

Abstract
Currently, reversed-phase high-performance liquid chromatography (HPLC) is the method of choice for determining the types and amounts of muropeptide subunits comprising bacterial peptidoglycan. Although effective and sensitive, the technique does not lend itself to high throughput screening, and its complexity and equipment requirements may dissuade some investigators from pursuing certain types of cell wall experiments. Previously, we showed that muropeptides can be labeled with a fluorescent dye and separated by fluorophore-assisted carbohydrate electrophoresis (FACE), a simple and rapid gel procedure that might serve as a prelude to more intense analysis by HPLC. To validate the utility of FACE, we used both techniques to perform a side-by-side analysis of the peptidoglycan of eight mutants and their Escherichia coli parent strain. FACE and HPLC both detected the seven major muropeptides, which represent more than 95% of the total muropeptides present in this organism. In addition, FACE returned the same relative and quantitative results in 92% of 72 measurements, indicating that the procedure gives an accurate overview of peptidoglycan composition. The results also suggest a possible biochemical activity for the AmpC and AmpH proteins of E. coli, and the use of FACE as an in vitro enzyme assay detected possible substrate preferences for the endopeptidase penicillin binding protein 4.
Keywords: Muropeptide, Peptidoglycan, Fluorophore-assisted carbohydrate electrophoresis, High-performance liquid chromatography, Penicillin binding protein, E. coli
Most eubacteria owe their shape and mechanical stability to a peptidoglycan wall which forms a monomolecular sacculus enclosing the cell. This material consists of a series of carbohydrate chains (polymers of alternating N-acetylglucosamine (NAG)2 and N-acetylmuramic acid) interconnected by covalent cross-links between short peptides [1,2]. Over 100 peptidoglycan types emerge from this basic structure because of variations in length and composition of the peptide side chains and because of differences in the way these peptides are covalently linked to one another [3]. When the sacculus is digested by lysozyme or muramidases, which cleave the glycan chains, mature peptidoglycan is seen to be composed of a range of subunits (muropeptides) [4,5]. For example, digests of Escherichia coli peptidoglycan typically yield 10–12 major muropeptides, although 30–40 minor compounds may be detected in some circumstances [4,5].
Because it is unique to bacteria and important for their survival, interfering with the integrity of the peptidoglycan sacculus forms the basis of many of our most valuable antimicrobial agents. For this reason and for understanding the fundamental biology of cell walls and cell shape, it is important to monitor and understand peptidoglycan structure in a variety of circumstances and mutants.
Currently, reversed-phase high-performance liquid chromatography (HPLC) is the most accurate method of analyzing the muropeptide composition of peptidoglycan [4,5]. However, because the technique can be demanding and limits the rapidity with which multiple samples can be processed, we adapted the technique of fluorophore-assisted carbohydrate electrophoresis (FACE) [6,7] as an alternate method for muropeptide analysis [8]. Although preliminary experiments indicated that FACE worked as well as HPLC in detecting the major muropeptides of E. coli [8], there is as yet no direct comparison of the analytical correspondence between the two procedures. Therefore, the objective of the present work was to optimize and validate the FACE procedure as a rapid and reliable method that could substitute for or complement the use of HPLC in certain circumstances. To make this comparison we employed both methods to analyze the muropeptide composition of E. coli and eight isogenic mutants. The results indicate that FACE detects the same major muropeptides as does HPLC, and both methods return similar relative concentrations of these subunits.
Materials and methods
Strains and growth conditions
E. coli strains and mutants are listed in Table 1. Bacteria were maintained by growth on Luria–Bertani (LB) medium, but were grown in M9 minimal glucose medium prior to peptidoglycan preparation [9]. B. Meberg constructed E. coli CS18-4 by inserting a dacD∷-Kan allele into E. coli CS109 by procedures described previously [10], and she constructed CS804-1K by P1 transduction of the dacD∷Kan allele from CS18-4 into CS703-1 [10].
Table 1.
E. coli strains
| Strains | Genotype | PBP(s) deleted | Source |
|---|---|---|---|
| CS109 | W1485, λ− F− thi rph-1 rpoS | None | C. Schnaitman |
| CS12-7 | CS109 dac A | 5 | [21] |
| CS13-2 | CS109 ΔmrcA-yrfEF | 1a | [21] |
| CS14-2 | CS109 ampC | AmpC | |
| CS15-3 | CS109 ampH | AmpH | [21] |
| CS18-4K | CS109 dacD∷Kan | DacD | B. Meberg |
| CS214-1 | CS109 ΔmrcA-yrfEF dacC | 1a 5 | |
| CS357-3 | CS109 ΔmrcA-yrfEF dacC ampH | 1a 5 AmpH | [21] |
| CS362-1 | CS109 ΔmrcA-yrfEF dacC ampC | 1a 5 AmpC | |
| CS420-2 | CS109 ΔmrcA-yrfEF dacC ampC ampH | 1a 5 AmpC AmpH | [21] |
| CS617-1K | CS109 mrcA∷Kan dacB dacC pbpG ampC ampH | 1a 4 6 7 AmpC AmpH | [10] |
| CS703-1 | CS109 mrcA dacB dacA dacC pbpG ampC ampH | 1a 4 5 6 7 AmpC AmpH | [10] |
| CS804-1K | CS109 mrcA∷Kan dacB dacA dacC pbpG dacD ampC ampH | 1a 4 5 6 7 DacD AmpC AmpH | B. Meberg |
Peptidoglycan preparation
Peptidoglycan was prepared as described previously [8,11]. Bacteria from a fresh overnight culture were diluted 1:200 into 400mlM9 minimal glucose medium and grown at 37 °C to an A600 of 0.5–0.7. Cells were cooled rapidly to 4 °C, harvested by centrifugation at 10,000g for 15 min at 4 °C, and then resuspended to 0.2 g/ml in distilled water. Resuspended cells were added dropwise, with vigorous stirring, to an equal volume of boiling 8% sodium dodecyl sulfate (SDS). The solution was boiled for 30 min, after which the lysate was allowed to cool overnight to room temperature. Insoluble peptidoglycan was pelleted by ultracentrifugation at 100,000g for 60 min at room temperature, and the pellet was washed, resuspended in distilled water, and repelleted at least six times until the SDS concentration fell below 1 μg/ml, as determined by the methylene blue assay [12]. Pelleted peptidoglycan was resuspended in 5ml of 10mM Tris–HCl, pH 7.0, plus 10mM NaCl and solubilized by 5-min sonication in closed microcentrifuge tubes. Glycogen contamination was removed by adding α-amylase (100 μg/ml final) and imidazole (0.32M final), and the mixture was incubated for 2 h at 37 °C. Afterward, Pronase (pretreated by incubation at 60 °C for 2 h to inactivate lysozyme) was added to 200 μg/ml final concentration, and the sample was incubated at 60 °C for 1.5 h to remove lipoprotein. The solution was added, with vigorous stirring, to an equal volume of boiling 8% SDS and boiled for 15 min. Insoluble peptidoglycan was pelleted, washed free of SDS as described above, and resuspended in a volume of 0.02% NaN3 so that the samples contained equal concentrations of peptidoglycan (by adjusting the volume so that the A208 of a 1:50 dilution was 0.3). Long-term storage was at −20 °C.
