

Abstract
Objective
Sorting mechanisms that cause the amyloid precursor protein (APP) and the β-secretases and γ-secretases to colocalize in the same compartment play an important role in the regulation of Aβ production in Alzheimer’s disease (AD). We and others have reported that genetic variants in the Sortilin-related receptor (SORL1) increased the risk of AD, that SORL1 is involved in trafficking of APP, and that under expression of SORL1 leads to overproduction of Aβ. Here we explored the role of one of its homologs, the sortilin-related VPS10 domain containing receptor 1 (SORCS1), in AD.
Methods
We analyzed the genetic associations between AD and 16 SORCS1–single nucleotide polymorphisms (SNPs) in 6 independent data sets (2,809 cases and 3,482 controls). In addition, we compared SorCS1 expression levels of affected and unaffected brain regions in AD and control brains in microarray gene expression and real-time polymerase chain reaction (RT-PCR) sets, explored the effects of significant SORCS1-SNPs on SorCS1 brain expression levels, and explored the effect of suppression and overexpression of the common SorCS1 isoforms on APP processing and Aβ generation.
Results
Inherited variants in SORCS1 were associated with AD in all datasets (0.001 < p < 0.049). In addition, SorCS1 influenced APP processing. While overexpression of SorCS1 reduced γ-secretase activity and Aβ levels, the suppression of SorCS1 increased γ-secretase processing of APP and the levels of Aβ.
Interpretations
These data suggest that inherited or acquired changes in SORCS1 expression or function may play a role in the pathogenesis of AD.
Amyloid β (Aβ), the putative culprit in Alzheimer’s disease (AD) is produced by successive β-secretase (BACE) cleavage of the amyloid precursor protein (APP) at the N-terminus of the Aβ peptide followed by γ-secretase cleavage of the membrane-bound C-terminal APP fragment.1 This mechanism explains Aβ accumulation in early-onset familial AD due to mutations in the APP2 presenilin 1 (PSEN1),3 and presenilin 2 (PSEN2)4 genes. The explanation for the accumulation of Aβ40 and Aβ42 in the common late-onset form of the disease remains largely unknown.
APP and the secretases are integral transmembrane proteins. They are dynamically sorted into the plasma membrane and the membranes of intracellular organelles.5,6 As a consequence, sorting mechanisms that cause APP and the secretases to colocalize in the same cellular compartment are expected to play important roles in the regulation of Aβ production. During the last 2 decades, the trans-Golgi network and the endosome were identified as the key organelles managing the complex movement of the transmembrane proteins via secretory and endocytic pathways.7,8
We and others have previously reported9–16 that variants in the Sortilin-related receptor (SORL1), which maps to chromosome 11q23.3, are associated with AD, that SORL1 is involved in trafficking of APP from the cell surface to the Golgi-endoplasmic reticulum complex, and that under expression of SORL1 leads to overexpression of Aβ and an increased risk of AD.9 SORL1 belongs to the mammalian Vps10p-domain sorting receptor family, which is a group of 5 type-I membrane homologs (SORL1, Sortilin, SorCS1, SorCS2, and SorCS3).17–20 All 5 proteins are characterized by a luminal, extracellular VPS10 domain. In sortilin, the VPS10 domain is the only luminal, extracellular module. In SORL1 the VPS10 domain is followed by low-density lipoprotein (LDL) receptor-like regions and fibronectin type-III domains. In the homologous SorCS1, SorCS2, and SorCS3 the transmembrane domain is preceded by a leucine-rich module.21 Following the extracellular and transmembrane segment, each receptor carries a short (40–80 amino acids) cytoplasmic domain comprising typical motifs for interaction with cytosolic adaptor molecules. All of these receptors are prominently expressed in the nervous system and particularly high in the brain, and may be important for neuronal activity,22,23 although their precise function remains elusive.
We hypothesized that genetic variants in SORL1 homologues, in particular SORCS1, might also affect the risk of AD. This was supported by the facts that: (1) we previously observed an association of single nucleotide polymorphism (SNP) rs7082289 in SORCS1 (p = 0.013) in late-onset AD families9; and that (2) Li et al.24 examined a Canadian dataset of 753 AD cases and 736 controls, and SNP rs601883 in SORCS1 was associated with AD risk (hazard ratio [HR], 3.41; 95% confidence interval [CI], 1.87–6.23), and SNP rs7907690 was also significant in logistic regression analyses (p = 0.05). In a chromosome 10–specific association study with 1,412 SNPs in 4 independent Caucasian datasets,25 SNP rs600879 in SORCS1 was significantly associated with AD in an exploratory sample set and an independent replication sample set, although the authors used replication rather than correction for multiple testing for evidence of association (uncorrected p-value across datasets: 0.0043). In a case-control dataset of 506 cases and 558 controls, rs17277986 in SORCS1 was significant in the overall datasets (p = 0.0025) and most significant in the female subset (allelic association p = 0.00002) in single-marker and haplotype analyses.26
The goal of this study was to analyze the genetic associations between AD and SORCS1 in 6 independent data sets that had sufficient power to detect modest gene effects. We focused on the SNPs that had been reported by the previous studies as described above, adding SNP rs7082289 at 108,357,010bp that was associated with AD in a previous report by us.9 We also compared SorCS1 expression levels of affected and unaffected brain regions in AD and control brains in expanded microarray gene expression and real-time polymerase chain reaction (RT-PCR) sets, explored the effects of significant SORCS1-SNPs on SorCS1 brain expression levels, and explored the effect of suppression and overexpression of the common SorCS1 isoforms on APP processing and Aβ generation.
Patients and Methods
Participants
Six datasets included: (1) 371 cases and 349 controls of European descent (Toronto dataset27); (2) 229 Caucasian cases and 301 controls from the NIA-LOAD study28; (3) 763 cases and 1,170 controls from the MIRAGE Caucasian dataset29,30; (4) 660 cases and 844 controls from the Miami Caucasian data-set30; (5) 310 cases and 327 controls from the MIRAGE African American dataset29,30; and (6) 476 cases and 491 controls from a Caribbean Hispanic dataset (see Supporting Information Methods). While mean age of onset and mean age of controls was slightly younger in the MIRAGE compared to the other datasets, frequency of the apolipoprotein E (APOE)-e4 allele was higher in the NIA-LOAD and MIRAGE African American datasets. The clinical characteristics of these datasets are summarized in Supporting Information Table 1. The diagnoses of “probable” or “possible” AD were defined according to the National Institute of Neurological and Communication Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) diagnosis criteria31 at clinics specializing in memory disorders or in clinical investigations. Persons were classified as “controls” when they were without cognitive impairment or dementia at last visit. Informed consent was obtained from all participants using procedures approved by institutional review boards at each of the clinical research centers collecting human subjects.
TABLE 1.
SORCS1 SNPs Genotyped in this Study
| Marker Number | db SNP rs Number | Alleles | MAF (HapMap CEU) | Orientation/Strand | Physical Map Location (bp) | Distance from Previous Marker (bp) | SNP Type | Location |
|---|---|---|---|---|---|---|---|---|
| 1 | rs7082289 | C/T | 0.09 | Fwd/B | 108,357,010 | Cds-synon | Exon 23 | |
| 2 | rs1572456 | C/G | 0.35 | Fwd/T | 108,557,366 | 200,106 | Intron | Intron 3 |
| 3 | rs6584769 | A/G | 0.01 | Fwd/T | 108,662,263 | 105,147 | Intron | Intron 2 |
| 4 | rs4918274 | A/G | 0.26 | Fwd/T | 108,719,950 | 57,687 | Intron | Intron 1 |
| 5 | rs2900717 | C/T | 0.24 | Fwd/B | 108,721,677 | 1,727 | Intron | Intron 1 |
| 6 | rs11193137 | G/T | 0.01 | Fwd/B | 108,722,481 | 804 | Intron | Intron 1 |
| 7 | rs12571141 | A/G | 0.32 | Fwd/T | 108,738,660 | 16,179 | Intron | Intron 1 |
| 8 | rs17277986 | C/T | 0.32 | Fwd/B | 108,748,716 | 9,806 | Intron | Intron 1 |
| 9 | rs6584777 | C/T | 0.31 | Fwd/B | 108,754,004 | 5,538 | Intron | Intron 1 |
| 10 | rs12251340 | G/T | 0.01 | Fwd/B | 108,754,129 | 125 | Intron | Intron 1 |
| 11 | rs1887635 | A/G | 0.25 | Fwd/T | 108,755,782 | 1,653 | Intron | Intron 1 |
| 12 | rs7907690 | A/G | 0.46 | Fwd/T | 108,852,117 | 96,335 | Intron | Intron 1 |
| 13 | rs7089127 | C/T | 0.06 | Fwd/B | 108,868,606 | 16,489 | Intron | Intron 1 |
| 14 | rs625792 | C/T | 0.25 | Rev/B | 108,869,599 | 993 | Intron | Intron 1 |
| 15 | rs601883 | C/G | 0.09 | Fwd/T | 108,904,435 | 34,836 | Intron | Intron 1 |
| 16 | rs600879 | A/G | 0.11 | Fwd/T | 108,913,108 | 8,673 | Intron | Intron 1 |
Marker intervals are calculated on the basis of NCBI locations. Throughout the article, SNPs are referred to by sequential numbers (marker number) reflecting their relative physical map positions. Orientation or strand information was obtained from NCBI: “Fwd/T” refers to forward or top strand; “Rev/B” refers to reverse or bottom strand. The linkage disequilibrium maps for these SNPs are given in Supporting Information Figure 1. See also Supporting Information Table 2 for information on platforms used for SORCS1 and APOE genotyping.
Cds-synon = coding sequence-synonymous; HapMap CEU = haplotype map from Utah residents with Northern and Western European ancestry, from the Centre d’Etude du Polymorphisme Humain (CEPH); MAF = minor allele frequency; NCBI = National Center for Biotechnology Information; SNP = single nucleotide polymorphism.
Genotyping
Each study site provided the results from genotyping of 16 SORCS1 SNPs and APOE. Information on genotyping platforms used in each study is provided in Supporting Information Table 2. The 16 SORCS1 SNPs included SNPs in the 5′ end of the gene that were significant in preliminary analyses and previous studies,24,25 and SNP rs7082289 that had been associated with AD our earlier report.9 In order to facilitate interpretation of the results for the reader, we refer to these 16 SNPs by sequential numbers (marker number) reflecting their relative physical map positions. Information on numbering, location, orientation, and type of the SNPs is given in Table 1.
TABLE 2.
