

Abstract
Silymarin, derived from the milk thistle plant Silybum marianum, is widely used for self-treatment of liver diseases, including hepatitis C virus (HCV), and its antiviral activity has been demonstrated in vitro and in HCV patients administered an intravenous formulation of the major silymarin flavonolignans, silybin A and silybin B. The safety and dose-exposure relationships of higher than customary oral doses of silymarin and its acute effects on serum HCV RNA were evaluated in noncirrhotic HCV patients. Four cohorts of 8 patients with well-compensated, chronic noncirrhotic HCV who failed interferon-based therapy were randomized 3:1 to silymarin or placebo. Oral doses of 140, 280, 560, or 700 mg silymarin were administered every 8 hours for 7 days. Steady-state exposures for silybin A and silybin B increased 11-fold and 38-fold, respectively, with a 5-fold increase in dose, suggesting nonlinear pharmacokinetics. No drug-related adverse events were reported, and no clinically meaningful reductions from baseline serum transaminases or HCV RNA titer were observed. Oral doses of silymarin up to 2.1 g per day were safe and well tolerated. The nonlinear pharmacokinetics of silybin A and silybin B suggests low bioavailability associated with customary doses of silymarin may be overcome with doses above 700 mg.
Keywords: Silymarin, pharmacokinetics, flavonolignans, hepatitis C virus (HCV), milk thistle, botanical drug, herbal
Approximately 180 million people worldwide and more than 4 million people in the United States are infected with hepatitis C (HCV), and approximately 80% of infected patients develop chronic HCV. However, only 50% of chronic HCV patients with genotype 1 achieve a sustained virologic response with peginterferon and ribavirin,1,2 the only clinically proven therapy.3 In addition, low tolerability and significant adverse effects often lead to treatment discontinuation, further decreasing response rates.4 Therefore, many patients have turned to complementary and alternative medicines to self-treat their disease. It has been estimated that up to 42% of Americans consume complementary and alternative medicines (CAM) annually,5 and approximately one third of patients seen in US liver clinics report the use of CAM.6
Silymarin, derived from the milk thistle plant, Silybum marianum, is an herbal remedy that has been used for centuries for diseases of the liver.7 Depending on geographic region, anywhere from 8% to 33% of patients in the United States use silymarin for the treatment of chronic HCV.8 Silymarin is a complex mixture of 6 major flavonolignans (silybins A and B, isosilybins A and B, silychristin, and silydianin), as well as other minor polyphenolic compounds.9 Antioxidant,10,11 anti-inflammatory/immunomodulatory,12,13 and antifibrotic14,15 properties of silymarin have been demonstrated in various in vitro and animal models. However, these potentially hepatoprotective effects of silymarin have not been firmly established in patients with liver disease using customary oral daily doses. Inconsistent findings from several randomized clinical trials may reflect differences in the trial designs and doses employed and in the silymarin preparations studied.16
Limited data from human and animal studies suggest silymarin flavonolignans undergo rapid and extensive conjugative metabolism with primarily biliary excretion, resulting in short half-lives and low systemic exposures when administered orally.17–21 Consequently, higher than customary doses of silymarin, administered orally or intravenously at least 2 to 3 times daily, may be necessary to overcome silymarin’s rapid first-pass metabolism. A randomized, double-blind, placebo-controlled study using 1200 mg of silymarin administered once daily to patients with chronic HCV failed to produce a significant effect on either serum transaminases or quality-of-life measures.22 Antiviral properties of silymarin have been demonstrated in a human HCV replicon system at concentrations that are 2 to 3 orders of magnitude higher than those achieved in plasma with customary oral doses.23,24 Recent results from a pilot study in patients with chronic HCV using an intravenous formulation of the dihydrosuccinate sodium salt forms of silybin A and silybin B, the 2 most abundant flavonolignans in milk thistle extracts, suggest that some silymarin flavonolignans may have antiviral activity.25 Whether or not the failure to observe hepatoprotective or antiviral effects in previous studies using oral doses of silymarin reflects the inability to achieve the necessary blood concentration of one or more of the flavonolignans primarily responsible for the pharmacological activity of silymarin is not known. In addition, only certain patients may derive benefit from chronic oral administration of high doses of silymarin because exposures can vary 3- to 5-fold higher depending on the type and grade/stage of liver disease.26 A high-dose study using a phospholipid formulation of silybin A and B attained exposures 2 orders of magnitude greater than those achieved with customary doses.27 However, grade 2/3 liver enzyme elevations with increased gastrointestinal complaints were reported. Together, these data suggest that higher exposures may also carry a greater risk for adverse effects.
