Int. Ed. 2001;40:4544. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. Doucet H. Eur. J. Org. Chem. 2008:2013.
- [14].a) Netherton MR, Dai C, Neuschütz K, Fu GC. J. Am. Chem. Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; b) Saito B, Fu GC. J. Am. Chem. Soc. 2007;129:9602. doi: 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Saito B, Fu GC. J. Am. Chem. Soc. 2008;130:6694. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- [15].Clark JH. Green Chem. 1999;1:1. [Google Scholar]
- [16].For comprehensive reviews on cyclopalladation reactions, see: Ryabov AD. Synthesis. 1985:233. Canty AJ. In: Comprehensive organometallic chemistry II: a review of the literature 1982-1994. Abel EW, Stone FGA, Wilkinson G, editors. Pergamon; 1995. pp. 225–255.
- [17].a) Cope AC, Siekman RW. J. Am. Chem. Soc. 1965;87:3272. For an early example of cyclopalladation of C(sp2)–H bonds directed by azo groups, see: [Google Scholar]; b) Constable AG, McDonald WS, Sawkins LC, Shaw BL. J. Chem. Soc. Chem. Commun. 1978:1061. For an early example of cyclopalladation of C(sp3)–H bonds directed by oximes see: [Google Scholar]
- [18].For early examples of exploiting cyclopalladation reactions in synthesis, see: Carr K, Sutherland JK. J. Chem. Soc. Chem. Commun. 1984:1227. Baldwin JE, Jones RH, Najera C, Yus M. Tetrahedron. 1985;41:699. Bore L, Honda T, Gribble GW. J. Org. Chem. 2000;65:6278. doi: 10.1021/jo0008688.
- [19].For development of mechanistic models for cyclopalladation, see: Ryabov AD, Sakodinskaya IK, Yatsimirsky AK. J. Chem. Soc. Dalton Trans. 1985:2629. Burger P, Bergman RG. J. Am. Chem. Soc. 1993;115:10462. Canty AJ, van Koten G. Acc. Chem. Res. 1995;28:406. Gómez M, Granell J, Martinez M. Organometallics. 1997;16:2539. Gómez M, Granell J, Martinez M. J. Chem. Soc., Dalton Trans. 1998:37. Vigalok A, Uzan O, Shimon LJW, Ben-David Y, Martin JML, Milstein D. J. Am. Chem. Soc. 1998;120:12539. Webster CE, Fan Y, Hall MB, Kunz D, Hartwig JF. J. Am. Chem. Soc. 2003;125:858. doi: 10.1021/ja028394c. Davies DL, Donald SMA, Macgregor SA. J. Am. Chem. Soc. 2005;127:13754. doi: 10.1021/ja052047w. Lafrance M, Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. García-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a.
- [20].For a recent review on C(sp3)–H bond functionalization, see: Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem.—Eur. J. 2010;16:2654. doi: 10.1002/chem.200902374.
- [21].For reviews, see: Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. Ackermann L. Top. Organomet. Chem. 2008;24:35. Kalyani D, Sanford MS. Top. Organomet. Chem. 2008;24:85.
- [22].Oi S, Fukita S, Inoue Y. Chem. Commun. 1998:2439. [Google Scholar]
- [23].Kakiuchi F, Kan S, Igi K, Chatani N, Murai S. J. Am. Chem. Soc. 2003;125:1698. doi: 10.1021/ja029273f. [DOI] [PubMed] [Google Scholar]
- [24].Sun C-L, Li B-J, Shi Z-J. Chem. Commun. 2010;46:677. doi: 10.1039/b908581e. [DOI] [PubMed] [Google Scholar]
- [25].a) Giri R, Chen X, Yu J-Q. Angew. Chem. Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; b) Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew. Chem. Int. Ed. 2005;44:7420. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- [26].Louie J, Hartwig JF. Angew. Chem. Int. Ed. 1996;35:2359. [Google Scholar]
- [27].Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]
- [28].Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- [29].Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J. Am. Chem. Soc. 2002;124:11856. doi: 10.1021/ja027311p. [DOI] [PubMed] [Google Scholar]
- [30].Chen X, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:12634. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]
- [31].(a) Uenishi J.-i., Beau J-M, Armstrong RW, Kishi Y. J. Am. Chem. Soc. 1987;109:4756. [Google Scholar]; (b) Frank SA, Chen H, Kunz RK, Schnaderbeck MJ, Roush WR. Org. Lett. 2000;2:2691. doi: 10.1021/ol0062446. [DOI] [PubMed] [Google Scholar]
- [32].Mei T-S, Giri R, Maugel N, Yu J-Q. Angew. Chem. Int. Ed. 2008;47:5215. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]
- [33].(a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Wang D-H, Mei T-S, Yu J-Q. J. Am. Chem. Soc. 2008;130:17676. doi: 10.1021/ja806681z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang D-H, Wasa M, Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- [35].Ohwada T, Nonomura T, Maki K, Sakamoto K, Ohya S, Muraki K, Imaizumi Y. Bioorg. Med. Chem. Lett. 2003;13:3971. doi: 10.1016/j.bmcl.2003.08.072. [DOI] [PubMed] [Google Scholar]
- [36].Bachrach SM. J. Org. Chem. 2008;73:2466. doi: 10.1021/jo702665r. [DOI] [PubMed] [Google Scholar]
- [37].Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem. Int. Ed. 2008;47:4882. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- [38].Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]