Results of Single-SNP Association Analyses
| SNP | NIA-LOAD
|
Toronto
|
Miami
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | F_A | F_U | A2 | χ2 | OR | L95 | U95 | p | A1 | F_A | F_U | A2 | χ2 | OR | L95 | U95 | p | A1 | F_A | F_U | A2 | χ2 | OR | L95 | U95 | p | |
| 1 | Not genotyped | Unreliable data | T | 0.08 | 0.1 | C | 2.1 | 0.83 | 0.65 | 1.07 | 0.15 | ||||||||||||||||
|
| |||||||||||||||||||||||||||
| 2 | C | 0.34 | 0.33 | G | 0.2 | 1.06 | 0.82 | 1.38 | 0.648 | C | 0.290 | 0.300 | G | 0.5 | 0.92 | 0.73 | 1.15 | 0.468 | C | 0.33 | 0.32 | G | 0.1 | 1.03 | 0.88 | 1.20 | 0.74 |
|
| |||||||||||||||||||||||||||
| 3 | Not polymorphic | G | 0.004 | 0.010 | A | 0.6 | 0.56 | 0.13 | 2.36 | 0.424 | Not polymorphic | ||||||||||||||||
|
| |||||||||||||||||||||||||||
| 4 | A | 0.22 | 0.29 | G | 5.1 | 0.72 | 0.54 | 0.96 | 0.024 | A | 0.290 | 0.220 | G | 8.3 | 1.42 | 1.12 | 1.80 | 0.004 | A | 0.24 | 0.28 | G | 5.1 | 0.83 | 0.70 | 0.98 | 0.025 |
|
| |||||||||||||||||||||||||||
| 5 | C | 0.22 | 0.28 | T | 3.9 | 0.75 | 0.57 | 1.00 | 0.049 | C | 0.280 | 0.220 | T | 6.9 | 1.38 | 1.09 | 1.76 | 0.008 | C | 0.23 | 0.27 | T | 6.7 | 0.80 | 0.68 | 0.95 | 0.009 |
|
| |||||||||||||||||||||||||||
| 6 | G | 0.01 | 0.01 | T | 0.9 | 0.52 | 0.13 | 2.02 | 0.335 | G | 0.0013 | 0.0014 T | 0.002 | 0.94 | 0.06 | 15.02 | 0.964 | G | 0.0007 | 0.001 T | 0.6 | 0.42 | 0.04 | 4.04 | 0.4383 | ||
|
| |||||||||||||||||||||||||||
| 7 | A | 0.27 | 0.34 | G | 5.2 | 0.73 | 0.56 | 0.96 | 0.023 | A | 0.320 | 0.270 | G | 4.9 | 1.30 | 1.03 | 1.63 | 0.026 | A | 0.28 | 0.33 | G | 8.4 | 0.79 | 0.68 | 0.93 | 0.004 |
|
| |||||||||||||||||||||||||||
| 8 | T | 0.27 | 0.33 | C | 5.4 | 0.73 | 0.56 | 0.95 | 0.02 | T | 0.320 | 0.260 | C | 5.1 | 1.30 | 1.03 | 1.64 | 0.024 | T | 0.28 | 0.33 | C | 9.1 | 0.79 | 0.67 | 0.92 | 0.003 |
|
| |||||||||||||||||||||||||||
| 9 | T | 0.30 | 0.37 | C | 5.4 | 0.74 | 0.57 | 0.95 | 0.021 | T | 0.330 | 0.280 | C | 4.7 | 1.28 | 1.02 | 1.61 | 0.03 | T | 0.29 | 0.34 | C | 8.9 | 0.79 | 0.67 | 0.92 | 0.003 |
|
| |||||||||||||||||||||||||||
| 10 | T | 0.01 | 0.02 | G | 0.01 | 0.95 | 0.35 | 2.56 | 0.914 | T | 0.010 | 0.010 | G | 0.2 | 0.78 | 0.24 | 2.58 | 0.686 | T | 0.001 | 0.002 G | 0.3 | 0.64 | 0.12 | 3.48 | 0.599 | |
|
| |||||||||||||||||||||||||||
| 11 | A | 0.21 | 0.2 | G | 0.2 | 1.07 | 0.79 | 1.45 | 0.647 | A | 0.190 | 0.240 | G | 4.7 | 0.76 | 0.59 | 0.97 | 0.03 | A | 0.23 | 0.2 | G | 2.4 | 1.15 | 0.96 | 1.38 | 0.123 |
|
| |||||||||||||||||||||||||||
| 12 | A | 0.46 | 0.47 | G | 0.1 | 0.97 | 0.76 | 1.24 | 0.785 | A | 0.460 | 0.490 | G | 0.8 | 0.91 | 0.74 | 1.12 | 0.364 | A | 0.5 | 0.47 | G | 2.8 | 1.13 | 0.98 | 1.31 | 0.096 |
|
| |||||||||||||||||||||||||||
| 13 | C | 0.06 | 0.09 | T | 3.2 | 0.65 | 0.40 | 1.05 | 0.073 | C | 0.070 | 0.060 | T | 0.6 | 1.17 | 0.77 | 1.78 | 0.455 | C | 0.07 | 0.09 | T | 3.4 | 0.77 | 0.58 | 1.02 | 0.066 |
|
| |||||||||||||||||||||||||||
| 14 | G | 0.3 | 0.25 | A | 2.8 | 1.26 | 0.96 | 1.66 | 0.096 | C | 0.270 | 0.280 | T | 0.5 | 0.92 | 0.73 | 1.16 | 0.492 | C | 0.27 | 0.26 | T | 0.2 | 1.04 | 0.88 | 1.22 | 0.647 |
|
| |||||||||||||||||||||||||||
| 15 | C | 0.12 | 0.14 | G | 0.7 | 0.86 | 0.59 | 1.24 | 0.411 | G | 0.130 | 0.130 | C | 0.1 | 1.04 | 0.77 | 1.42 | 0.797 | G | 0.12 | 0.14 | C | 3.6 | 0.81 | 0.65 | 1.01 | 0.058 |
|
| |||||||||||||||||||||||||||
| 16 | A | 0.11 | 0.12 | G | 0.5 | 0.87 | 0.60 | 1.28 | 0.49 | T | 0.110 | 0.120 | C | 0.2 | 0.93 | 0.67 | 1.29 | 0.663 | Not genotyped | ||||||||
| MIRAGE Whites
|
MIRAGE African Americans
|
Caribbean Hispanics
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | A1 | F_A | F_U | A2 | GEE BETA | L95 | U95 | p | A1 | F_A | F_U | A2 | GEE BETA | L95 | U95 | p | A1 | F_A | F_U | A2 | χ2 | OR | L95 | U95 | P | |
| 1 | T | 0.09 | 0.08 | C | 0.159 | −0.06 | 0.38 | 0.214 | T | 0.27 | 0.28 | C | −0.065 | −0.31 | 0.18 | 0.627 | T | 0.16 | 0.17 | C | 0.2 | 0.95 | 0.74 | 1.21 | 0.646 | |
|
| ||||||||||||||||||||||||||
| 2 | G | 0.32 | 0.31 | C | −0.048 | −0.17 | 0.07 | 0.478 | G | 0.33 | 0.36 | C | 0.103 | −0.10 | 0.31 | 0.356 | C | 0.32 | 0.31 | G | 0.4 | 1.07 | 0.88 | 1.30 | 0.527 | |
|
| ||||||||||||||||||||||||||
| 3 | Not genotyped | Not genotyped | G | 0.06 | 0.05 | A | 0.3 | 1.13 | 0.76 | 1.69 | 0.6 | |||||||||||||||
|
| ||||||||||||||||||||||||||
| 4 | A | 0.25 | 0.26 | G | −0.067 | −0.20 | 0.07 | 0.368 | A | 0.08 | 0.07 | G | 0.075 | −0.28 | 0.43 | 0.712 | A | 0.21 | 0.2 | G | 1.2 | 1.09 | 0.87 | 1.36 | 0.281 | |
|
| ||||||||||||||||||||||||||
| 5 | C | 0.24 | 0.25 | T | −0.047 | −0.18 | 0.09 | 0.535 | C | 0.07 | 0.07 | T | 0.097 | −0.29 | 0.49 | 0.657 | C | 0.18 | 0.19 | T | 0.2 | 0.95 | 0.75 | 1.19 | 0.653 | |
|
| ||||||||||||||||||||||||||
| 6 | Not genotyped | Not genotyped | G | 0.06 | 0.07 | T | 0.1 | 0.96 | 0.66 | 1.37 | 0.741 | |||||||||||||||
|
| ||||||||||||||||||||||||||
| 7 | A | 0.29 | 0.3 | G | −0.056 | −0.18 | 0.07 | 0.429 | A | 0.08 | 0.07 | G | 0.133 | −0.24 | 0.51 | 0.529 | A | 0.2 | 0.19 | G | 0.2 | 1.04 | 0.83 | 1.31 | 0.694 | |
|
| ||||||||||||||||||||||||||
| 8 | T | 0.28 | 0.3 | C | −0.075 | −0.20 | 0.05 | 0.287 | T | 0.08 | 0.07 | C | 0.116 | −0.26 | 0.49 | 0.58 | T | 0.2 | 0.19 | C | 0.4 | 1.07 | 0.85 | 1.34 | 0.517 | |
|
| ||||||||||||||||||||||||||
| 9 | T | 0.31 | 0.32 | C | −0.03 | −0.15 | 0.09 | 0.658 | T | 0.28 | 0.28 | C | 0.033 | −0.18 | 0.25 | 0.789 | T | 0.44 | 0.42 | C | 1.4 | 1.12 | 0.93 | 1.34 | 0.242 | |
|
| ||||||||||||||||||||||||||
| 10 | T | 0.01 | 0.01 | G | Unreliable data | T | 0.33 | 0.33 | G | −0.005 | −0.27 | 0.26 | 0.974 | T | 0.09 | 0.11 | G | 1.1 | 0.88 | 0.65 | 1.18 | 0.302 | ||||
|
| ||||||||||||||||||||||||||
| 11 | G | 0.23 | 0.21 | A | −0.077 | −0.21 | 0.06 | 0.313 | G | 0.11 | 0.15 | A | 0.297 | 0.03 | 0.56 | 0.053 | A | 0.19 | 0.25 | G | 11 | 0.69 | 0.55 | 0.85 | 0.001 | |
|
| ||||||||||||||||||||||||||
| 12 | A | 0.44 | 0.46 | G | −0.079 | −0.19 | 0.03 | 0.239 | A | 0.2 | 0.2 | G | −0.043 | −0.30 | 0.21 | 0.763 | A | 0.33 | 0.36 | G | 1.7 | 0.90 | 0.75 | 1.09 | 0.188 | |
|
| ||||||||||||||||||||||||||
| 13 | C | 0.07 | 0.07 | T | −0.07 | −0.32 | 0.18 | 0.615 | C | 0.05 | 0.06 | T | −0.109 | −0.60 | 0.38 | 0.668 | C | 0.04 | 0.05 | T | 0.1 | 0.92 | 0.60 | 1.42 | 0.739 | |
|
| ||||||||||||||||||||||||||
| 14 | C | 0.22 | 0.25 | T | −0.208 | −0.34 | −0.07 | 0.007 | C | 0.07 | 0.06 | T | 0.138 | −0.26 | 0.54 | 0.542 | G | 0.19 | 0.21 | A | 2.1 | 0.84 | 0.67 | 1.05 | 0.146 | |
|
| ||||||||||||||||||||||||||
| 15 | G | 0.12 | 0.12 | C | −0.02 | −0.20 | 0.16 | 0.838 | G | 0.23 | 0.25 | C | −0.109 | −0.36 | 0.14 | 0.442 | C | 0.15 | 0.16 | G | 0.2 | 0.95 | 0.75 | 1.22 | 0.636 | |
|
| ||||||||||||||||||||||||||
| 16 | Not genotyped | Not genotyped | A | 0.2 | 0.19 | G | 0.3 | 1.07 | 0.86 | 1.35 | 0.595 | |||||||||||||||
All p values of p ≤ 0.05 are in bold. All p values that are close to statistical significance (0.05 ≤ p ≤ 0.1) are in italics. SNPs that are associated with a changed risk for AD in at least 2 independent data sets are underlined. For the characteristics of the study populations, see Supporting Information Table 1. For the association of genotypes of SNPs with brain SORCS1 expression levels, see Supporting Information Table 3. For SORCS1 linkage disequilibrium maps of the individual datasets, see Supporting Information Figure 1.