Preparation and fluorescent labeling of muropeptides
Muropeptides were produced by digesting peptidoglycan with N-acetylmuramidase SG (USB Specialty Biochemicals, Amersham Life Sciences, Cleveland, OH), which was added directly to thawed peptidoglycan aliquots to give a final concentration of 133 μg/ml. The reaction consisted of 50 μl peptidoglycan (A208 ~0.3), 7.5 μl of 0.2 M phosphate buffer, pH 5.5, 2.5 μl of 0.2% NaN3, 5.0 μl dH2O, and 10 μl N-acetylmuramidase SG (1 mg/ml stock solution). Digestion was accomplished by incubation at 37 °C overnight, after which the sample was dried under centrifugal vacuum. The reducing ends of the resulting muropeptides were labeled with one of the following fluorescent compounds: 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) (Molecular Probes, Inc., Eugene, OR), 7-aminonaphthalene-1,3-disulfonic acid (ANDA) (Aldrich Chemical Co., Milwaukee, WI.), or 4-aminonaphthalene-1-sulfonic acid (ANSA) (Aldrich Chemical Co.), as described [6–8]. Stock solutions (0.2 M) of the above dyes were prepared by adding the following amounts of each fluorophore to 500 μl of acetic acid:water (3:17 v/v): ANTS (0.0428 g), ANDA (0.0402 g), and ANSA (0.0230 g). To each dried muropeptide sample was added 5 μl 0.2M fluorescent label (ANTS, ANDA, or ANSA) plus 5 μl 1.0M NaCNBH3. The mixture was incubated at 45 °C for 3 h, after which 10 μl SDS–PAGE loading buffer (without SDS or dithiothreitol) was added, and the muropeptides were separated by polyacrylamide gel electrophoresis.
Separation and visualization of labeled muropeptides
Fluorophore-assisted carbohydrate electrophoresis was performed in a minigel apparatus as described [8]. At first, labeled muropeptides were separated by using commercially prepared high-percentage (40%) acrylamide gels and reagents from Glyko, Inc. [8]. However, during the course of these experiments the company discontinued its business and these electrophoresis product lines, although the Glyko ANTS labeling reagents continue to be supplied by ProZyme (San Leandro, CA). Therefore, we systematically varied the gel composition, running buffer, and electrophoresis conditions to determine which combination of conditions gave optimal band separation. The results are summarized in Table 2. Thereafter, samples were separated by electrophoresis for 90 min in 0.5-mm-thick minigels composed of 35% polyacrylamide containing urea and glycerol, using a Tris–Tricine (0.5 M:0.5M) running buffer. The exact gel formulation per liter was 350 g acrylamide, 1.6 g bis-acrylamide, 384 ml 1M Tris–HCl, pH 8.8, the final volume being brought to 1000 ml by adding a 1:1 mixture of 8M urea:glycerol. Muropeptide bands were visualized by exposure to UV irradiation and photographed by a charge-coupled device camera in a Fluor-S MultiImager (Bio-Rad Laboratories, Inc., Hercules, CA), and the relative amounts of fluorescence were measured and computed by using Quantity One (v 4.0.3) software (Bio-Rad).
Table 2.
Optimization of electrophoresis conditions for FACE
| Electrophoresis conditions | Resolution |
|---|---|
| Polyacrylamide gel | |
| Glyko gel cassette (commercially prepared) | +++++ |
| 30% Polyacrylamide | ++ |
| 35% Polyacrylamide | ++ |
| 30% Polyacrylamide + glycerol:urea | +++ |
| 35% Polyacrylamide + glycerol:urea | ++++ |
| Running buffer (using 35% polyacrylamide gel + glycerol:urea) | |
| Glyko running buffer | ++++ |
| Tris–glycine (0.25 M:0.25M) | + |
| Tris–glycine (0.5 M:0.5M) | + |
| Tris–tricine (0.25 M:0.25M) | ++ |
| Tris–tricine (0.5 M:0.5M) | ++++ |
| Tris–tricine (0.75 M:0.75M) | +++ |
| Gel size (35% gel + glycerol:urea, using Tris–tricine (0.5 M:0.5M) buffer) | |
| Minigel (90×100 mm)/0.5-mm spacers | ++++ |
| Minigel (90×100 mm)/0.75-mm spacers | ++ |
| Large gel (185×195 mm)/1.0-mm spacers | + |
The procedure described under Materials and methods was altered by varying the polyacrylamide gel composition, the electrophoresis running buffer, and the gel size. Resolution was determined by visual inspection of band sharpness and distance from neighboring bands and ranked on a five-point scale from poor (+) to excellent (+++++).
Separation of muropeptides by reversed-phase HPLC was performed as described previously [4,5,13].
Mass spectrometry
Muropeptides were isolated from a one-dimensional lane of a FACE gel by excising individual bands and eluting the muropeptides overnight into a small amount of distilled water. Contaminating buffer was removed by passing the samples through ZipTipC18 tips according to the manufacture's instructions (Millipore Corp., Bedford, MA). The samples were concentrated by centrifugal evaporation, and resuspended in water, and their masses were determined by MALDI mass spectrometry by S. Hoffmann-Benning of the Mass Spectrometry Facility, Michigan State University.
Size exclusion chromatography
Muropeptides were size-fractionated by passing samples through a 40-cm column (20-ml bed volume) of Biogel P6 (Bio-Rad). The flow rate was 10 ml/h of 100 mM LiCl in 20 mM potassium phosphate buffer, pH 6.0, and 0.6-ml fractions were collected. Fractions were dried under vacuum centrifugation, labeled with ANTS, and analyzed by FACE.
Purification of penicillin binding protein 4 (PBP 4)
To express PBP 4 for partial purification, the PBP 4 (dacB) gene was placed under control of the arabinose promoter by cloning it into pBAD18-Cam [14], creating plasmid pSL4, using procedures described previously [15]. The dacB gene was amplified by polymerase chain reaction from E. coli CS109 genomic DNA by using primers supplied by MWG Biotech, Inc. (High Point, NC): a forward primer, 5′-CTCTCTGCTAGCAATTCATTATGCGATTTTCCAG-3′ (the NheI site is double underlined, the Shine–Dalgarno site is denoted by a single wavy underline, and the ATG start codon is denoted by a single underline), and a reverse primer, 5′-GCGCAAGCTTAGTGGTGGTGGTGGTGGTGATTGTTCTGATAAATATCTTTATACAAAGGGC-3′ (the HindIII site is double underlined and a 6-codon histidine tag is denoted by a single underline). PCR was performed by preincubating the primers with genomic DNA and Deep Vent Taq polymerase (New England Biolabs, Beverly, MA) for 5 min at 94 °C, followed by 30 cycles of 94 °C (1 min), 55 °C (1.5 min), 72 °C (2 min). PCR was terminated by a 10-min incubation at 72 °C and the sample was stored at 4 °C. The amplified DNA fragment was purified by using a QIAquick column (Qiagen, Inc., Studio City, CA), digested with NheI and HindIII, ligated into similarly digested pBAD18-Cam, and the resulting recombinant plasmid was electroporated into E. coli. The sequence of the cloned gene was confirmed by sequencing performed at MWG Biotech, Inc.