To optimize dose selection for phase II studies,28 this ascending multiple oral dose phase I, double-masked, placebo-controlled study was conducted to evaluate the safety, tolerability, and dose-exposure relationships of a standardized extract of silymarin at higher than customary daily doses in a carefully defined population of noncirrhotic HCV patients. An understanding of the pharmacokinetics of each of the 6 principal flavonoligans in silymarin is essential for identifying flavonolignans with favorable pharmacologic profiles and to minimize potential risks with long-term oral maintenance therapy.
MATERIALS AND METHODS
Participants
Thirty-two male and female patients ≥18 years of age with chronic noncirrhotic HCV who previously failed interferon-based therapy were recruited into the study. Patients were required to have serologic evidence of HCV infection documented by quantifiable HCV ribonucleic acid (RNA) in serum by polymerase chain reaction (PCR) assay and elevated alanine aminotransferase (ALT) levels ≥65 IU/L within 1 year prior to screening and ALT ≥45 IU/L during screening. Patients were required to have a platelet count ≥130 cells/mm3, a creatinine clearance (calculated according to Cockcroft-Gault) >60 mL/min, serum albumin ≥3.2 g/dL, total bilirubin ≤1.5 mg/dL, prothrombin time (PT)/international normalized ratio (INR) ≤1.3 times normal at screening, and negative urine pregnancy screen for women of childbearing potential who were also required to use barrier methods of contraception during the study. Use of standard doses of over-the-counter multivitamins or cough/cold preparations was allowed.
Patients were excluded if they had evidence of portal hypertension or other chronic liver disease, had serologic evidence of infection with human immunodeficiency virus, or received antiviral therapy for HCV within 6 months of screening. Patients were excluded for the following reasons: allergy to milk thistle or its preparations; use of silymarin or other milk thistle preparations or use of antioxidants within 30 days of screening; chronic use of acetaminophen >2 g/d; use of oral contraceptive, warfarin, or metronidazole; concurrent use of the following cytochrome P450 3A4 inducers: aminoglutethimide, aprepitant, carbamazepine, dexamethasone, efavirenz, ethosuximide, garlicsupplements, glucocorticoids, glutethimide, griseofulvin, modafinil, nafcillin, nevirapine, oxcarbazepine, phenobarbital, phenytoin, primidone, rifabutin, rifampin, rifapentine, and St. John’s wort; historical liver biopsy demonstrating the presence of cirrhosis (Ishak stage 5 or 6), ≥15% steatosis, or evidence of steatohepatitis; positive urine drug screen for drugs of abuse; alcohol consumption of >12 g/d for ≥6 months prior to screening; or other evidence of alcohol or drug abuse within 6 months of screening. Women who were pregnant or breastfeeding were also excluded. All patients agreed not to consume alcohol 48 hours prior to study randomization through study completion.