A1 = minor allele; A2 = wild-type allele; AD = Alzheimer’s disease; F_A = frequency of minor allele in affecteds; F_U = frequency of minor allele in unaffecteds; SNP = single-nucleotide polymorphism; χ2 = chi-square test statistic.
Microarray Gene Expression
Expression profiling was performed separately for the cerebellum, parietal-occipital neocortex, and amygdala regions from 19 AD and 10 control brains from the New York Brain Bank (www.nybb.hs.columbia.edu). This 3-region approach allowed us to enhance the signal-to-noise ratio,32 and to determine those changes in expression patterns of candidate genes that are specific for late-onset AD and consistent with distribution of AD pathology.
For the expression profiling of AD and control brains, the Affymetrix GeneChip® Human Exon 1.0 ST Arrays were used. Frozen brain tissue was ground over liquid nitrogen and stored at −80°C until use. Total RNA was extracted and purified using TRIzol Plus RNA purification kit (Invitrogen). Quantification and qualification of all RNA preparations was performed using an Agilent Bioanalyzer (RNA 6000 nano-kit) and only samples with RNA integrity number (RIN) > 8 were used in the subsequent RNA amplification and hybridization steps. The GeneChip expression 2-cycle target labeling kit (Affymetrix) was used for all samples according to Affymetrix protocols. Briefly, the procedure consists of an initial ribosomal RNA (rRNA) reduction step and 2 cycles of reverse transcription followed by in vitro transcription (IVT). For each sample, 1 μg of total RNA is initially subjected to removal of rRNA using the RiboMinus™ Transcriptome Isolation Kit (Invitrogen) and spiked with Eukaryotic PolyA RNA controls (Affymetrix). The rRNA-depleted fraction was used for complementary DNA (cDNA) synthesis by reverse-transcription primed with T7-random hexamer primers, followed by second-strand synthesis. This cDNA serves as template for in vitro transcription to obtain amplified antisense complementary RNA (cRNA). Subsequently cRNA from the first round was reverse-transcribed using random primers to obtain single-stranded sense DNA. In this second reverse-transcription, dUTP is incorporated into the DNA to allow for subsequent enzymatic fragmentation using a combination of UDG and APE1. All reverse and in vitro transcription steps were performed using the GeneChip WT cDNA synthesis and amplification Kit (Affymetrix). The resulting fragmented DNA was labeled with Affymetrix DNA Labeling Reagent. Labeled fragmented DNA was hybridized to Affymetrix Human Exon 1.0 ST arrays, then washed and stained using the GeneChip Hybridization, Wash and Stain Kit. Fluorescent images were recorded on a GeneChip scanner 3000 and analyzed with the GeneChip operating software.
RT-PCR
Candidate genes that showed changes in expression patterns were confirmed by RT-PCR using TaqMan Gene Expression Assays (Applied Biosystems), which included specific primers and fluorogenic probes for each gene. The TaqMan Assays use targeted regions spanning exon-exon boundaries. Each cDNA sample from the same brain regions used in the microarray gene expression analysis was analyzed in triplicate 20-μl reactions and only 1 gene assayed per reaction. The Assays Hs00364666_m1 and Hs01016139_m1 (Applied Biosystems) were used to confirm SorCS1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and bACTIN TaqMan Endogenous controls (Applied Biosystems) were used for normalization. In addition to all the individual samples, a calibrator sample comprised of a pool of all cDNA samples was assayed in every plate for inter-run calibration. Reactions were run on an iQ5 Real-Time Detection System (Bio-Rad). Efficiencies for each assay were calculated from a standard curve using serial dilutions of the calibrator sample. Results from these curves were used to determine appropriate cDNA sample dilution. Relative levels of gene expression were obtained by the 2ΔΔCt method using iQ5 software (Bio-Rad).
Construction of Expression Plasmids and RNA Interference
The cDNAs encoding 2 common constitutively expressed SorCS1 isoforms with different cytoplasmic tails (SorCS1a, NM_052918; and SorCS1b, NM_001013031) were purchased from OriGene Technology. Both isoforms were cloned into the pCMV6-XL4 vector (OriGene Technology). In addition, short hairpin RNA (shRNA) against SorCS1 and the control scrambled sequence shRNA (TRC library; Open Biosystems) were used in the APP processing assays.20 At least 3 of 5 shRNA were able to knockdown SorCS1-gene expression (by 60–80%) by western blot analysis.
Cell Culture and Transfection
HEK293 stably or transiently transfected with the APP Swedish mutation (APPsw),33 M17, and SH-SY5Y cells were maintained in Dulbecco’s modified Eagle’s media (Invitrogen) containing 10% fetal bovine serum and penicillin and streptomycin. Transfections were performed by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. In the case of shRNA transfections, HEK293 cells were transfected and then media was changed the day after. After culturing for an additional 24 hours, the media was changed and cells were incubated for additional 48 hours.9 Conditioned media was collected for the Aβ enzyme-linked immunoassay (ELISA) and cells were harvested for western blotting. For the shRNAs, the HEK293 or M17 cells were transiently transfected and assayed after 5 days.
APP-GV Assay
The γ-secretase activity and nuclear translocation of the APP/ Fe65/TIP60 protein complex was monitored with the APP-GV assay.34 The APP-GV assay is a luciferase-based assay34 consisting of the APP gene’s C-terminus (AICD) fused to a transcription factor composed of the GAL4 DNA binding domain with VP16 transcriptional activator (GV). In addition, the AICD fragment is fused to the GV domains as a positive control of AICD generation and allows for the evaluation of the AICD-specific contribution to the observed modulation in the APP-GV assay. Briefly, SorCS1 cDNA or SorCS1 shRNAs transiently transfected were evaluated in either the APP-GV or the AICD-GV assay, as previously described34 in the cell lines HEK293 and the neuroblastoma M17.
Western Blotting and Immunoprecipitation
Antibodies against SorCS1 were purchased from R&D systems and Santa Cruz Biotechnology. Anti-β-tubulin and anti-APP C-terminal (anti-APPc, 8717 and anti-APPn, 22C11) were from Sigma. Normal goat immunoglobulin G (IgG) and rabbit IgG were from R&D systems and Sigma-Aldrich, respectively. Anti-Aβ (6E10) was obtained from Covance. For the immunoprecipitation, 5ml of the antibody was incubated with 500μg to 750μg of protein lysates and 20μl (bed volume) of protein G-Agarose beads (Sigma) at 4°C overnight with rotation. After extensive washing with the lysis buffer (3 times diluted radio immunoprecipitation assay [RIPA] buffer from Cell Signaling Technology), the bound proteins were separated by boiling in 1× lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) and subjected to western blot analysis. All immunoblots were visualized by enhanced chemiluminescence (ECL; GE Healthcare).
Colocalization Double-Labeling Immunohistochemical Study
SH-SY5Y cells plated on coverslips were fixed with 10% buffered formaldehyde followed by multiple washes in phosphate buffered saline (PBS) and a 15–30-minute incubation in 0.01% triton-100, 2% bovine serum albumin (BSA; Sigma; B4287) in PBS. This was followed by a 3-hour incubation at room temperature in the SorCS1 antibody (1:200; R&D Systems) and anti-APP C-terminal (1:500; Sigma; #8717). A cocktail of secondary antibodies containing donkey-anti-goat-Alexa Fluor 488 (1:100, Molecular Probes) and donkey-anti-mouse Alexa-Fluor 594 (1:100; Molecular Probes) for SorCS1 and APP, respectively, were applied for 1 hour at room temperature. Sections were then rinsed in PBS 3 times and mounted with the proper medium (Vectashield; Vector Labs). Confocal fluorescence images were acquired with a confocal laser-scanning microscope (Fluoview FV300-TP; Olympus).
Aβ Assays
Levels of Aβ40 and Aβ42 were measured using ELISA kits (Invitrogen), following the manufacturer’s protocols.
Statistical Methods
First, SNP marker data were assessed for deviations from Hardy-Weinberg equilibrium (HWE). Then, logistic regression models in PLINK (http://pngu.mgh.harvard.edu/~purcell/plink) were used to assess genotypic and allelic associations with AD. Multivariate logistic regression analysis, using PLINK, was performed to adjust for APOE-ε4, sex and age-at-onset or age-at-examination. The false discovery rate (FDR),35 which controls the expected proportion of incorrectly rejected null hypotheses (type-I errors), was used to account for the error in multiple comparisons. At a minor allele frequency (MAF) of 0.1 and a 0.05 α-level, we had 80% power to detect an effect size of odds ratio [OR] 1.2.
Linkage disequilibrium (LD) structure was examined using Haploview (http://www.broad.mit.edu/mpg/haploview/index.php). Haplotype blocks were defined using the confidence intervals algorithm. The default settings were used in these analyses, which create 95% confidence bounds on D′ to define SNP pairs in strong LD. Haplotype analyses were carried out using a window of 3 contiguous SNPs using Haplo.stats v1.1.1 for case-control data.