We attached six histidine codons to the 3′ end of the cloned PBP 4 gene in hopes that the His-tagged product could be purified by metal chelation chromatography. However, this procedure gave poor yields. Therefore, PBP 4 was purified as follows. Plasmid pSL4 was transformed into E. coli CS804-1K, from which all low-molecular-weight PBP genes were deleted (Table 1), so that PBP 4 could be purified in the absence of contaminating PBPs. CS804 pSL4 was grown at 37 °C in 2L of LB broth containing chloramphenicol (20 μg/ml) to an A600 of ~0.3. At this point, PBP 4 expression was induced by adding arabinose to a final concentration of 0.01% [16]. Growth was continued until the culture reached an A600 of ~0.9, and the cells were harvested by centrifugation at 10,000g for 15 min at 4 °C. Approximately 15 g (wet weight) of cells were suspended in 30 ml of digestion solution (0.2M Tris–HCl, pH 8.0, 0.01M MgCl2, 120 μg/ml lysozyme, 1 μg/ml DNase) containing 40 μg/ml of phenylmethylsulfonyl fluoride. Cells were disrupted by three passages through an Aminco pressure cell (Aminco, Urbana, IL) at 16,000 lb/in2. The homogenate was cleared by centrifugation at 100,000g for 1 h at 4 °C, and the supernatant was collected.
Procion Blue Navy H-ER dye (Pro Chemical and Dye, Inc., Somerset, MA) was coupled to Fractogel TSK HW-65F (Pierce, Rockford, IL) and used to purify PBP 4 by dye-affinity chromatography as described [17]. PBP 4-containing supernatant (30 ml) was mixed with 10 ml bed volume of dye-substituted resin that had been equilibrated previously with 50 mM Tris–HCl, pH 8.9. The mixture was incubated with shaking at 4 °C for 4 h, after which the gel was collected by low-speed centrifugation and poured into a column. Unbound material was removed by four washing steps: two 15-ml washes composed of a 1:1 ratio of 50 mM Tris–HCl, pH 8.9, and 50 mM glycine–NaOH, pH 10.5, followed by two 15-ml washes of 50 mM glycine–NaOH, pH 10.5. PBP 4 was eluted from the column by a solution of 2M NaCl in 50 mM glycine–NaOH, pH 10.5. Collected fractions were dialyzed against a solution of 5 mM MgSO4 containing 0.02% NaN3, and stored at 4 °C. The purity of PBP 4 was confirmed by sodium dodecyl sulfate–poly-acrylamide gel electrophoresis (SDS–PAGE). Total protein was determined by staining with Gel-Code (Pierce) and the presence of active PBP 4 was detected by labeling with 125I-penicillin X [18].
Digestion of muropeptides by PBP 4
Peptidoglycan (50-μl aliquots) were digested overnight at 37 °C with N-acetylmuramidase SG and dried by vacuum centrifugation. Samples were resuspended in a final volume of 50 μl 10 mM Tris–maleate, pH 6.8, 1 mM MgCl2, plus PBP 4. As a control, PBP 4 was inactivated by a 3-h pretreatment with penicillin-X (1 mg/ml) prior to adding the enzyme to the reaction. After digestion at 37 °C for 4 or 12 h, the samples were dried by centrifugation under vacuum, labeled with ANTS dye, and analyzed by FACE.
Results
Muropeptide separation and labeling
After determining the best acrylamide gel and buffer combinations for separating muropeptides by FACE (Table 2), we labeled digested peptidoglycan with three fluorophores to see which resolved the greatest number of muropeptide bands in polyacrylamide gels. Labeling with ANSA imparts one negative charge to each muropeptide, ANDA contributes two negative charges, and ANTS adds three negative charges. An example of the structure of one muropeptide–ANTS compound is illustrated in Fig. 1. Fluorophore-labeled muropeptides gave similar patterns in FACE gels, with band migration distance increasing in the order of ANSA < ANDA < ANTS, consistent with the number of negative charges contributed by each dye (Fig. 2). Though the general order of bands was similar for muropeptides labeled by each dye, putative trimer muropeptides were resolved better when labeled with ANSA (Fig. 2). Relative amounts of the three main muropeptide categories were equivalent when labeled by any of the dyes (Table 3). However, the separation distance between the monomer and the dimer muropeptides was greater when samples were labeled with ANTS. In addition, separation of bands representing dimer–tetramer–tetramer (D44) and dimer–tetramer–trimer-Dap (D43Dap) was much improved in ANTS-labeled samples compared to that in ANSA-labeled samples, in which we could not see the latter compound (see below). Therefore, because monomers and dimers constitute the major muropeptide components, for routine analysis in this work we labeled with ANTS to obtain the greatest separation of these compounds. The increased separation also simplified the removal of gel fragments for isolating individual muropeptides.
Fig. 1.
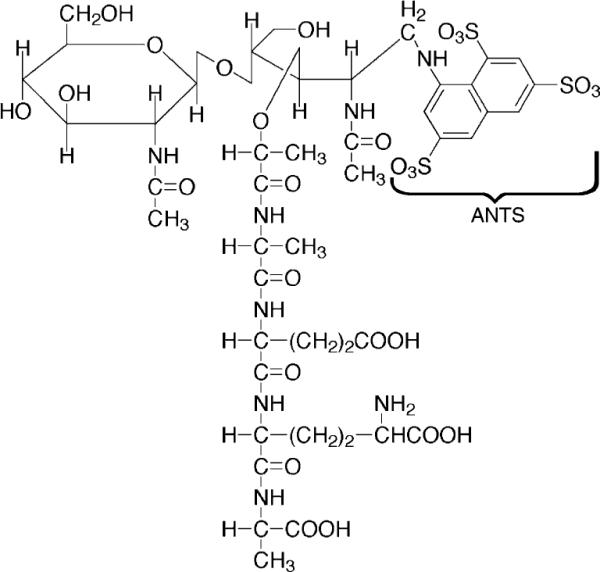
Structure of a muropeptide coupled to ANTS. A muropeptide monomer consisting of N-acetylglucosamine, N-acetylmuramic acid, and a four-amino-acid side chain is shown with ANTS dye covalently bound to the reducing end of the N-acetylmuramic acid.
Fig. 2.

Muropeptides labeled with different fluorophores. E. coli CS109 peptidoglycan was digested with N-acetylmuramidase, and the resulting muropeptides were labeled with the fluorophores, ANSA (lane 1), ANDA (lane 2), and ANTS (lane 3). The samples were separated by using the optimal conditions established for FACE gel electrophoresis as described under Materials and methods. The brackets in each lane denote the approximate positions of trimers, dimers, and monomers, respectively. Monomer muropeptides are composed of a single N-acetylglucosamine-N-acetylmuramic acid (NAG–NAM) glycan subunit plus an amino acid side chain of two to five residues, dimer muropeptides are composed of two NAG–NAM glycan subunits cross-linked to one another via their peptide side chains, and trimer muropeptides are composed of three NAG–NAM glycan subunits cross-linked to one another via their peptide side chains. Bands migrating faster than monomers are low-molecular-mass constituents of the fluorescent dye solutions.
Table 3.
Muropeptide classes detected by labeling with different fluorophores
| Muropeptide class | Fluorophore (%) |
||
|---|---|---|---|
| ANSA | ANDA | ANTS | |
| Trimers | 9.3±1.1 | 111.7±0.7 | 9.8±0.6 |
| Dimers | 34.7±2.9 | 33.1±4.1 | 35.2±1.9 |
| Monomers | 44.8±3.6 | 45.4±4.0 | 45.6±1.8 |
E. coli CS109 peptidoglycan was labeled as described in the legend to Fig. 2, and the relative percentages of muropeptide bands of each size class in FACE gels were quantified by using the one-dimensional band analysis module of Quantity One software. Data represent the mean±standard deviation of two separate experiments.