Trial Design
Four dose cohorts of 8 patients each were randomized 3:1, via a Web-based randomization system used by each site’s pharmacist, to receive oral silymarin or placebo every 8 hours for 7 days. Patients and all study personnel, except for pharmacists, were blinded to treatment assignments until trial completion. No significant adverse events have been observed in prior studies with silymarin at oral doses up to 1200 mg per day. Therefore, to verify the safety of oral doses of silymarin ~2 times higher than previously studied, we chose a simple, 3:1 randomized active to placebo, sequential dose cohort escalation design with a sample size that would provide information on safety, tolerability, and pharmacokinetics, which is customary for phase I investigations. Cohorts were enrolled sequentially at doses of 140 mg, 280 mg, 560 mg, or 700 mg Legalon. Legalon (Madaus, Germany [now Rottapharm|Madaus, Italy]) brand of silymarin was selected as the clinical trial material for the Silymarin Product Development Program for use in National Institutes of Health (NIH)–sponsored clinical trials for liver diseases from competing bids in response to a Notice of Opportunity by the National Center for Complementary and Alternative Medicine and the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH. The 140-mg dose is the customary dose of Legalon and was included in the trial design as a reference to previous investigations.
Patients were recruited from December 2006 to July 2008, and no protocol deviations were granted. Twenty-one doses were dispensed to patients upon randomization. The first dose was self-administered under direct supervision in a clinical research. The last study dose was administered on day 8 with 240 mL of water to patients fasted overnight. Patients remained in the unit for 48 hours for collection of blood and urine samples. Three daily meals were served in the unit with breakfast served ≥2 hours postdose on day 8. Fourteen serial blood samples were collected at 0 (predose), 0.5, 1, 1.5, 2, 4, 6, 8, 12, 15, 18, 24, 32, and 48 hours postdose. On day 8, 3 timed urine collections were obtained at 0 to 12 hours, 12 to 24 hours, and 24 to 48 hours postdose determined in urine collections from patients in the 140-, 280-, and 560-mg dose groups. Eight-hour post-dose trough plasma samples were collected during safety visits on days 1, 4, and 6. The phase I trial design also included a single-dose pharmacokinetic study that was performed for each participant prior to the start of the 7-day dosing period. The single-dose pharmacokinetic data obtained from this phase I trial will be described in a separate report.
Serum HCV RNA level was determined on day 1 prior to the first dose and following the final dose on day 8 by quantitative PCR (Roche AMPLICOR assay, Nutley, New Jersey). Patient adherence was assessed by patient drug diary, pill counts, and by maintaining records of drugs dispensed and returned.
The study was conducted at 4 clinical centers: University of North Carolina at Chapel Hill, Beth Israel Deaconess Medical Center, University of Pennsylvania, and Thomas Jefferson University. Institutional review boards of participating centers approved the protocol; all patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki and guidelines on good clinical practice.
Safety Assessment
Safety was assessed during safety visits on study days 1, 4, 6, and 8, which consisted of clinical laboratory tests and reports of clinical adverse events using a standardized symptom assessment questionnaire. In addition, on day 8, the questionnaire was completed at predose (0 hours) and at approximately 24 and 48 hours postdose. Common Terminology Criteria for Adverse Events (CTCAE v3.0) was used to grade the severity of adverse events. Physical examinations and electrocardiograms were completed at baseline and at end of study. Decisions to dose escalate were made after a safety evaluation by a safety committee masked to treatment, which consisted of the principal investigators from the 4 clinical centers and an external safety monitor.
Study Drug
Silymarin (Legalon) and matching placebo were manufactured in hard capsules by Madaus Rottapharm Group (Cologne, Germany); all study doses were administered from Lot No. 0418901. Each dose consisted of 5 silymarin and/or placebo capsules packaged in single-use medicine dose cups. The flavonolignan content of each capsule was determined according to previously published liquid chromatography/mass spectrometry (LC/MS) methods as follows: 23.2 mg, silybin A; 32.0 mg, silybin B; 11.8 mg, isosilybin A; 6.6 mg, isosilybin B; 24.9 mg, silychristin; and 29.0 mg, silydianin.17 These 6 flavonolignans account for 70.8% (127.5 mg silymarin equivalent to 140 mg of silymarin as determined by the manufacturer’s 2,4-dinitrophenylhydrazine [DNPH] method) of the 180-mg milk thistle extract contained in each capsule. Based on interim stability testing results performed by the manufacturer, Legalon 140 capsules are stable under normal conditions (25°C, 60% relative humidity) for at least 9 months. For the purpose of this report, we refer to the 1-, 2-, 4-, or 5-capsule doses of Legalon as equal to 140, 280, 560, and 700 mg of silymarin, respectively.