We also performed a meta-analysis of all datasets. To determine the strength of associations between the individual SORCS1 SNPs and AD, we calculated a pooled OR for each marker using fixed and random effects models using PLINK. We first performed meta-analyses of unadjusted results from the individual datasets, and then repeated the meta-analyses using the results from the individual datasets adjusted for APOE-ε4, sex and age-at-onset or age-at-examination. The p values for each SNP were corrected for multiple testing (ie, analysis of 16 SNPs in total) using the FDR.35 Between-data-set heterogeneity was quantified using the I2 metric for inconsistency36 and its statistical significance was tested with the chi-square distributed Q statistic.37 I2 is provided by the ratio of (Q-df)/Q, where df = the number of degrees of freedom (1 less than the number of combined datasets); it is considered large for values above 50% and Q is considered statistically significant for p = 0.10.36,37
For analysis to determine genes in which expression levels differ between affected and unaffected brain regions and AD and control brains, we performed both within-group and between-group factors analysis of variance (ANOVA) using Partek Genomics Suite 6.4 (http://www.partek.com/partekgs). Prior to analysis, we log10-transformed rank invariant normalized expression data. Also in these analyses, the FDR was used to account for the error in multiple comparisons.
Statistical Analysis for the Cell Biology and Assays
For these analyses, an ANOVA with post hoc correction was performed using Graphpad Statistical software (Graphpad, Inc., San Diego, CA) to compare mean expression levels. All of the data was normalized to transfection efficiency (eg, green fluorescent protein [GFP]), and then to the control values on each plate for every assay to allow for comparisons across experiments.
Results
Association of SORCS1 SNPs with AD
Table 2 shows the single-SNP associations of the 16 genotyped SNPs with AD across the various datasets. Five out of 6 datasets (all 4 Caucasian datasets and the Caribbean Hispanic dataset) showed an association with at least 1 allele but the specific alleles and the direction of the association differed, likely due to differences in LD patterns and allele frequencies. In particular, the block of SNPs 4–9 in the 5′ end of SORCS1 was associated with AD in 3 of the Caucasian datasets (NIA-LOAD, Toronto, and Miami datasets). In the NIA-LOAD and Miami data-sets, alleles G, T, G, C, and C at markers 4, 5, 7, 8, and 9 were associated with an increased risk of AD (0.003 < p < 0.049), while in the Toronto dataset alleles A, C, A, T, and T were associated with AD risk (0.004 < p < 0.03). Within this block, SNP 6 did not reach statistical significance, probably due to very low allele frequency.
In addition, in each of these Caucasian datasets, several of the remaining markers toward the 5′ end (SNPs 10–16) showed associations that were marginally significant. In the Caribbean Hispanic (p = 0.001) and the Toronto dataset (p = 0.03), the A allele of SNP 11 was associated with AD, and this SNP showed also a trend toward a lower AD risk in the MIRAGE African American dataset (p = 0.053). In the fourth Caucasian dataset, the MIRAGE Caucasian sample, none of the markers of the block 4–9 reached statistical significance, but the T allele of SNP 14 was associated with an increased risk of AD (p = 0.007). These effects were independent of age, sex, and APOE status (Supporting Information Table 6). Supporting Information Figure 3 shows the LD pattern of SORCS1 in Caucasians and the relative location of the 16 genotyped SNPs. Supporting Information Table 5 shows the genotype counts for each SNP.
Consistent with the single-marker results for block 4–9, haplotypes T-T-G at SNPs 5-6-7 and C-C-G at SNPs 8-9-10 in the 3-SNP-window haplotype analyses were associated with an increased risk of AD in the NIA-LOAD and Miami datasets and a decreased risk of AD in the Toronto dataset (0.001 < p < 0.02; Table 3). In addition, a second set of haplotypes (C-T-A at SNPs 5-6-7 and T-T-G at SNPs 8-9-10) was associated with AD risk in an inverse fashion. In addition, consistent with the single-marker results for the A allele of SNP 11, haplotype AAT at SNPs 11|12|13 was associated with a lower risk of AD in the MIRAGE African American and the Caribbean Hispanics datasets (0.001 < p < 0.03), and haplotype AGT was significantly associated or close to significance in the Toronto and MIRAGE African American datasets (0.02 < p < 0.06).
TABLE 3.
Results of Haplotype Association Analyses
| SNPs | Block | NIA-LOAD
|
Toronto
|
Miami
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype Frequency |
Case Frequency |
Control Frequency |
χ2 | p | Haplotype Frequency |
Case Frequency |
Control Frequency |
χ2 | p | Haplotype Frequency |
Case Frequency |
Control Frequency |
χ2 | p | ||
| 2|3|4 | Block 1 | |||||||||||||||
|
| ||||||||||||||||
| 2|3|4 | GAG | 0.482 | 0.503 | 0.472 | 0.969 | 0.32 | 0.509 | 0.487 | 0.533 | 3.157 | 0.07 | 0.49619 | 0.50437 | 0.48965 | 1.0257 | 0.30 |
|
| ||||||||||||||||
| 2|3|4 | CAG | 0.241 | 0.258 | 0.223 | 1.791 | 0.18 | 0.235 | 0.225 | 0.245 | 0.821 | 0.36 | 0.24138 | 0.2541 | 0.23171 | 1.2882 | 0.19 |
|
| ||||||||||||||||
| 2|3|4 | GAA | 0.171 | 0.151 | 0.188 | 2.494 | 0.11 | 0.194 | 0.226 | 0.16 | 10.084 | 0.001 | 0.18055 | 0.16918 | 0.18956 | −1.7851 | 0.07 |
|
| ||||||||||||||||
| 2|3|4 | CAA | 0.093 | 0.081 | 0.101 | 1.22 | 0.26 | 0.056 | 0.058 | 0.054 | 0.109 | 0.74 | 0.08188 | 0.07235 | 0.08908 | −1.5111 | 0.13 |
|
| ||||||||||||||||
| 5|6|7 | Block 2 | |||||||||||||||
|
| ||||||||||||||||
| 5|6|7 | TTG | 0.676 | 0.714 | 0.65 | 4.941 | 0.02 | 0.704 | 0.678 | 0.731 | 4.899 | 0.02 | 0.68789 | 0.71781 | 0.66455 | 3.0689 | 0.002 |
|
| ||||||||||||||||
| 5|6|7 | CTA | 0.255 | 0.230 | 0.276 | 2.928 | 0.08 | 0.247 | 0.276 | 0.216 | 7.132 | 0.007 | 0.24936 | 0.22646 | 0.26725 | −2.5127 | 0.01 |
|
| ||||||||||||||||
| 5|6|7 | TTA | 0.06 | 0.050 | 0.064 | 0.933 | 0.33 | 0.048 | 0.044 | 0.052 | 0.423 | 0.51 | 0.06029 | 0.05413 | 0.06507 | −1.2384 | 0.21 |
|
| ||||||||||||||||
| 8|9|10 | Block 3 | |||||||||||||||
|
| ||||||||||||||||
| 8|9|10 | CCG | 0.649 | 0.687 | 0.622 | 4.864 | 0.02 | 0.694 | 0.668 | 0.721 | 4.866 | 0.02 | 0.6787 | 0.7089 | 0.65507 | 3.11341 | 0.001 |
|
| ||||||||||||||||
| 8|9|10 | TTG | 0.306 | 0.274 | 0.328 | 3.68 | 0.05 | 0.289 | 0.315 | 0.261 | 4.972 | 0.02 | 0.30409 | 0.27725 | 0.32509 | −2.80155 | 0.005 |
|
| ||||||||||||||||
| 8|9|10 | CTG | 0.027 | 0.025 | 0.029 | 0.185 | 0.66 | 0.011 | 0.011 | 0.011 | 0.015 | 0.90 | 0.01276 | 0.01079 | 0.0143 | −0.85168 | 0.39 |
|
| ||||||||||||||||
| 8|9|10 | CTT | 0.015 | 0.015 | 0.015 | 0.016 | 0.90 | — | — | ||||||||
|
| ||||||||||||||||
| 11|12|13 | Block 4 | |||||||||||||||
|
| ||||||||||||||||
| 11|12|13 | GGT | 0.4 | 0.402 | 0.397 | 0.029 | 0.86 | 0.37 | 0.393 | 0.345 | 3.634 | 0.05 | 0.37021 | 0.36336 | 0.37732 | −1.33175 | 0.18 |
|
| ||||||||||||||||
| 11|12|13 | AAT | 0.087 | 0.089 | 0.086 | 0.017 | 0.89 | 0.084 | 0.079 | 0.089 | 0.407 | 0.52 | 0.09806 | 0.10699 | 0.09193 | 1.55811 | 0.11 |
|
| ||||||||||||||||
| 11|12|13 | AGC | 0.016 | 0.012 | 0.019 | 0.826 | 0.36 | — | 0.01542 | 0.01964 | 0.0131 | 0.08679 | 0.93 | ||||
|
| ||||||||||||||||
| 11|12|13 | AGT | 0.091 | 0.105 | 0.08 | 1.969 | 0.16 | 0.12 | 0.101 | 0.14 | 5.184 | 0.02 | 0.09722 | 0.09985 | 0.09423 | 0.83569 | 0.40 |
|
| ||||||||||||||||
| 11|12|13 | GAC | 0.026 | 0.023 | 0.028 | 0.193 | 0.66 | 0.022 | 0.020 | 0.025 | 0.471 | 0.49 | 0.02801 | 0.03368 | 0.02542 | −0.24364 | 0.80 |
|
| ||||||||||||||||
| 11|12|13 | GAT | 0.345 | 0.345 | 0.345 | 0 | 0.99 | 0.36 | 0.355 | 0.365 | 0.152 | 0.69 | 0.35542 | 0.36102 | 0.34939 | 1.04121 | 0.29 |
|
| ||||||||||||||||
| 11|12|13 | GGC | 0.027 | 0.018 | 0.035 | 2.775 | 0.09 | 0.031 | 0.038 | 0.024 | 2.302 | 0.12 | 0.0329 | 0.01545 | 0.0446 | −3.11308 | 0.001 |
|
| ||||||||||||||||
| 11|12|13 | AAC* | — | 0.01 | 0.010 | 0.01 | 0.031 | 0.86 | — | ||||||||
|
| ||||||||||||||||