Assignment of muropeptides to specific bands
In FACE gels, approximately 14–15 muropeptide bands are visible in digests of peptidoglycan from E. coli [8,19]. Identification of the muropeptides comprising individual bands was accomplished by a combination of methods, including size-fractionation chromatography, the behavior of PBP mutants, mass spectrometry, and the concordance of HPLC and FACE results. The first step was to determine which bands represented monomers, dimers, and trimers. Monomers are single NAG–NAM subunits (N-acetylglucosamine linked to N-acetylmuramic acid) with peptides of various lengths attached to NAM (e.g., Fig. 1), dimers are formed by cross-linking two monomers via their peptide side chains, and trimers represent three cross-linked subunits [4,5]. These categories were resolved by fractionation of muropeptides through a Biogel P6 column, labeling with ANTS, and separation by FACE (Fig. 3). Trimers eluted first (Fig. 3, fraction 32), appearing as a shoulder preceding the major dimer peak (Fig. 3, fractions 38 and 41), which was followed by a peak of monomer muropeptides (Fig. 3, fractions 46–52). This simplified subsequent band identification. Note that the monomer and dimer peaks from the fractionation column each contained one band that migrated in FACE gels at a position consistent with that of trimer muropeptides. The identity of these two bands has not been established.
Fig. 3.
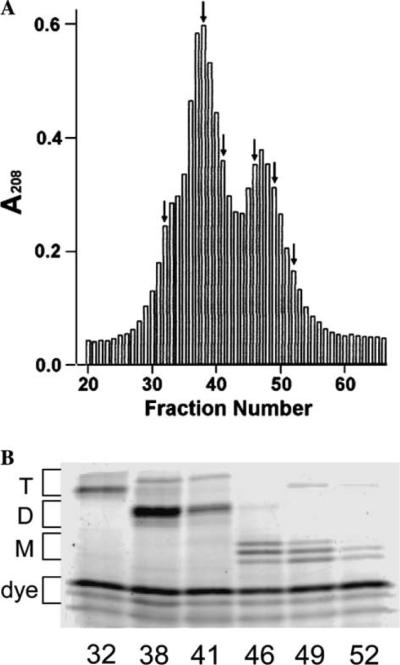
Identification of muropeptide classes by size-exclusion chromatography. Peptidoglycan was digested with N-acetylmuramidase SG and labeled with ANTS. The resulting muropeptides were applied to a Biogel P-6 column, fractionated, and the muropeptides were detected as described under Materials and methods. (A) Muropeptides from digested peptidoglycan of E. coli CS703-1 were eluted from the column and detected by absorbance at 208 nm. Arrows indicate fractions selected for FACE analysis shown in B. (B) Muropeptide composition of six fractions from A, detected by FACE. The fraction number is given below each lane. T, trimer muropeptides; D, dimer muropeptides; M, monomer muropeptides; dye, background of residual ANTS fluorophore common to each sample.
The identities of two bands fractionating as monomer muropeptides (Fig. 4, lanes 3 and 4) and that of one dimer muropeptide (Fig. 4, lane 5) were confirmed by eluting individual bands from a FACE gel and determining their molecular masses by MALDI mass spectrometry (Fig. 5 and Table 4). Unfortunately, despite numerous attempts under many conditions of sample preparation, further mass spectrometry of additional bands was unsuccessful due to contamination by extraneous compounds. This apparently was caused by a complication of ANTS behavior during mass spectrometry and by contamination with carbohydrates derived from the acrylamide gel itself. Nonetheless, positive identification of these three muropeptides was sufficient to create an interpretive baseline from which other bands could be identified by complementary means.
Fig. 4.

Isolation of muropeptides after separation by FACE. Individual bands were cut from a FACE gel, eluted into distilled water, concentrated by evaporation, resuspended in water, and applied to separate lanes of a second FACE gel. Lane 1 is a standardized ladder of fluorescent glucose oligomers, with the least abundant band (third from bottom) representing a polymer of four glucose residues. The muropeptides in lanes 3, 4, and 5 were eluted and identified by mass spectrometry as the monomer-tetrapeptide, monomer-pentapeptide, and dimer-tetrapeptide-tetrapeptide, respectively (Table 4).
Fig. 5.
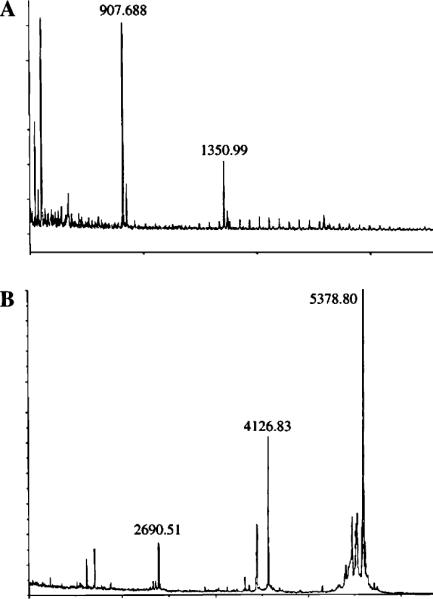
Mass spectrometry of two muropeptide bands. A. Monomer–tetrapeptide (M4)–ANTS. The band shown in Fig. 4, lane 3, was eluted and subjected to mass spectrometry. The M4–ANTS signal is at 1350.99. A contaminating peak at 907.688 originated after passage of the material through the Millipore ZipTip matrix used for sample preparation (data not shown). (B) Dimer–tetrapeptide–tetrapeptide (D44)–ANTS. The band shown in Fig. 4, lane 5, was eluted and repurified by HPLC to be certain that only a single component was analyzed by mass spectrometry. The peak at 2690.51 represents the dimer, that at 5378.80 represents a condensation of two dimers, and the peak at 4126.88 is an unidentified breakdown product.
Table 4.
Molecular mass of muropeptides labeled with ANTS
| Muropeptides | Molecular mass |
Compounds | |||
|---|---|---|---|---|---|
| Calculated | Measured (mass spec) | Difference | Error (%) | ||
| M3 | 1351.19 | 1350.99 | −0.2 | −0.015 | Monomer–tetra |
| M4 | 1422.26 | 1422.30 | +0.06 | +0.004 | Monomer–penta |
| D44 | 2684.36 | 2690.51 | +6.15 | +0.23 | Dimer–tetra–tetra |
Muropeptides shown in Fig. 4, lanes 3, 4, and 5, were eluted from the FACE gel and analyzed by mass spectrometry. The mass of the D44 muropeptide was determined only after further purification of the band by HPLC prior to mass spectrometry, to be certain that a single compound was analyzed.
Comparison of muropeptide profiles determined by HPLC and FACE
Reversed-phase high-performance liquid chromatography is the best and most well-established method for separating muropeptides [4,5]. Therefore, we compared the muropeptide composition of peptidoglycan from nine different strains of E. coli by analyzing identical samples with HPLC and FACE (Table 5).
Table 5.