Analysis of Silymarin Flavonolignans
Whole-blood samples were collected in two 3-mL ethylenediaminetetraacetic acid–lined tubes (K2-EDTA tubes; BD, Franklin Lakes, New Jersey) and centrifuged at 2400 rpm for 10 minutes at 4°C. Plasma was aspirated and transferred to polypropylene tubes. Urine weight and volume for each of the 3 timed intervals were recorded, and four 10-mL aliquots for each interval were stored in polypropylene tubes. Plasma and urine samples were temporarily stored at −70°C by each clinical site for <30 days prior to shipment to the University of North Carolina, where they were acidified by addition of glacial acetic acid (final concentration 1% acetic acid) and stored at −70°C until analysis.
For the determination of parent (ie, nonconjugated) flavonolignan concentrations in plasma, a 125-μL aliquot of each patient sample was incubated for 6 hours at 37°C without hydrolytic enzymes. A second 125-μL aliquot was incubated with a mixture of sulfatase (80 U/mL) and β-glucuronidase (8000 U/mL) (Sigma-Aldrich, St. Louis, Missouri) for the determination of total (ie, parent + conjugates) flavonolignan concentrations, which were expressed as “parent flavonolignan equivalents.” After incubation, 50 ng of naringenin (internal standard) in 25 μL of 50% MeOH was added to the samples, which were then deproteinized and processed using a high-throughput protein filtration procedure. Briefly, 140-μL aliquots of the samples (equal to 100 μL plasma) were transferred to preequilibrated 96-well protein filtration plates (0.2 μm cutoff, Orochem Technologies, Lombard, Illinois) containing 700 μL ice-cold acetonitrile/1% HAc in each well. Filtration plates were placed on the top of a CaptiVac (Varian, Palo Alto, California) vacuum collar with a 96-well collection plate on the bottom. After ~5 minutes at room temperature, the filtration was performed under a slight vacuum over 5 to 8 minutes until all filtration plate wells were dried. Collection plates were removed, sealed, and kept at room temperature for 30 minutes before the filtrates were dried using a Zipvap 96-well evaporator (ChromTech, Apple Valley, Minnesota) at ~45°C under a stream of nitrogen. To reconstitute the dried sample residues, we added 100 μL of MeOH/1% HAc (40:60, v/v) to each well, and then collection plates were vortexed on a microplate shaker for ~2 minutes at 1600 rpm. Each sample was then transferred to 0.5-mL microfuge tubes and centrifuged at 15 000 g and 4°C for 10 minutes. Finally, 75 μL of the supernatants was transferred to glass high- performance liquid chromatography (HPLC) vials.