| 14|15|16 | Block 5 | |||||||||||||||
|
| ||||||||||||||||
| 14|15|16 | AGG | 0.534 | 0.534 | 0.53 | 0.018 | 0.89 | 0.099 | 0.099 | 0.098 | 0.006 | 0.93 | |||||
|
| ||||||||||||||||
| 14|15|16 | ACA* | — | 0.109 | 0.107 | 0.112 | 0.085 | 0.77 | |||||||||
|
| ||||||||||||||||
| 14|15|16 | ACG | 0.083 | 0.068 | 0.096 | 2.668 | 0.10 | 0.516 | 0.525 | 0.507 | 0.459 | 0.49 | SNP #16 not genotyped | ||||
|
| ||||||||||||||||
| 14|15|16 | AGA | 0.113 | 0.100 | 0.123 | 1.415 | 0.23 | — | |||||||||
|
| ||||||||||||||||
| 14|15|16 | GCG | 0.047 | 0.053 | 0.042 | 0.672 | 0.41 | 0.245 | 0.236 | 0.255 | 0.679 | 0.41 | |||||
|
| ||||||||||||||||
| 14|15|16 | GGG | 0.217 | 0.235 | 0.206 | 1.24 | 0.26 | 0.026 | 0.029 | 0.023 | 0.675 | 0.41 | |||||
| SNPs | Block | MIRAGE Whites
|
MIRAGE African Americans
|
Caribbean Hispanics
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | S-E(S) | Var(s) | Z | p | Frequency | S-E(S) | Var(s) Z | p | Haplotype | Case | Control | χ2 | p | |||
| 2|3|4 | Block 1 | |||||||||||||||
|
| ||||||||||||||||
| 2|3|4 | GAG | 0.503 | 0.490 | 0.518 | 1.524 | 0.22 | ||||||||||
|
| ||||||||||||||||
| 2|3|4 | CAG | SNP3 not genotyped | SNP3 not genotyped | 0.248 | 0.252 | 0.245 | 0.15 | 0.70 | ||||||||
|
| ||||||||||||||||
| 2|3|4 | GAA | 0.14 | 0.143 | 0.132 | 0.534 | 0.47 | ||||||||||
|
| ||||||||||||||||
| 2|3|4 | CAA | 0.057 | 0.058 | 0.056 | 0.069 | 0.79 | ||||||||||
|
| ||||||||||||||||
| 2|3|4 | GGG | 0.034 | 0.036 | 0.033 | 0.109 | 0.74 | ||||||||||
|
| ||||||||||||||||
| 2|3|4 | CGG | 0.011 | 0.013 | 0.009 | 0.895 | 0.34 | ||||||||||
|
| ||||||||||||||||
| 5|6|7 | Block 2 | |||||||||||||||
|
| ||||||||||||||||
| 5|6|7 | TTG | 0.733 | 0.734 | 0.735 | 0.002 | 0.97 | ||||||||||
|
| ||||||||||||||||
| 5|6|7 | CTA | SNP 6 not genotyped | SNP 6 not genotyped | 0.183 | 0.180 | 0.182 | 0.007 | 0.93 | ||||||||
|
| ||||||||||||||||
| 5|6|7 | TTA | 0.015 | 0.020 | 0.01 | 3.265 | 0.07 | ||||||||||
|
| ||||||||||||||||
| 5|6|7 | TGG | 0.064 | 0.064 | 0.066 | 0.061 | 0.81 | ||||||||||
|
| ||||||||||||||||
| 8|9|10 | Block 3 | |||||||||||||||
|
| ||||||||||||||||
| 8|9|10 | CCG | 0.68 | 2.619 | 48.103 | 0.378 | 0.71 | 0.277 | −3.6 | 16.796 | −0.878 | 0.38 | 0.565 | 0.552 | 0.579 | 1.451 | 0.23 |
|
| ||||||||||||||||
| 8|9|10 | TTG | 0.291 | −2.314 | 43.815 | −0.35 | 0.73 | 0.0765 | 3.833 | 5.917 | 1.576 | 0.12 | 0.195 | 0.198 | 0.187 | 0.332 | 0.56 |
|
| ||||||||||||||||
| 8|9|10 | CTG | 0.0199 | 0.181 | 3.956 | 0.091 | 0.93 | 0.327 | 1.683 | 21.356 | 0.364 | 0.72 | 0.136 | 0.151 | 0.122 | 3.517 | 0.06 |
|
| ||||||||||||||||
| 8|9|10 | CTT | 0.32 | −1.917 | 22.65 | −0.403 | 0.69 | 0.102 | 0.097 | 0.109 | 0.815 | 0.37 | |||||
|
| ||||||||||||||||
| 11|12|13 | Block 4 | |||||||||||||||
|
| ||||||||||||||||
| 11|12|13 | GGT | 0.386 | 5.708 | 75.413 | 0.657 | 0.51 | 0.655 | 4.185 | 23.135 | 0.87 | 0.38 | 0.521 | 0.542 | 0.502 | 3.065 | 0.08 |
|
| ||||||||||||||||
| 11|12|13 | AAT | 0.0776 | 4.871 | 23.676 | 1.001 | 0.32 | 0.0213 | −2.83 | 1.777 | −2.123 | 0.03 | 0.097 | 0.076 | 0.118 | 9.634 | 0.001 |
|
| ||||||||||||||||
| 11|12|13 | AGC | 0.011 | 0.011 | 0.011 | 1.393 | 0.24 | ||||||||||
|
| ||||||||||||||||
| 11|12|13 | AGT | 0.122 | −2.73 | 32.492 | −0.479 | 0.63 | 0.105 | −5.576 | 9.019 | −1.857 | 0.06 | 0.111 | 0.101 | 0.118 | 1.393 | 0.24 |
|
| ||||||||||||||||
| 11|12|13 | GAC | 0.025 | −2.35 | 7.168 | −0.878 | 0.38 | 0.0133 | −0.375 | 0.846 | −0.407 | 0.68 | 0.015 | 0.015 | 0.015 | 0.001 | 0.98 |
|
| ||||||||||||||||
| 11|12|13 | GAT | 0.344 | −4.082 | 70.196 | −0.487 | 0.63 | 0.165 | 4.388 | 15.454 | 1.116 | 0.26 | 0.227 | 0.238 | 0.216 | 1.375 | 0.24 |
|
| ||||||||||||||||
| 11|12|13 | GGC | 0.0268 | 0.941 | 7.154 | 0.352 | 0.73 | 0.0358 | −0.699 | 2.928 | −0.408 | 0.68 | 0.014 | 0.015 | 0.013 | 0.063 | 0.80 |
|
| ||||||||||||||||
| 11|12|13 | AAC | |||||||||||||||
|
| ||||||||||||||||
| 14|15|16 | Block 5 | |||||||||||||||
|
| ||||||||||||||||
| 14|15|16 | AGG | 0.522 | 0.526 | 0.515 | 0.229 | 0.63 | ||||||||||
|
| ||||||||||||||||
| 14|15|16 | ACA | SNP 16 not genotyped | SNP 16 not genotyped | 0.046 | 0.046 | 0.047 | 0.011 | 0.92 | ||||||||
|
| ||||||||||||||||
| 14|15|16 | ACG | 0.091 | 0.092 | 0.092 | 0.004 | 0.95 | ||||||||||
|
| ||||||||||||||||
| 14|15|16 | AGA | 0.141 | 0.150 | 0.134 | 1.036 | 0.31 | ||||||||||
|
| ||||||||||||||||
| 14|15|16 | GCG | 0.018 | 0.016 | 0.02 | 0.533 | 0.47 | ||||||||||
|
| ||||||||||||||||
| 14|15|16 | GGG | 0.18 | 0.169 | 0.189 | 1.301 | 0.25 | ||||||||||
All p 6values of ≤0.05 are in bold. All p values that are close to statistical significance (0.05 ≤ p ≤ 0.1) are in italics. SNPs that are associated with a changed risk for AD in at least 2 independent data sets are underlined.
χ2 = chi square test statistic;AD = Alzheimer’s disease; SNP = single-nucleotide polymorphism.
Finally, we performed a meta-analysis of the single-marker associations in which we first excluded the Toronto dataset due to the differences in allele frequencies (Table 4). Because there was no evidence for between-dataset heterogeneity of fixed effects estimates and the random effects estimates across datasets were similar, we adopted the pooled estimate derived by the fixed effects model. In these analyses, the block of SNPs 4–9 was significantly associated with AD, with p values ranging from 0.0008 to 0.007. In addition, SNP 13 was significantly associated with AD (p = 0.03). When the Toronto dataset was included in the analyses, the associations were attenuated but remained significant for SNPs 7–9 (0.01 < p < 0.05).
TABLE 4.
Meta-Analysis of Single-SNP Associations in the Caucasian Datasets
| All Caucasian Datasets Excluding Toronto Dataset
|
All Caucasian Datasets
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Allele1 | Allele2 | Weight | Zscore | p | Direction | I2 | p(Q)a | Allele1 | Allele2 | Weight | Zscore | p | Directiona | I2 | p(Q) |
| 1 | Not genotyped in NIA-LOAD | Not genotyped in NIA-LOAD | ||||||||||||||
|
| ||||||||||||||||
| 2 | c | g | 3967 | 0.881 | 0.37 | +++ | 0.22 | 0.92 | c | g | 4639 | 1.018 | 0.3 | ++++ | 0.23 | 0.92 |
|
| ||||||||||||||||
| 3 | Not polymorphic | Not polymorphic | ||||||||||||||
|
| ||||||||||||||||
| 4 | a | g | 3967 | −2.769 | 0.005 | − − − | 0.34 | 0.9 | a | g | 4639 | −1.75 | 0.08 | − − −+ | 4.21 | 0.9 |
|
| ||||||||||||||||
| 5 | t | c | 3967 | 2.693 | 0.007 | +++ | 0.12 | 0.9 | t | c | 4639 | 1.618 | 0.1 | +++− | 3.12 | 0.9 |
|
| ||||||||||||||||
| 6 | Not genotyped in MIRAGE Caucasian | Not genotyped in MIRAGE | ||||||||||||||
|
| ||||||||||||||||
| 7 | a | g | 3967 | −3.153 | 0.001 | − − − | 0.32 | 0.9 | a | g | 4639 | −2.182 | 0.03 | − − −+ | 6.14 | 0.9 |
|
| ||||||||||||||||
| 8 | t | c | 3967 | −3.334 | 0.0008 | − − − | 0.24 | 0.9 | t | c | 4639 | −2.405 | 0.01 | − − −+ | 5.27 | 0.9 |
|
| ||||||||||||||||
| 9 | t | c | 3967 | −2.895 | 0.003 | − − − | 5.23 | 0.7 | t | c | 4639 | −1.909 | 0.05 | − − −+ | 6.48 | 0.7 |
|
| ||||||||||||||||
| 10 | t | g | 3967 | −0.319 | 0.74 | +− − | 9.47 | 0.4 | t | g | 4639 | −0.359 | 0.71 | +333 | 8.84 | 0.4 |
|
| ||||||||||||||||
| 11 | a | g | 3967 | 1.892 | 0.06 | +++ | 7.86 | 0.5 | a | g | 4639 | 1.348 | 0.17 | +++− | 7.96 | 0.5 |
|
| ||||||||||||||||
| 12 | a | g | 3967 | 0.15 | 0.88 | −+ − | 16.13 | 0.3 | a | g | 4639 | −0.06 | 0.95 | −+ − − | 15.42 | 0.3 |
|
| ||||||||||||||||
| 13 | t | c | 3967 | 2.168 | 0.03 | +++ | 14.1 | 0.3 | t | c | 4639 | 1.784 | 0.07 | +++− | 14.14 | 0.3 |
|
| ||||||||||||||||
| 14 | t | c | 3967 | 0.902 | 0.36 | +− − | 18.26 | 0.3 | t | c | 4639 | 1.047 | 0.29 | +− −+ | 16.93 | 0.3 |
|
| ||||||||||||||||
| 15 | c | g | 3967 | 0.999 | 0.31 | ++− | 0.43 | 0.9 | c | g | 4639 | 0.806 | 0.42 | ++− | 2.31 | 0.9 |
|
| ||||||||||||||||
| 16 | Not genotyped in MIRAGE Caucasian/Miami | Not genotyped in MIRAGE Caucasian/Miami | ||||||||||||||
All p values of ≤0.05 are in bold. All p values that are close to statistical significance (0.05 ≤p ≤ 0.1) are in italics. SNPs that are associated with a changed risk for AD in at least 2 independent data sets are underlined.