Muropeptide percentage composition of E. coli strains as determined by HPLC and FACE
| Muropeptides | CS109 wild type |
CS12-7 Δ PBP 5 |
CS13-2 Δ PBP la |
CS214-1 Δ PBP la 5 |
CS14-2 Δ ampC |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| HPLC | FACE | HPLC | FACE | HPLC | FACE | HPLC | FACE | HPLC | FACE | |
| M3 | 9.8±0.1 | 8.2±0.8 | 7.9±0.1 | 7.7±0.6 | 9.6±0.4 | 8.0±0.8 | 8.0±0.5 | 7.5±0.8 | 9.6±1.1 | 7.8±0.8 |
| M4 | 43.9±0.9 | 41.4±3.1 | 42.2±1.7 | 32.1±2.8 | 44.8±1.1 | 41.1±1.6 | 41.6±1.0 | 38.0±5.6 | 46.6±7.5 | 38.4±4.5 |
| M-5 | 0.1±0.0 | 0.5±0.3 | 5.0±1.1 | 6.0±1.3 | 0.1±0.0 | 0.6±0.1 | 4.2±0.2 | 4.9±0.8 | 0.1±0.0 | 1.7±0.1 |
| M3KR | 1.3±0.1 | ND | 1.0±0.4 | ND | 1.7±0.4 | ND | 1.2±0.3 | ND | 1.5±0.3 | ND |
| D44 | 34.3±1.1 | 38.2±2.3 | 31.2±3.1 | 40.9±0.9 | 36.0±0.7 | 36.8±1.6 | 36.9±1.1 | 36.8±8.4 | 33.4±5.5 | 40.0±4.5 |
| D45 | 1.4±0.0 | 7.6±0.1 | ||||||||
| D43 + D43Dap | 2.4±0.3 | 2.5±0.3 | 2.5±0.6 | 1.9±0.3 | 2.4±0.1 | 3.1±1.1 | 2.9±0.6 | 3.2±1.3 | 2.0±0.5 | 1.7±0.4 |
| D43KR | 1.6±0.5 | 1.6±0.6 | 1.4±0.1 | 1.4±0.9 | 1.9±0.3 | 1.9±1.2 | 1.5±0.2 | 1.9 | 1.8±0.7 | 3.1±2.8 |
| Unknown | 2.8±0.9 | 4.7±1.3 | 2.2±0.6 | |||||||
| T444 | 4.3±1.0 | 2.7±0.1 | 2.6±1.2 | 1.9±0.2 | 3.0±1.0 | 3.7±1.6 | 3.1±0.5 | 1.2±0.1 | 3.9±1.3 | 2.4±1.7 |
| T443 + T443Dap | 0.4±0.1 | 0.4±0.1 | 0.4±0.3 | 0.4±0.1 | 0.3±0.2 | 0.5±0.4 | 0.5±0.3 | 0.4±0.0 | 0.6±0.6 | 0.7±0.3 |
| Trimer-KR | 0.2±0.1 | 0.2±0.0 | 0.3±0.0 | 0.2±0.0 | 0.4±0.1 | |||||
| Totals | 99.5±1.1 | 98.1±0.2 | 98.7±0.2 | 96.7±0.6 | 97.9±0.1 | 97.6±0.1 | 98.7±0.2 | 95.7±0.4 | 97.9±0.2 | 94.1±0.5 |
| CS109 wild type |
CS15-3 Δ ampH |
CS362-1 Δ PBP 1a 5 ampC |
CS357-3 Δ PBP 1a 5 ampH |
CS420-2 Δ PBP 1a 5 ampCH |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HPLC | FACE | HPLC | FACE | HPLC | FACE | HPLC | FACE | HPLC | FACE | |
| M3 | 9.8±0.1 | 8.2±0.8 | 8.6±0.5 | 7.2±0.3 | 7.5±2.0 | 6.5±0.4 | 7.6±1.3 | 7.1±1.1 | 4.3±0.4 | 3.0±0.9 |
| M4 | 43.9±0.9 | 41.4±3.1 | 42.2±3.1 | 37.8±3.6 | 45.1±1.5 | 38.1±7.1 | 42.3±2.1 | 37.6±4.0 | 38.0±7.8 | 34.5±5.5 |
| M-5 | 0.1±0.0 | 0.5±0.3 | 0.1±0.0 | 1.1±0.7 | 4.8±0.5 | 5.5±1.1 | 4.8±0.2 | 4.9±0.1 | 8.5±2.2 | 6.9±0.4 |
| M3KR | 1.3±0.1 | ND | 1.4±0.1 | ND | 1.4±0.2 | ND | 1.4±0.2 | ND | 2.0±0.6 | ND |
| D44 | 34.3±1.1 | 38.2±2.3 | 37.1±1.0 | 41.4±2.2 | 34.9±4.1 | 40.0±4.6 | 22.8±4.4 | 38.2±4.1 | 30.5±1.2 | 31.0±0.8 |
| D45 | 1.4±0.0 | 11.4±0.2 | 7.0±0.1 | |||||||
| D43 + D43Dap | 2.4±0.3 | 2.5±0.3 | 2.7±0.4 | 3.2±1.2 | 2.2±0.2 | 3.6±1.6 | 2.2±0.4 | 3.2±1.1 | 2.9±1.4 | 1.9±0.6 |
| D43KR | 1.6±0.5 | 1.6±0.6 | 2.1±0.6 | 0.8±0.5 | 1.9±0.8 | ND | 1.4±0.0 | ND | 1.7±0.7 | ND |
| Unknown | 2.8±0.9 | |||||||||
| T444 | 4.3±1.0 | 2.7±0.1 | 4.5±1.2 | 5.2±1.3 | 3.0±0.3 | 3.8 | 3.3±0.1 | 1.6±1.5 | 4.4±1.4 | 4.5±0.1 |
| T443 + T443Dap | 0.4±0.1 | 0.4±0.1 | 0.7±0.8 | 0.5±0.2 | 0.3±0.0 | 0.6 | 0.7±0.6 | 0.5±0.2 | 0.9±0.3 | 1.3±0.8 |
| Trimer-KR | 0.2±0.1 | 0.3±0.1 | 0.6±0.6 | 0.2±0.0 | 0.3±0.2 | |||||
| Totals | 99.5±1.1 | 98.1±0.2 | 97.9±0.3 | 96.0±2.9 | 94.8±1.5 | 90.3±6.8 | 94.2±0.3 | 88.9±2.5 | 89.7±2.1 | 82.9±6.4 |
PBP genes deleted from each strain are listed below the strain name. Muropeptide designations: monomers (M), dimers (D), and trimers (T) are classified as those having tripeptide (three), tetrapeptide (four), or pentapeptide (five) side chains attached to the muramic acid residue. The muropeptides are designated according to the abbreviations described in Table 6. Muropeptides with lipoprotein attached to the side chain had this moiety removed by proteolysis during peptidoglycan preparation but retained the Lys-Arg (KR) residues. The peptide linkages between dimers and trimers were either alanine-to-diaminopimelic acid (no suffix) or DAP-to-DAP (“Dap” suffix). HPLC data are the mean±standard deviation (SD) of measurements from two different gels. For comparison purposes, the amount of anhydromuramic acid muropeptides (<5% of the total) was omitted from the HPLC data because it could not be detected by the FACE technique. The FACE data for CS109, CS14-2, and CS15-3 are the mean±SD of measurements from three different gels. FACE data for all other strains are the mean±SD of measurements from two different gels. Percentages enclosed in solid boxes denote those that are significantly higher than those in the wild type. Percentages enclosed in the dotted boxes denote those that are significantly lower than those in the wild type. Underlined percentages denote results given by the FACE procedure that are significantly different from those given by HPLC analysis. Unknown, a band appearing in some FACE gels but whose chemical identity is not known; ND, not detected.
Peptidoglycan was prepared from each strain, and digested with muramidase, and the muropeptides were processed in parallel for HPLC and FACE analyses. Examples of HPLC results for muropeptides derived from wild-type E. coli and three mutants are shown in Fig. 6. The amounts of material in each HPLC peak were quantified and expressed as a percentage of the total muropeptides. Likewise, intensities of monomer, dimer, and trimer bands in FACE gels were measured, and the amount of material in each band was reported as a percentage of the total. The bands present in FACE gels were aligned with the HPLC results in three ways—first, by matching FACE bands identified by mass spectrometry; second, by matching the positions of the three muropeptide size classes; and third, by matching the relative percentages measured for each band. These procedures were mutually supportive of muropeptide-to-band assignments and allowed us to identify and compare 10 major muropeptides between the two techniques (Tables 5 and 6).