Plasma concentrations of silymarin flavonolignans were quantified by liquid chromatography electron spray ionization mass spectrometry (LC-ESI-MS), as previously described using a Luna C18 analytical column (50 × 2.0 mm i.d., 3 μm; Phenomenex, Torrance, California); an isocratic mobile phase consisting of 43% methanol, 56% water, and 1% glacial acetic acid (pH 2.8); a flow rate of 0.3 mL/min; a 25-μL injection volume; and a 13-minute run time.17
For the analysis of flavonolignans in urine, 0.5 mL of urine sample was transferred into a 1.5-mL vial and centrifuged at 3000 g for 5 minutes. Aliquots of 100 μL of urine were then transferred to 1.5-mL polypropylene tubes, buffered using sodium acetate (pH 5.0, 0.125 M in a final incubation volume of 120 μL), and incubated at 37°C with gentle shaking for 6 hours with or without a mixture of sulfatase (80 U/mL) and β-glucuronidase (8000 U/mL) for the determination of total (parent + conjugates) or parent (nonconjugated) flavonolignans, respectively. After incubation, 250 ng of naringenin (internal standard) was added to each sample, followed by addition of 380 μL of ice-cold methanol containing 0.1% acetic acid to precipitate protein. Tubes were then vortexed and centrifuged at 15 000 g and 4°C for 15 minutes, and then 100 μL of the supernatants was transferred to LC vials, diluted with 100 μL of MeOH/H2O (50/50, v/v), and vortexed before 25 μL was injected for LC-ESI-MS analysis (final dilution ratio of urine sample: 1/10). Calibration curves in blank urine were treated similarly. For authentic reference standards, the composition of silybin (silibinin, Sigma-Aldrich, St. Louis, Missouri) was confirmed to be a mixture of silybin A (SA) and silybin B (SB) by LC-ESI-MS, and the specific contents of SA and SB were analyzed to be 48% and 52%, respectively. Silychristin (SC) was obtained from ChromaDex (Santa Ana, California), and silydianin (SD) was purchased from U.S. Pharmacopoeia (USP, Rockville, Maryland). Isosilybin A (ISA) and isosilybin B (ISB) reference standards were obtained as a generous gift from Ulrich Mengs (Madaus GmbH).
Separation of silymarin flavonolignans in urine samples was performed using an Agilent HP 1050 LC system (Palo Alto, California) with a BrownLee RP C18 guard column (15 × 3.2 mm i.d., 7 μm; PerkinElmer, Shelton, Connecticut) and a AQUA C18 (2) analytical column (150 × 4.6 mm i.d., 5 μm; Phenomenex, Torrance, California) and a methanol/0.1% acetic acid mobile phase gradient from 55% methanol to 60% methanol over 20 minutes followed by a column wash with 80% methanol, a flow rate of 0.4 mL/min (70 bars pressure), an injection volume of 25 μL, and a run time of 25 minutes. Typical retention times for SC, SD, SBA, SBB, ISBA, ISBB, and naringenin were 8.3, 10.1, 15.0, 16.4, 19.4, 20.5, and 17.4 minutes, respectively. MS detection was conducted using a PE Sciex API 100 LC/MS system (PE Sciex, Toronto, Canada) with a TurboIonspray (Applied Biosystems, Foster City, California) interface in the negative ESI ionization mode. MS parameters used for qualitative analysis included the following: ionspray voltage, −3100 V; ion-spray temperature, 450°C; orifice voltage, −30 V; focusing ring voltage, −200 V; entrance potential (EP), −10 V; nebulizer gas, 10 L/min; curtain gas, 8 L/min; channel electron multiplier, 2400; dwell time, 300 ms; scan speed, 0.61 s; and scan mode, selective ion monitoring (SIM) with [M-H]− for silymarin flavonolignans (m/z 481), silymarin sulfates (m/z 561), silymarin glucuronides (m/z 657), and naringenin (m/z 271).
Concentrations of silymarin flavonolignans in the samples were estimated with 1/x2 weighted least squares regression equations derived from the peak area ratios of individual silymarin flavonolignans to that of the internal standard. For each silymarin flavonolignan, the limit of detection was 20 ng/mL, and the quantitative ranges for parent and for total flavonolignan were 50 to 2500 ng/mL and 100 to 20 000 ng/ml, respectively. Intra- and interday precisions were 1.7% to 11% and 4.5% to 14%, respectively.