Heterogeneity (Cochran’s Q) p-value.
χ2 = chi square test statistic; AD = Alzheimer’s disease; SNP = single-nucleotide polymorphism.
SorCS1 Gene Expression
Microarray expression analyses of the amygdala tissue of the 19 AD and 10 control brains showed significantly lower expression of SorCS1 in AD brains compared to control brains (mean gene expression intensity: 3.69 ± 0.03 vs 4.35 ± 0.04, p = 0.00006; Figs 1A, B). When we used tissue from regions that are less affected by the AD process (ie, cerebellum and occipital lobe), SorCS1 expression did not differ between AD cases and controls (p = 0.8 and p = 0.9, respectively).
FIGURE 1.

(A) View of SORCS1 exon expression profile in 19 AD (red triangles) and 10 control (blue squares) amygdala tissue. Each triangle dot represents least squares mean expression of an exon in AD tissue; each square dot represents least squares mean expression of an exon in control tissue. The mean gene expression intensity of AD vs controls was 3.69 ± 0.03 vs 4.35 ± 0.04 (p = 0.00006) across all exons. The top part of the graph shows the structure of the SorCS1b (above) and SorCS1a (below) isoforms in this region retrieved from the UCSC browser. (B) Dot plot showing the 2D distribution of amygdala expression levels of the 19 AD (red) and 10 control (blue) samples. 2D = 2-dimensional; AD = Alzheimer’s disease; UCSC = University of California, Santa Cruz.
Quantitative RT-PCR of the amygdala brain tissue of the 29 brain samples confirmed these results for SorCS1 (mean expression intensity: 2.4 ± 0.64 in AD brains vs 3.0 ± 0.46 in control brains, p = 0.05) and the SorCS1b isoform (mean expression intensity: 1.6 ± 0.37 in AD brains vs 3.1 ± 0.37 in control brains, p < 0.0001).
Finally, in order to determine whether any of the disease-associated SNPs affect SorCS1 expression levels, we genotyped these 29 Caucasian brain samples for the 16 SORCS1 SNPs. In these analyses, several SNPs were associated with statistically significant decreases (~40%) in expression levels at probe sites closely located to these markers (Supporting Information Table 3). When the analyses were adjusted for affection status the results remained essentially unchanged (Supporting Information Table 4). There was no evidence that these associations were due to outliers (Supporting Information Fig 4). Supporting Information Figure 5 shows gene expression levels of the commonly used housekeeping genes ACTB and RPL5 for comparison. Due to the small sample size of gene expression data, multivariable linear regression analyses that can control for confounders such as age at death, gender, or RNA integrity number (RIN) was not possible.
FIGURE 4.
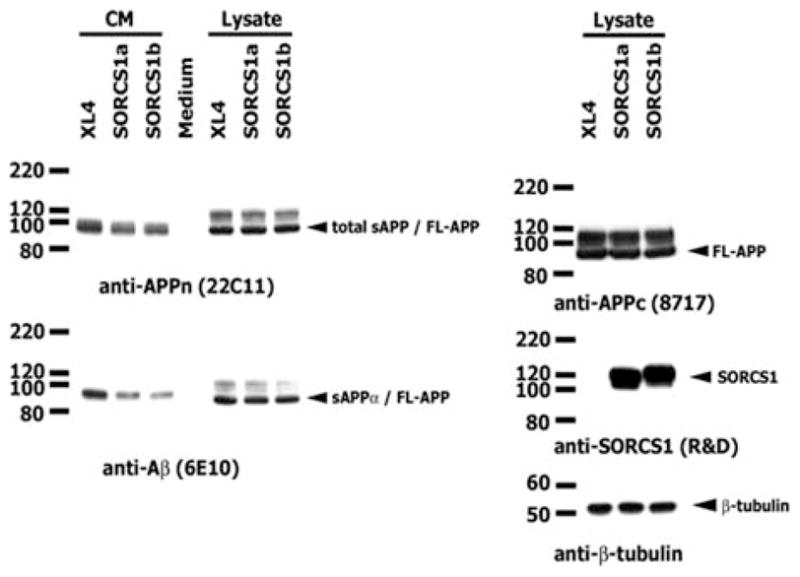
Overexpression of SorCS1 decreased the secretion of total APPs and sAPPα into culture media. CM and total cell lysate from HEK293 APPsw transfected with SorCS1 isoforms were analyzed by western blot with antibodies against APP (22C11 and 8717), Aβ (6E10), and SorCS1 (R&D). Total sAPP and sAPPα from CM were decreased compared with XL4 in spite of similar FL-APP expression in cell lysate. Western blot was done on the same samples with β -tubulin antibody (loading control). The expression of both SorCS1 isoforms in lysate was similar. APP = amyloid precursor protein; APPsw = amyloid precursor protein Swedish mutation; CM = conditioned media; FL-APP = APP holoprotein; sAPPα = α-secretase cleavage; XL4 = empty vector.
FIGURE 5.
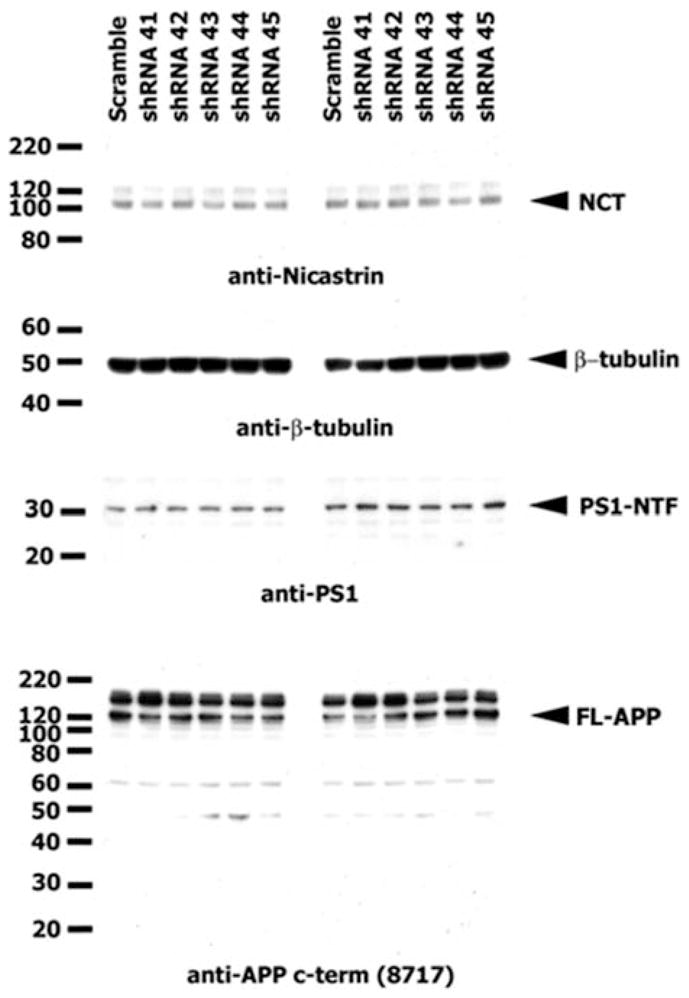
Knockdown of SorCS1 using shRNAs (41–45) did not affect the levels of transiently transfected FL-APP, NCT, PS1-NTF, or β-tubulin. (See also Supporting Table 7 for sequence information on shRNAs used and Supporting Fig 2 for western blot confirmation of endogenous expression of SorCS1 protein). APP = APP = amyloid precursor protein; FL-APP = APP holoprotein; NCT = nicastrin; PS1-NTF = pre-senilin1 N-terminal fragment; shRNA = short hairpin RNA.
Evaluation of the Role of SorCS1 in APP Metabolism
OVEREXPRESSION OF SORCS1
HEK293 cells stably transfected with the APP Swedish mutation (293APPsw) were transiently transfected with SorCS1 isoforms a or b. Expression of an ~130-kDa protein was detected by western blotting with 2 SorCS1 antibodies in transfected cell lysates (Fig 2A). Overexpression of SorCS1 did not alter the expression of APP holoprotein or its maturation (see Fig 2B) as compared to beta tubulin levels (see Fig 2C).
FIGURE 2.

Overexpression of SorCS1 in HEK293 APPsw cells (XL4 transfected samples). (A) SorCS1 expression was confirmed by 2 different SorCS1 antibodies (R&D and H120). (B) Expression of APP in SorCS1 transfectants. Overexpression of SorCS1 did not have an effect on FL-APP expression or its maturation. (C) Western blot done on the same samples with β-tubulin antibody (loading control). APP = amyloid precursor protein; APPsw = APP Swedish mutation; FL-APP = APP holoprotein; XL4 = empty vector.
To determine whether the overexpression of SorCS1 affects Aβ secretion, we measured Aβ40 and Aβ42 levels in conditioned media of HEK293APPsw cells transiently transfected with SorCS1a and SorCS1b (Fig 3). Overexpression of both SorCS1 isoforms resulted in a significant ~30% decrease of both Aβ40 (0.0001 < p < 0.0034) and Aβ42 secretion (0.0004 < p < 0.0321). The same conditioned media was analyzed using western blotting to detect the secreted forms of APP and SorCS1. The results showed that the soluble N-terminal ectodomain of APP (total sAPP) and the fragment generated by α-secretase cleavage (sAPPα) were decreased in conditioned media of transfectants with the SorCS1 isoforms (Fig 4).
FIGURE 3.