Fig. 6.

Reverse-phase HPLC analysis of muropeptides derived from the peptidoglycan of wild-type E. coli CS109 and three mutants. The muropeptide peaks are labeled according to the abbreviations defined in Table 6. In B, C, and D, only those peaks that changed substantially from the preceding panel are labeled. (A) E. coli CS109. (B) E. coli CS12-7 (PBP 5 deleted). (C) E. coli CS357-3 (PBP 1a, PBP 5, and AmpH deleted). (D) E. coli CS420-2 (PBP 1a, PBP 5, AmpC, and AmpH deleted).
Table 6.
Muropeptide abbreviations
| Monomers | |
| M2, | Dipeptide |
| M3, | Tripeptide |
| M4, | Tetrapeptide |
| M5, | Pentapeptide |
| M4G, | Tetrapeptide(glycine end) |
| M3KR, | Tripeptide Lppa |
| Dimers | |
| D43, | Tetrapeptide–tripeptide |
| D44, | Tetrapeptide–tetrapeptide |
| D45, | Tetrapeptide–pentapeptide |
| D4G3Dap, | Tetrapeptide(glycine end)–tripeptide (Dap–Dap)b |
| D33Dap, | Tripeptide–tripeptide (Dap–Dap) |
| D43Dap, | Tetrapeptide–tripeptide (Dap–Dap) |
| D44G, | Tetrapeptide–tetrapeptide(glycine end) |
| D4G5, | Tetrapeptide(glycine end)–pentapeptide |
| D33KRDap, | Tripeptide–tripeptide Lpp (Dap–Dap) |
| D43KR, | Tetrapeptide–tripeptide Lpp |
| Trimers | |
| T444 | Tetrapeptide–tetrapeptide–tetrapeptide |
| T443 | Tetrapeptide–tetrapeptide–tripeptide |
| T443Dap | Tetrapeptide–tetrapeptide–tripeptide (Dap–Dap) |
| Anhydro muropeptide compounds b | |
| A1, D33KRA | Dimer tripeptide–tripeptide Lpp ahn |
| A2, T444A | Trimer tetrapeptide–tetrapeptide–tetrapeptide anh |
| A3, T443A | Trimer tetrapeptide–tetrapeptide–tripeptide anh |
| A4, T443DapA | Trimer tetrapeptide–tetrapeptide–tripeptide (Dap–Dap) anh |
| A5, D44A | Dimer tetrapeptide–tetrapeptide-anh |
| A6, D43A | Dimer tetrapeptide–tripeptide anh |
| A7, D43DapA | Dimer tetrapeptide–tripeptide (Dap–Dap) anh |
| A8, M4A | Monomer tetrapeptide anh |
| A9, M3A | Monomer tripeptide anh |
Lpp, muropeptide with two amino acid residues (“KR” = Lys–Arg) linked to DAP, which represent the remnant of Braun's lipoprotein that was removed during sample processing; Dap–Dap, peptide crosslink formed between two diaminopimelic acids in side chains.
Anhydro compounds (“anh”) are muropeptides containing 1,6-anhydromuramic acid, in which the reducing end of the muramic acid moiety is blocked.
Nine major bands accounted for 89–98% of the total muropeptide fluorescence present in FACE gels. This compared very well with the HPLC technique, in which the 10 major muropeptides represented 90–99% of the total muropeptides detected by HPLC (Table 5). One common muropeptide resolved by HPLC but not by FACE was the monomer-Lys-Arg (M3KR, note abbreviations in Table 6), which represents the site at which lipoprotein was attached before its removal from peptidoglycan by protease during sample preparation. Also, one dimer (the tetrapeptide–pentapeptide, D45) could not be distinguished from the major dimer (the tetrapeptide–tetrapeptide, D44), probably because the high concentration of the D44 product obscured the less abundant D45 band. Therefore, the results for these two compounds were combined in the FACE results (Table 5). Likewise, 2 other muropeptides (T443 and T443Dap) could not be distinguished from one another, possibly because they migrated as one band under the conditions employed. Nonetheless, FACE did detect the 7 most abundant muropeptides as well as did HPLC, which is significant because these represent 95–97% of the components in each sample.
A quantitative comparison between the techniques was also favorable. For the seven most abundant muropeptides, within the margins of error, FACE and HPLC detected the same relative and numerical amounts of each muropeptide in 66 of 72 measurements (seven muropeptides for each of nine strains), giving a correspondence of 92%. The 6 instances in which FACE returned measurements at odds with the HPLC data were confined to the monomer–pentapeptide (3 cases) and dimer–KR (3 cases). These muropeptides represented only 1–2% of the compounds in these samples, so that inaccuracies in measuring band fluorescence in FACE gels may explain the discrepancies.
Overall, compared to HPLC, the FACE technique detected the same major components of peptidoglycan and returned the same relative and quantitative results with a 92% correspondence. Thus, the FACE technique accurately characterized the numbers and amounts of the major muropeptides compared to those detected by HPLC. Fig. 7 summarizes the locations of the major bands and how they were identified.
Fig. 7.
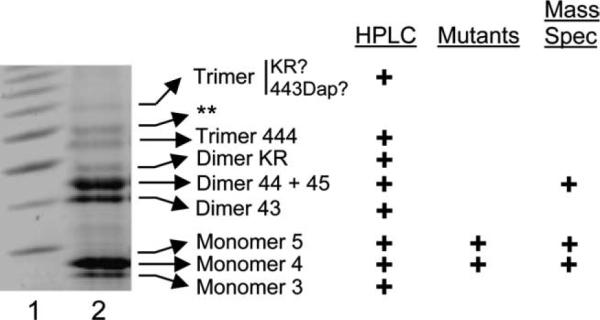
Location and identification of muropeptides in FACE gels. Lane 1, carbohydrate reference ladder (Glyko, Inc.). Lane 2, E. coli CS109 muropeptides labeled with ANTS and separated by FACE. Arrows indicate the identities of bands as determined by correspondence with muropeptide results when fractionated by reversed-phase HPLC (HPLC), by the behavior of bands in E. coli mutants (Mutants), or by mass spectrometry (Mass Spec). The double asterisk (**) denotes an unidentified band.
Comparison of muropeptide differences among E. coli mutants
The E. coli strains in the above experiment differed from one another in the numbers of penicillin binding proteins present, and some of these mutants displayed altered ratios of muropeptides in peptidoglycan digests. Thus, it was possible to determine whether the FACE technique performed as well as HPLC in detecting changes in muropeptide levels. The greatest alteration was an increase in monomer–pentapeptide in strains from which the gene encoding PBP 5 was deleted. In the affected strains (CS12-7, CS214-1, CS362-1, CS357-3, and CS420-2), the level of monomer–pentapeptide increased from 0.1–0.5% to 5–9% (Table 5, solid boxes). In all cases, FACE and HPLC measurements were equivalent in direction of change and in numerical amount. In addition, both techniques detected the same intriguing result in the quadruple mutant, CS420-2. The amount of monomer–pentapeptide in this strain was higher than that in other strains lacking PBP 5 (7–9% versus ~5%) (Table 5, CS420-2 solid box), and the amount of monomer–tripeptide was much less than that in any other strain (3–4% versus 7–10%) (Table 5, CS420-2 dotted box). In this mutant, simultaneously eliminating PBP 5, AmpC, and AmpH evidently increases the level of pentapeptides and decreases the level of tripeptides beyond that effected by the loss of PBP 5 alone.