Data Analysis
Pharmacokinetic parameters, including area under plasma concentration-time curve from time 0 to 48 hours (AUC0–48 h), maximum plasma concentration (Cmax), time to Cmax (tmax), renal clearance (CLR), apparent clearance (total oral clearance divided by bioavailability, [CL/F]), and terminal half-life (t1/2), were calculated using noncompartmental methods, WinNonlin-Pro (Version 5.2; Pharsight Corp, Mountain View, California). A constant dosing interval (tau) of 8 hours was assumed for the calculation of steady-state AUC0–8 h using the linear up/log down trapezoidal method. To obtain pharmacokinetic parameters for the sum of all parent flavonolignan concentrations and for the sum of all total flavonolignan concentrations, we summed the parent or total flavonolignan concentration for each of the 6 flavonolignans at each time point over the entire sampling period prior to performing a pharmacokinetic analysis. For CL/F calculations, the dose of each silymarin flavonolignan was determined from their specific content in the milk thistle product as described above. Apparent CLR(0–8 h) was approximated from the amount of flavonolignans excreted in a 12-hour urine collection divided by the AUC0–8 h for each patient. All pharmacokinetic parameters are reported as medians with minimum and maximum values, except where indicated as a mean ± standard deviation. Pharmacokinetic parameter estimates across more than 2 dose groups were compared using a permutation test based on the Kruskal-Wallis statistic. For pairwise comparison, a permutation test based on 2-sample Wilcoxon’s statistic was used. For multiple comparisons, P values were adjusted using Holm’s step-down method. P < .05 was used for statistical significance. All of the analyses were done using SAS Version 9.1.3 (SAS Institute, Inc, Cary, North Carolina).
RESULTS
Patients
Baseline demographics are presented in Table I. Study participants were primarily male and white, and ages ranged from 30 to 64 years across all cohorts. Patients were characterized by well-compensated, noncirrhotic liver disease, as evidenced by total bilirubin, which ranged between 0.3 and 1.4 mg/dL, platelet counts between 135 and 402 cells/mm3, and normal INRs. Patients in the 560-mg dose cohort had higher median body mass indexes (BMI), whereas patients in the 700-mg dose cohort had lower median BMIs.
Table I.
Participant Baseline Demographics
| Dose Cohort
|
|||||
|---|---|---|---|---|---|
| Placebo | 140 mg | 280 mg | 560 mg | 700 mg | |
| Male/female, n | 7:1 | 5:1 | 4:2 | 5:1 | 3:3 |
| White/black, n | 5:2 | 2:4 | 5:1 | 4:2 | 5:1 |
| Age, y | 51 (30, 57) | 55 (49, 64) | 52 (50, 59) | 51 (43, 54) | 55 (50, 60) |
| Weight, kg | 93 (61, 121) | 86 (61, 96) | 93 (71, 99) | 104 (86, 123) | 69 (49, 102) |
| BMI, kg/m2 | 30 (22, 49) | 27 (26, 30) | 28 (25, 42) | 33 (26, 41) | 25.2 (21.1, 36.4) |
| Total bilirubin, mg/dL | 0.6 (0.3, 0.9) | 0.7 (0.5, 1.0) | 0.8 (0.3, 1.0) | 0.6 (0.3, 1.4) | 0.6 (0.4, 0.8) |
| ALT, U/L | 77 (63, 163) | 73 (51, 124) | 152 (70, 288) | 113 (81, 214) | 95 (48, 164) |
| AST, U/L | 50 (39, 162) | 83 (40, 117) | 140 (61, 271) | 89 (46, 110) | 71 (47, 116) |
| Platelets, cells/mm3 | 225 (148, 402) | 175 (135, 229) | 196 (177, 308) | 192 (150, 225) | 192 (139, 276) |
| INR | 1 (0.9, 1.1) | 1.1 (1.0, 1.2) | 1.1 (1.0, 1.2) | 1.0 (1.0, 1.1) | 1.0 (0.9, 1.2) |
Data are presented as medians (minimum, maximum). ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; INR, international normalized ratio.
Compliance and Safety
Adherence to the total number of doses during the study was >98% across all study cohorts, with 84% of patients compliant with the entire 21-dose regimen.
Only 1 of the 24 patients (4%) receiving silymarin reported any adverse events, and no adverse events were reported by the 8 placebo patients. The 2 reported adverse events at a dose of 280 mg were classified as gastrointestinal (nausea) and headache and appeared mild to moderate, resolved within 24 hours, and were believed to be unrelated to treatment.