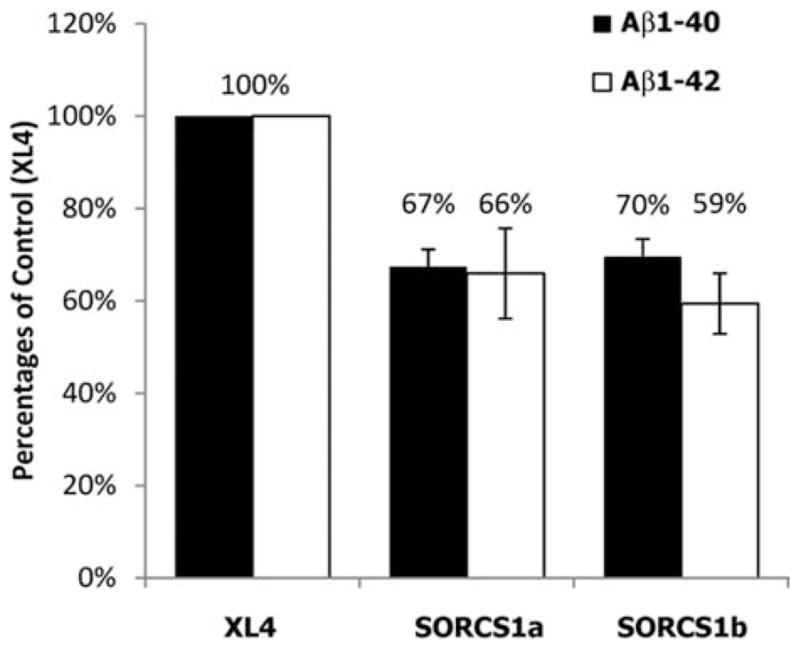
Measurement of Aβ1–40 and Aβ1–42 from conditioned media of HEK293 APPsw overexpressing SorCS1 isoforms. Aβ levels normalized to total protein were measured. Data shown as percentages of control values (XL4; n = 3 per experiment, 3 independent experiments). APP = amyloid precursor protein; APPsw = amyloid precursor protein Swedish mutation; FL-APP = APP holoprotein; XL4 = empty vector.
KNOCKDOWN OF SORCS1
HEK293 and M17 cell lines were used for the knockdown experiments. Our western blot confirmed the endogenous expression of SorCS1 protein (Supporting Information Fig 2). Five shRNAs targeting SorCS1 (shRNAs 41–45) and control shRNA (scrambled shRNA) were obtained from Open Biosystems (Supporting Information Table 7). To examine the knockdown efficiency, the HEK293 cell line was transfected with each shRNA and then total cell lysates were analyzed by western blotting with SorCS1 antibodies. We observed a reduction of endogenous SorCS1 protein using the SorCS1 targeting shRNAs but not the control scrambled shRNA.
The effect of SorCS1 knockdown on Aβ40 secretion was measured in conditioned media from HEK293 cells co-transfected with SorCS1 shRNAs 41–45 and APP that effectively reduced the expression of SorCS1 protein, but did not affect the levels of APP holoprotein, presenilin1 or nicastrin (Fig 5). Knockdown of SorCS1 also showed 2-fold to 3-fold elevation of Aβ40 levels (p < 0.01 to p < 0.001 as compared to scrambled shRNA condition (ANOVA with Bonferroni correction; Graph-Pad Software, La Jolla, CA) in conditioned media from transfected HEK293 cells (Fig 6). The level of Aβ42 was under the detection limit.
FIGURE 6.
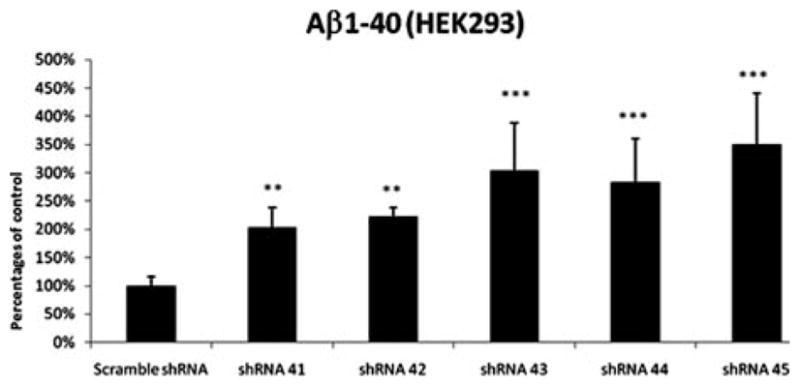
Knockdown of SorCS1 by shRNAs did affect the level of Aβ40. shRNA 41, 42, 43, 44, and 45, or scrambled sequence shRNA were transfected in HEK293 cells and 3 days after transfection, the cells were incubated for additional 48 hours. Data are expressed as percentages of control values. Aβ1–40 secretion was measured in conditioned media and normalized to total protein levels. The data are representative for the Aβassays and the assay has been performed in at least 3 separate experiments in replicates of 8 samples per condition (24-well format), standard deviation error bars are shown, **p < 0.01, ***p < 0.001 as compared to scrambled shRNA condition (ANOVA with Bonferroni correction; GraphPad Software, La Jolla, CA). ANOVA = analysis of variance; shRNA = short hairpin RNA.
In addition to the Aβ ELISA measurements, we have also utilized the APP-GV assay to monitor gamma secretase activity and nuclear translocation of the APP/ Fe65/TIP60 protein complex.34 The shRNAs against SorCS1 were also used to further investigate the role of SorCS1 in APP processing. The APP-GV assay was developed as a sensitive method to detect γ-secretase processing of APP and is based upon the nuclear translocation of the APP C-terminus fragment (AICD) to activate a transcriptional reporter.34 In both HEK293 and M17 (Fig 7), 4 of the 5 SorCS1-shRNAs caused a significant increase greater than 3-fold in APP processing (p < 0.05 to p < 0.01) as compared to scrambled shRNA condition (ANOVA with Bonferroni correction; Graph-Pad Software) while not affecting the nuclear translocation of the control AICD-GV only-fragment.
FIGURE 7.

Gamma secretase activity and nuclear translocation of APP assays with Sorcs1 shRNAs. (A) Both the APP-GV and AICD-GV assay were performed in HEK293 cells. The data from SorCS1a cDNA and the 5 shRNA SorCS1 was normalized to either APP-GV only or AICD-GV with the scrambled sequence shRNA (shRNA-scrambled), which was included as a negative control. (B) APP-GV assay was also performed in the human neuroblastoma cell line, M17 with SorCS1a cDNA or 5 of the Sorcs1 shRNAs along with the shRNA-sc. The data are representative for the APP-GV assays and the assay has been performed in at least 3 separate experiments in replicates of 8 samples per condition (96-well format), standard deviation error bars are shown, *p < 0.05, **p < 0.01 as compared to APP-GV only (ANOVA; GraphPad Software, La Jolla, CA). AICD = APP gene C-terminus; GV = transcription factor composed of the GAL4 DNA binding domain with VP16 transcriptional activator; ANOVA = analysis of variance; APP = APP = amyloid precursor protein; cDNA = complementary DNA; shRNA = short hairpin RNA.
INTERACTION OF APP AND SORCS1
To assess the potential mechanism by which SorCS1 might modulate APP processing, we next investigated if it was mediated by a direct interaction between APP and SorCS1. We performed co-immunoprecipitation experiments using HEK293APPsw overexpressing SorCS1a or SorCS1b. Immunoprecipitation of SorCS1 using the SorCS1 antibody (R&D) and western blotting for SorCS1 (H120) or APP (anti-APPn, 22C11) revealed a ~130-kDa band (SorCS1) or ~100-kDa band (FL-APP) only in SorCS1-transfectants but not in the cells transfected with empty vector (XL4), which express only low levels of endogenous SorCS1 (Fig 8). In addition, an endogenous interaction between APP and SorCS1 was observed in SH-SY5Y cells (Supporting Information Fig 6).
FIGURE 8.

Interaction between APP and SorCS1 in HEK293 APPsw cells overexpressing SorCS1 isoforms. HEK293 APPsw cells transfected with SorCS1 isoform or XL4 were lysed using one-third-diluted RIPA buffer and then immunoprecipitated with (A) SorCS1 or (B) APP. (A) Immunoprecipitation for SorCS1 was successful (left). FL-APP was coimmunoprecipitated with SorCS1 (middle). (B) SorCS1 was coimmunoprecipitated with APP. The right immunoblot shows the expression levels of (A) SorCS1 and (B) APP in cell lysate. APP = amyloid precursor protein; APPsw = amyloid precursor protein Swedish mutation; FL-APP = APP holoprotein; XL4 = empty vector.
Finally, confocal analysis supported the colocalization of SorCS1 and APP in SH-SY5Y cells. SorCS1 and APP immunoreactive elements were identified in the perinuclear area by Alexa Fluor 488 (green), and 594 (red) fluorescence, respectively (Supporting Information Fig 7).
Discussion
The accumulated findings reported here suggest that variation in SORCS1 sequence, expression, and function may influence the development of AD. Although the identity of the specific AD-associated sequence variations in SORCS1 remains to be determined, our results imply that (1) there are different AD-associated allelic variants in the SORCS1 gene in different populations; (2) these variants are likely to be in intronic regulatory sequences that effect cell type–specific or tissue-specific expression of SorCS1; and (3) that genetic variation in SORCS1 might affect AD risk by altering the physiological role of SorCS1 in the processing of APP holoprotein.
Similar to observations on the SORCS1 homologue SORL19 no single SNP or haplotype was associated with AD in all datasets. In addition, the direction of the effect of some of the disease-associated alleles differed between some of the datasets, which we attribute to population differences in LD with the true genetic effector. Another potential explanation for these series-related differences could be confounding genetic or environmental factors that are influential in 1 dataset but not the others. Several issues diminish the possibility that the association between SORCS1 and AD is spurious. First, the association was initially identified in the NIA-LOAD family 6k dataset (rs7082289; SNP 1) using conservative family-based association tests, which are less sensitive to confounding due to population stratification. Second, several alleles and haplotypes were associated with altered risk for AD in at least 2 unrelated data sets. Third, the associations of several SNPs remained significant in meta-analyses that included the Toronto dataset. Differences in the LD patterns between the datasets are a possible explanation for the inverse direction of the association with AD in the Toronto dataset as compared to the other Caucasian datasets. It is possible that the minor alleles of significant SNPs are in linkage with risk alleles in some datasets or populations but in linkage with protective variants in others. Of note, the direction of the effect of SNP 11 in the Toronto dataset was similar to the direction of the effect in the Caribbean Hispanic and MIRAGE African American datasets. Alternatively, the genotyped variants are not the disease-causing variants but rather identify protective or harmful disease-modifying variations in SORCS1. Fourth, the finding of association with different SNPs in different ethnic groups is a not an unusual observation in complex diseases.38 The occurrence of pathogenic mutations across multiple domains of disease genes (allelic heterogeneity) and the absence of these variants in some datasets or ethnic groups (locus heterogeneity) are frequently observed in both monogenic and complex traits.39 Because the 16 genotyped SNPs do not cover the whole genetic variation in the SORCS1 gene (see Supporting Information Fig 3), it is possible that additional polymorphisms in nontagged regions of the gene are associated with AD risk.