As a further test of the utility of FACE, we examined the muropeptide composition of two additional mutants lacking six and seven PBPs. In both strains, the proportion of dimer and trimer muropeptides increased, with a concomitant decrease in monomer muropeptides (Table 7). The result is consistent with what is known about the biochemical activities of the deleted PBPs. PBPs 4 and 7 are endopeptidases [1,20], so deletion of these proteins would be expected to reduce the number of dimers and trimers degraded to monomer muropeptides. The same six PBPs absent from CS617-1K are missing from CS703-1, but this latter strain also lacks PBP 5. This protein is the major DD-carboxypeptidase in E. coli, so in its absence the amount of monomer–pentapeptide increases dramatically in CS703-1 compared to either the wild-type parent or the mutant lacking six PBPs (Table 7). This is consistent with the results described above for mutants lacking PBP 5.
Table 7.
Muropeptide composition of E. coli PBP mutants
| Muropeptides | CS109 | CS617-1K | CS703-1 |
|---|---|---|---|
| Wild type | Δ1 a 4 6 7 CH | Δ1 a 4 5 6 7 CH | |
| Monomers | 43.2±2.6 | 26.8±1.9 | 23.0±2.1 |
| Dimers | 34.9±2.9 | 47.2±1.9 | 48.8±2.9 |
| Trimers | 10.5±1.4 | 15.2±1.3 | 15.1±0.7 |
| Monomers | |||
| Tripeptide | 11.8±0.7 | 5.5±0.4 | 7.1±0.5 |
| Tetrapeptide | 30.7±1.4 | 20.7±0.9 | 11.1±0.4 |
| Pentapeptide | 0.7±0.2 | 0.6±0.1 | 4.8±0.3 |
The percentages of muropeptides in these E. coli strains represent the mean±standard deviation from measurements of three different FACE gels. Amounts of muropeptides that are significantly different from those of E. coli CS109 are underlined: solid underline (higher), dotted underline (lower). The PBPs deleted from each strain are listed under the strain name: numbers represent specific PBP designations; C, AmpC; H, AmpH.
FACE as an in vitro enzyme assay
Because PBP 4 cleaves cross-linked peptide side chains in peptidoglycan [1], the enzyme is expected to digest muropeptide dimers and trimers, converting them into monomers. To determine whether the FACE technique could be used to detect the enzymatic activity of PBP 4, we incubated muropeptides from three E. coli strains with active and inactive PBP 4 and separated the resulting muropeptides by FACE (Table 8). In all cases, PBP 4 activity removed about 50% of dimer muropeptides with a concomitant increase in monomers (Table 8). On the other hand, trimer muropeptides were not decreased to a significant degree in muropeptides derived from CS109 unless incubation was prolonged to 12 h, and even then the effect was relatively small (a 20–30% reduction). A potential, but slight, decrease in trimers also occurred during incubation of PBP 4 with muropeptides from CS703-1. All types of dimers appeared to be reduced equally (data not shown), but only the tetrapeptide and the pentapeptide monomers increased (by approximately 100% each). The one exception to the increase of monomer–pentapeptide was when PBP 4 was incubated with muropeptides from CS703-1, in which the amount of this muropeptide was already elevated. Interestingly, the level of monomer–tripeptide remained unchanged in the presence or absence of PBP 4.
Table 8.
PBP 4 activity assayed by FACE
| Muropeptides | CS109 (4 h) |
CS109 (12 h) |
CS617-1K (4 h) |
CS703-1 (4 h) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| − | PBP 4 | PBP 4 + Pen-X | − | PBP 4 | PBP 4 + Pen-X | − | PBP 4 | PBP 4 + Pen-X | − | PBP 4 | PBP 4 + Pen-X | |
| Monomers | 44.0±2.7 | 67.2±4.7 | 48.9±3.4 | 43.8±2.7 | 76.7±3.4 | 45.0±3.1 | 29.2±3.2 | 53.6±3.5 | 28.0±2.5 | 25.1±3.2 | 52.8±3.5 | 23.7±3.2 |
| Tri | 11.3±0.4 | 11.7±0.6 | 11.0±0.7 | 12.3±0.3 | 13.9±0.6 | 11.7±0.4 | 5.6±0.2 | 5.1±0.5 | 5.2±0.3 | 8.0±0.4 | 7.3±0.3 | 7.3±0.2 |
| Tetra | 31.8±0.6 | 53.7±0.4 | 35.9±0.5 | 30.3±0.5 | 60.2±0.6 | 32.1±0.4 | 22.7±1.6 | 46.7±1.7 | 21.8±2.4 | 11.7±0.6 | 39.1±0.4 | 10.6±0.6 |
| Penta | 0.6±0.1 | 1.4±0.3 | 0.5±0.1 | 0.5±0.2 | 2.0±0.3 | 0.5±0.2 | 0.6±0.1 | 1.2±0.2 | 0.4±0.1 | 4.9±0.4 | 5.7±0.2 | 5.3±0.3 |
| Dimers | 36.9±1.3 | 18.0±0.9 | 39.8±0.6 | 37.3±2.4 | 9.9±0.7 | 39.0±0.8 | 49.4±2.3 | 27.7±1.2 | 45.7±2.3 | 45.5±0.9 | 23.4±1.5 | 45.7±1.0 |
| Trimers | 8.8±0.7 | 8.3±0.4 | 10.6±0.4 | 9.6±0.8 | 6.4±0.5 | 10.2±0.6 | 14.7±1.0 | 12.0±0.8 | 14.9±1.3 | 16.2±0.8 | 12.6±0.4 | 17.4±0.3 |
The percentage of muropeptides in peptidoglycan digests from three E. coli strains. The results represent the mean±standard deviation of measurements from two different FACE gels. Reactions were incubated for 4 or 12 h with muropeptide preparations, plus the following: no additions (−), PBP 4 (PBP 4), and PBP 4 inactivated by penicillin-X (PBP 4 + Pen-X). In the PBP 4 reaction (center column for each strain), amounts of muropeptides that increased significantly are denoted by a solid underline and those that decreased are denoted by a dotted underline.
Incubation of PBP 4 with undigested peptidoglycan produced a ladder of bands that probably represents murein strands of different lengths (data not shown). A similar distribution of glycan chain lengths has been observed after amidase cleavage of peptidoglycan, as detected by HPLC [11]. We also purified and tested the activity of a second endopeptidase, PBP 7, for its ability to degrade muropeptides. No activity was observed on these compounds (data not shown), consistent with the previous report showing that PBP 7 prefers an intact sacculus as substrate [20]. In summary, these results indicate that the FACE technique can be used, at least in a qualitative way, to detect the activity and potential substrate specificities of peptidoglycan hydrolases.
Discussion
The workhorse of peptidoglycan analysis has been the separation of peptidoglycan fragments by reversed-phase HPLC. In an effort to devise a simplified method for screening the peptidoglycan composition of multiple mutants under numerous conditions, we established that the muropeptide profile of E. coli could be detected by an adaptation of the FACE technique for separating carbohydrates [8,19]. By direct comparison, we now show that the FACE procedure detects and quantifies major muropeptides with an accuracy approaching that of HPLC. In particular, quantitation of total amounts of monomers, dimers, and trimers by FACE coincides with amounts detected by HPLC. Thus, FACE can be used as a screening tool to obtain a general overview of peptidoglycan composition and to monitor changes in its major constituents.