Steady-State Pharmacokinetics for Major Silymarin Flavonolignans
Pharmacokinetic data for the 6 parent flavonolignans after 7 days of chronic dosing are presented in Table II. Silybin A and silybin B were the only flavonolignans detected in plasma at doses from 140 to 560 mg, with silybin A accounting for approximately 84% ± 5% (mean ± standard deviation [SD]) of measurable flavonolignans. In contrast, 6 silymarin flavonolignans were detected in plasma at the 700-mg dose, where silybin A and silybin B accounted for 54% and 21%, respectively, whereas isosilybin A, isosilybin B, silychristin, and silydianin comprised 4%, 8%, 11%, and 2%, respectively. When compared to their AUC0–8 h at 140 mg, silybin A exposures increased approximately 2-, 5-, and 11-fold, whereas silybin B exposures increased disproportionately by approximately 5-, 8-, and 38-fold at doses of 280, 560, and 700 mg, respectively. Thus, nonlinear pharmacokinetics was observed for silybin B and especially for silychristin, where at a dose of 700 mg, the AUC0–8 h ranged as high as 1555 ng·h/mL while not detected in plasma at lower doses. For all doses, peak plasma concentrations were achieved by 2 hours, and elimination half-lives ranged from 0.8 to 2.4 hours among the 6 flavonolignans.
Table II.
Steady-State Pharmacokinetics of Parent Silymarin Flavonolignans
| Cohort | Pharmacokinetic Parameter | SA | SB | ISA | ISB | SC | SD |
|---|---|---|---|---|---|---|---|
| 140 mg | AUC0–8 h, ng· h/mL | 136 (0, 318)a | 16 (0, 94)c | ND | ND | ND | ND |
| Cmax, ng/mL | 40 (0, 296)a | 8 (0, 104)c | ND | ND | ND | ND | |
| t1/2, h | 0.8 (0.6, 1.8)b | 1.3 (0.4, 2.5)c | ND | ND | ND | ND | |
| CL/F, L/h | 130 (0, 276)a | 170 (0, 1000)c | ND | ND | ND | ND | |
| 280 mg | AUC0–8 h, ng· h/mL | 334 (331,553) | 87 (58, 122) | ND | ND | ND | ND |
| Cmax, ng/mL | 163 (77, 261) | 53 (17, 72) | ND | ND | ND | ND | |
| t1/2, h | 1.3 (0.6, 1.9) | 0.9 (0.3, 1.2)b | ND | ND | ND | ND | |
| CL/F, L/h | 139 (84, 149) | 736 (525, 1103) | ND | ND | ND | ND | |
| 560 mg | AUC0–8 h, ng· h/mL | 623 (466, 1148) | 135 (76, 317) | ND | ND | ND | ND |
| Cmax, ng/mL | 364 (144, 586) | 132 (34, 174) | ND | ND | ND | ND | |
| t1/2, h | 1.6 (1.1, 2.3) | 1.1 (0.5, 2.5) | ND | ND | ND | ND | |
| CL/F, L/h | 149 (81, 191) | 948 (404, 1684) | ND | ND | ND | ND | |
| 700 mg | AUC0–8 h, ng· h/mL | 1550 (586, 2591) | 609 (194, 1193) | 123 (18, 233) | 222 (72, 423) | 308 (104, 1555) | 66 (0, 103)a |
| Cmax, ng/mL | 580 (253, 1510) | 224 (94, 681) | 56 (13, 113) | 87 (36, 223) | 95 (29, 223) | 22 (0, 61)a | |
| t1/2, h | 1.2 (0.5, 2.4) | 1.3 (0.4, 2.5) | 1.1 (0.5, 1.5) | 1.1 (0.5, 1.9) | 2.4 (1.9, 6.6)b | 2.0 (0.3, 2.3)c | |
| CL/F, L/h | 75 (45, 198) | 267 (134, 825) | 499 (265, 3278) | 151 (78, 458) | 409 (207, 1197) | 2016 (0, 3919)a |
Data are presented as medians (minimum, maximum) for n = 6 participants, except for the 560-mg cohort, where n = 5. One participant was dropped from the pharmacokinetic analysis for the 560-mg cohort due to incorrect dosing for pharmacokinetic sampling on day 8. Superscript letters denote the number of participants in each cohort with quantifiable plasma concentrations:
5 of 6 participants,
4 of 6 participants, and
3 of 6 participants. Not detectable (ND) indicates plasma concentrations were below the limit of quantitation or where only 1 or 2 of 6 participants in a cohort had quantifiable plasma concentrations. SA, silybin A; SB, silybin B; ISA, isosilybin A; ISB, isosilybin B; SC, silychristin; SD, silydianin.