Also, it needs to be acknowledged that the sample sizes of the individual datasets were modest. Thus, it remains possible that larger individual datasets would have detected additional genotype-phenotype associations with smaller effect sizes or allele frequencies. Our meta-analyses of all Caucasian datasets, which included in total 2,309 cases and 3,482 controls, confirmed the findings for SNPs 4–9 derived from the individual study samples, and in addition pointed to SNP 13 as significantly associated with AD.
Our finding of a role of SORCS1 in AD is also supported by the results of our RT-PCR and brain microar-ray analyses. We found significantly lower SorCS1 expression levels in amygdala from AD brains compared to the controls, and decreases in SorCS1 expression levels at several probe sites were associated with closely located SNPs. In contrast, when tissue from regions that are less affected by the AD process (ie, cerebellum and occipital lobe) was used, SorCS1 expression did not differ between AD cases and controls. However, while plausible and consistent with our results from the genetic association studies and cell biological experiments, alternative explanations for these differences in expression levels must be discussed. It remains possible that SORCS1 expression levels in the amygdala were influenced by differential cell loss that is more pronounced in AD brains. Alternatively, differential expression between AD and control brains exists throughout the brain but could be more pronounced in the amygdale, and smaller differences in the control brain regions (ie, cerebellum and occipital cortex) may not have been detected due to small sample size.
Our data also revealed that high levels of SorCS1 result in modest (~30%) reductions in both Aβ40 and Aβ42, which would be protective. Conversely, the RNAi knockdown of SorCS1 on APP processing had the inverse observation of an increase in Aβ levels (2–3-fold). In addition, the APP-GV assay, which detects γ-secretase activity,34 demonstrated that the reduction of SorCS1 leads to a greater than 3-fold increase of γ-secretase activity on APP processing. Reminiscent of AD, the APP protein level and maturation was found to be unaffected, suggesting altered trafficking and/or increased activity of γ-secretase, leading to the generation of Aβ as likely mechanisms.
The mechanisms by which overexpression of SorCS1 transcripts modulates Aβ production is not immediately clear, but likely involves binding of SorCS1 to the APP holoprotein or to its processing enzymes, possibly separating APP away from BACE1 and γ-secretase cleavage.
In summary, while the genetic and biochemical data both infer a relationship between SorCS1 and the AD process, it is presently unclear how this is mediated. It is tempting to speculate that intragenic, noncoding polymorphisms in SORCS1 might account for the modest, yet consistent association with risk for AD, and might act by modulating SorCS1 expression. In particular, we hypothesize that high basal levels of SorCS1 expression in some individuals might have a protective effect, whereas low levels of expression may lead to elevated APP processing into Aβ as found in AD. Additional studies will be needed to determine whether carriers of alleles associated with differential risk for AD are indeed protected and that protection arises because of high levels of expression of SorCS1.
Acknowledgments
This research was supported by grants from the National Institutes of Health, National Institute on Aging (R37-AG15473; P01-AG07232 to R.M.; R01-AG09029 to L.A.F.; R01-AG025259 to L.A.F.; P30-AG13846); Blanchette Hooker Rockefeller Foundation (to R.M.); Charles S. Robertson Gift from the Banbury Fund (to R.M.); Merrill Lynch Foundation (to R.M.); an anonymous foundation (to L.A.F.); Asklepios-Med (Hungary) (to A.P.).
A subset of the participants in the Miami dataset was ascertained while M.A.P.-V. was a faculty member at Duke University. We thank Dr. Thomas Sudhof for the invaluable reagents related to the assays performed.
Footnotes
Additional Supporting Information can be found in the online version of this article.
Potential Conflict of Interest
C.C. received grants from The Rosalinde and Arthur Gilbert Foundation/AFAR New Investigator Award in Alzheimer’s Disease, The Taub Institute for Research on Alzheimer’s disease and the Aging Brain, and Columbia University. L.A.F. received grants from an anonymous foundation and the National Institutes of Health, National Institute on Aging. L-N.H. received grants from the Alzheimer Society of Canada, the Canadian Institutes of Health Research (CIHR), Alzheimer Society of Ontario, Howard Hughes Medical Institute, The Wellcome Trust, and the Ontario Research Fund. The laboratory under the direction of P.StG-H. received support from the Alzheimer Society of Canada, CIHR, Alzheimer Society of Ontario, Howard Hughes Medical Institute, The Wellcome Trust, and the Ontario Research Fund. R.M. received grants from the National Institutes of Health, National Institute on Aging, the Blanchette Hooker Rockefeller Foundation, the Charles S. Robertson Gift from the Banbury Fund, and the Merrill Lynch Foundation. A.P. received a grant from Asklepios-Med (Hungary). M.A.P.-V. received grants from the National Institutes of Health, National Institute on Aging, the Alzheimer’s Association; and the Louis D. Scientific Award of the Institut de France. C.R. received a Paul B. Beeson Career Development Award. E.R. received grants from CIHR. The remaining authors had nothing to report.
References
- 1.Edbauer D, Winkler E, Regula JT, et al. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 2.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 3.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 4.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 5.Harter C, Reinhard C. The secretory pathway from history to the state of the art. Subcell Biochem. 2000;34:1–38. doi: 10.1007/0-306-46824-7_1. [DOI] [PubMed] [Google Scholar]
- 6.Le Borgne R, Hoflack B. Protein transport from the secretory to the endocytic pathway in mammalian cells. Biochim Biophys Acta. 1998;1404:195–209. doi: 10.1016/s0167-4889(98)00057-3. [DOI] [PubMed] [Google Scholar]
- 7.Ponnambalam S, Baldwin SA. Constitutive protein secretion from the trans-Golgi network to the plasma membrane. Mol Membr Biol. 2003;20:129–139. doi: 10.1080/0968768031000084172. [DOI] [PubMed] [Google Scholar]
- 8.Gleeson PA, Lock JG, Luke MR, Stow JL. Domains of the TGN: coats, tethers and G proteins. Traffic. 2004;5:315–326. doi: 10.1111/j.1398-9219.2004.00182.x. [DOI] [PubMed] [Google Scholar]
- 9.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettens K, Brouwers N, Engelborghs S, et al. SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Hum Mutat. 2008;29:769–770. doi: 10.1002/humu.20725. [DOI] [PubMed] [Google Scholar]
- 11.Lee JH, Cheng R, Honig LS, et al. Association between genetic variants in SORL1 and autopsy-confirmed Alzheimer disease. Neurology. 2008;70:887–889. doi: 10.1212/01.wnl.0000280581.39755.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JH, Cheng R, Schupf N, et al. The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arch Neurol. 2007;64:501–506. doi: 10.1001/archneur.64.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Rowland C, Catanese J, et al. SORL1 variants and risk of late-onset Alzheimer’s disease. Neurobiol Dis. 2008;29:293–296. doi: 10.1016/j.nbd.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan EK, Lee J, Chen CP, et al. SORL1 haplotypes modulate risk of Alzheimer’s disease in Chinese. Neurobiol Aging. 2009;30:1048–1051. doi: 10.1016/j.neurobiolaging.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Kimura R, Yamamoto M, Morihara T, et al. SORL1 is genetically associated with Alzheimer disease in a Japanese population. Neurosci Lett. 2009;461:177–180. doi: 10.1016/j.neulet.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Kölsch H, Jessen F, Wiltfang J, et al. Association of SORL1 gene variants with Alzheimer’s disease. Brain Res. 2009;1264:1–6. doi: 10.1016/j.brainres.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 17.Hermey G, Riedel IB, Hampe W, et al. Identification and characterization of SorCS, a third member of a novel receptor family. Biochem Biophys Res Commun. 1999;266:347–351. doi: 10.1006/bbrc.1999.1822. [DOI] [PubMed] [Google Scholar]
- 18.Jacobsen L, Madsen P, Moestrup SK, et al. Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem. 1996;271:31379–31383. doi: 10.1074/jbc.271.49.31379. [DOI] [PubMed] [Google Scholar]
- 19.Kikuno R, Nagase T, Ishikawa K, et al. Prediction of the coding sequences of unidentified human genes. XIV. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 1999;6:197–205. doi: 10.1093/dnares/6.3.197. [DOI] [PubMed] [Google Scholar]
- 20.Rezgaoui M, Hermey G, Riedel IB, et al. Identification of SorCS2, a novel member of the VPS10 domain containing receptor family, prominently expressed in the developing mouse brain. Mech Dev. 2001;100:335–338. doi: 10.1016/s0925-4773(00)00523-2. [DOI] [PubMed] [Google Scholar]
- 21.Marchler-Bauer A, Anderson JB, Derbyshire MK, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermey G, Plath N, Hubner CA, et al. The three sorCS genes are differentially expressed and regulated by synaptic activity. J Neurochem. 2004;88:1470–1476. doi: 10.1046/j.1471-4159.2004.02286.x. [DOI] [PubMed] [Google Scholar]
- 23.Hermey G, Riedel IB, Rezgaoui M, et al. SorCS1, a member of the novel sorting receptor family, is localized in somata and dendrites of neurons throughout the murine brain. Neurosci Lett. 2001;313:83–87. doi: 10.1016/s0304-3940(01)02252-2. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 25.Grupe A, Li Y, Rowland C, et al. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet. 2006;78:78–88. doi: 10.1086/498851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang X, Slifer M, Martin ER, et al. Genomic convergence to identify candidate genes for Alzheimer disease on chromosome 10. Hum Mutat. 2009;30:463–471. doi: 10.1002/humu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogaeva EA, Premkumar S, Grubber J, et al. An alpha-2-macro-globulin insertion-deletion polymorphism in Alzheimer disease. Nat Genet. 1999;22:19–22. doi: 10.1038/8729. [DOI] [PubMed] [Google Scholar]
- 28.Lee JH, Cheng R, Graff-Radford N, et al. Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol. 2008;65:1518–1526. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graff-Radford NR, Green RC, Go RC, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594–600. doi: 10.1001/archneur.59.4.594. [DOI] [PubMed] [Google Scholar]
- 30.Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287:329–336. doi: 10.1001/jama.287.3.329. [DOI] [PubMed] [Google Scholar]
- 31.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 32.Lewandowski NM, Small SA. Brain microarray: finding needles in molecular haystacks. J Neurosci. 2005;25:10341–10346. doi: 10.1523/JNEUROSCI.4006-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu G, Nishimura M, Arawaka S, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 34.Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 35.Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 36.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127:820–826. doi: 10.7326/0003-4819-127-9-199711010-00008. [DOI] [PubMed] [Google Scholar]
- 38.Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant… or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 39.Vermeire S, Rutgeerts P. Current status of genetics research in inflammatory bowel disease. Genes Immun. 2005;6:637–645. doi: 10.1038/sj.gene.6364257. [DOI] [PubMed] [Google Scholar]