Although in many cases FACE may substitute for HPLC analysis of muropeptides, the technique does have two drawbacks. First, FACE does not detect anhydromuropeptides because they have no reducing ends for labeling. These compounds represent approximately 2–5% of the total muropeptides detected by HPLC and reflect the number of glycan chain ends within the bacterial sacculus. Second, although many other minor muropeptides are detected by FACE, they are diffcult to quantitate and we have not been able to confirm their identities. Even so, FACE can still detect significant changes in these minor bands and therefore can serve as a preliminary screen to identify samples requiring more thorough analysis by HPLC.
As an example of such screening potential, the muro-peptide profile of E. coli CS420-2 was shown to be enriched in monomer–pentapeptide beyond that observed in mutants lacking PBP 5 alone. In addition, of the strains tested, this mutant contained the lowest concentration of monomer–tripeptides. These observations suggest that AmpC and AmpH may be inefficient DD-carboxypeptidases or that they may positively influence the DD-carboxypeptidase activity of other enzymes. An alternate possibility is that AmpC and AmpH have a LD-carboxypeptidase activity normally required for degrading tetra- or pentapeptides to tripeptides. Although they are members of the well-characterized class C β-lactamases, neither AmpC nor AmpH has a known function in vivo, yet they must contribute to the normal physiology of E. coli because mutants lacking these two proteins and PBP 5 are morphologically aberrant [18]. In any case, the results emphasize that FACE screening can uncover biochemical clues worth pursuing in more detail.
Another potentially valuable use of FACE is as an in vitro enzyme assay for peptidoglycan synthetic and hydrolytic enzymes. As an example, we showed that PBP 4 activity was detected easily by changes in the relative proportions of muropeptides. In addition, by using a mixture of natural muropeptides as competing targets, the assay suggests that PBP 4 may have a preferential substrate. First, the enzyme appears to cleave dimer cross-links more efficiently than the peptide side chains of trimers. Second, the amounts of monomer–tetra- and pentapeptides increased significantly after incubation with PBP 4, but the level of monomer–tripeptides did not increase. This implies one of two things: either few tripeptide monomers cross-link with other monomers to form dimers or such compounds are not cleaved by PBP 4. Therefore, trimers and/or tripeptide-containing dimers may constitute a peptidoglycan component that is more resistant to endopeptidase activity, with as yet unknown consequences for cell wall structure and function.
In summary, the FACE technique is an accurate and biologically informative method for approaching questions about bacterial cell wall structure, and its use may precede or complement the classical HPLC procedure.
Acknowledgments
We especially thank Bernadette Meberg for strain constructions, Margaret Larson for laboratory assistance, and Susanne Hoffmann-Benning of the Mass Spectrometry Facility at Michigan State University for performing the mass spectrometry. This work was supported by Grant MCB-9982157 from the National Science Foundation.
Footnotes
Abbreviations used: NAG, N-acetylglucosamine; NAM, N-acetylmuramic acid; HPLC, high-performance liquid chromatography; FACE, fluorophore-assisted carbohydrate electrophoresis; ANTS, 8-aminonaphthalene-1,3,6-trisulfonic acid; ANDA, 7-aminonaphthalene-1,3-disulfonic acid; ANSA, 4-aminonaphthalene-1-sulfonic acid; MALDI, matrix-assisted laser desorption ionization; PBP, penicillin binding protein.
References
- [1].Höltje J-V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Labischinski H, Maidhof H. Bacterial peptidoglycan: overview and evolving concepts. In: Ghuysen J-M, Hakenbeck R, editors. Bacterial Cell Wall. Elsevier Science B.V; Amsterdam: 1994. pp. 23–38. [Google Scholar]
- [3].Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972;36:407–477. doi: 10.1128/br.36.4.407-477.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Glauner B. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 1988;172:451–464. doi: 10.1016/0003-2697(88)90468-x. [DOI] [PubMed] [Google Scholar]
- [5].Glauner B, Höltje J-V, Schwarz U. The composition of the murein of Escherichia coli. J. Biol. Chem. 1988;263:10088–10095. [PubMed] [Google Scholar]
- [6].Jackson P. The use of polyacrylamide-gel electrophoresis for the high-resolution separation of reducing saccharides labelled with the fluorophore 8-aminonaphthalene-1,3,6-trisulphonic acid. Biochem. J. 1990;270:705–713. doi: 10.1042/bj2700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stack RJ, Sullivan MT. Electrophoretic resolution and fluorescence detection of N-linked glycoprotein oligosaccharides after reductive amination with 8-aminonaphthalene-1,3,6-trisulphonic acid. Glycobiology. 1992;2:85–92. doi: 10.1093/glycob/2.1.85. [DOI] [PubMed] [Google Scholar]
- [8].Young KD. A simple gel electrophoretic method for analyzing the muropeptide composition of bacterial peptidoglycan. J. Bacteriol. 1996;178:3962–3966. doi: 10.1128/jb.178.13.3962-3966.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory Press; Plainview, NY: 1992. [Google Scholar]
- [10].Meberg BM, Sailer FC, Nelson DE, Young KD. Reconstruction of Escherichia coli mrcA (PBP 1a) mutants lacking multiple combinations of penicillin binding proteins. J. Bacteriol. 2001;183:6148–6149. doi: 10.1128/JB.183.20.6148-6149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Harz H, Burgdorf K, Höltje J-V. Isolation and separation of the glycan strands from murein of Escherichia coli by reversed-phase high-performance liquid chromatography. Anal. Biochem. 1990;190:120–128. doi: 10.1016/0003-2697(90)90144-x. [DOI] [PubMed] [Google Scholar]
- [12].Hayashi K. A rapid determination of sodium dodecyl sulfate with methylene blue. Anal. Biochem. 1975;67:503–506. doi: 10.1016/0003-2697(75)90324-3. [DOI] [PubMed] [Google Scholar]
- [13].Glauner B, Schwarz U. The analysis of murein composition with high-pressure-liquid chromatography. In: Hackenbeck R, Höltje J-V, Labischinski H, editors. The Target of Penicillin. Walter de Gruyter; New York: 1983. pp. 29–34. [Google Scholar]
- [14].Guzman L-M, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nelson DE, Young KD. Contributions of PBP 5 and DD-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J. Bacteriol. 2001;183:3055–3064. doi: 10.1128/JB.183.10.3055-3064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nelson DE, Ghosh AS, Paulson AL, Young KD. Contribution of membrane-binding and enzymatic domains of penicillin binding protein 5 to maintenance of uniform cellular morphology of Escherichia coli. J. Bacteriol. 2002;184:3630–3639. doi: 10.1128/JB.184.13.3630-3639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Henderson TA, Templin M, Young KD. Identification and cloning of the gene encoding penicillin-binding protein 7 of Escherichia coli. J. Bacteriol. 1995;177:2074–2079. doi: 10.1128/jb.177.8.2074-2079.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Henderson TA, Young KD, Denome SA, Elf PK. AmpC and AmpH proteins related to the class C b-lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli. J. Bacteriol. 1997;179:6112–6121. doi: 10.1128/jb.179.19.6112-6121.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Young KD. Techniques for analysis of peptidoglycan. In: Williams P, Ketley J, Salmond G, editors. Bacterial Pathogenesis. vol. 27. Academic Press; London: 1998. pp. 277–286. [Google Scholar]
- [20].Romeis T, Höltje J-V. Penicillin-binding protein 7/8 of Escherichia coli is a DD-endopeptidase. Eur. J. Biochem. 1994;224:597–604. doi: 10.1111/j.1432-1033.1994.00597.x. [DOI] [PubMed] [Google Scholar]
- [21].Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 1999;181:3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]