The apparent oral clearances (CL/F) for the 6 flavonolignans are also presented in Table II. At the 700-mg dose, CL/F for silybin A was 75 L/h, which approximated that of hepatic blood flow. However, CL/Fs for other flavonolignans ranged between 151 and 2016 L/h, suggesting very low oral bioavailability.
Steady-state pharmacokinetic parameters for the total flavonolignan concentration for each flavonolignan are presented in Table III. As defined in the Materials and Methods section, the total flavonolignan concentration represents the concentration of parent flavonolignan plus all conjugate species of parent flavonolignan in plasma. In contrast to parent flavonolignans, total concentrations of each of the 6 flavonolignans were quantifiable in plasma at all doses, and total silybin B concentrations accounted for the majority of total flavonolignans in plasma at approximately 38% ± 4% (mean ± SD) for all doses. The proportions of the other flavonolignans also remained relatively constant across all doses at 16% ± 2%, 17% ± 3%, 14% ± 4%, 12% ± 3%, and 3% ± 0.4% for silybin A, isosilybin A, isosilybin B, silychristin, and silydianin, respectively. For each flavonolignan, steady-state total flavonolignan exposures increased approximately 3-, 4-, and 8-fold at doses of 280, 560, and 700 mg, respectively, when compared to their AUC0–8 h at 140 mg. Peak total flavonolignan concentrations for most flavonolignans occurred by 4 hours, and their elimination half-lives ranged from 3 to 6 hours.
Table III.
Steady-State Pharmacokinetics of Total (Parent + Conjugates) Silymarin Flavonolignans
| Cohort | Pharmacokinetic Parameter | SA | SB | ISA | ISB | SC | SD |
|---|---|---|---|---|---|---|---|
| 140 mg | AUC0–8 h, ng· h/mL | 1098 (492, 2033) | 2716 (1140, 4595) | 1279 (468, 2663) | 877 (405, 2195) | 756 (147, 909) | 212 (0, 233)a |
| Cmax, ng/mL | 278 (111, 521) | 679 (268, 846) | 276 (86, 393) | 182 (77, 341) | 108 (23, 150) | 39 (0, 53)a | |
| t1/2, h | 3.8 (1.4, 4.9)a | 3.3 (1.8, 3.7)a | 6.3 (2.4, 9.2) | 5.3 (1.2, 6.1) | 8.2 (6.2, 10.5)b | 4.8 (4.5, 8.2)c | |
| 280 mg | AUC0–8 h, ng· h/mL | 2776 (1627, 3256) | 7851 (4992, 9669) | 2279 (1617, 2943) | 2008 (1232, 2470) | 3014 (1408, 5088) | 620 (265, 911) |
| Cmax, ng/mL | 552 (333, 720) | 1280 (1100, 1820) | 377 (297, 532) | 320 (241, 425) | 478 (290, 294) | 128 (78, 190) | |
| t1/2, h | 3.9 (1.5, 5.4) | 5.0 (2.0, 6.3) | 3.6 (2.1, 9.7) | 4.9 (1.2, 7.7) | —ϕ | 4.0 (1.8, 6.1) | |
| 560 mg | AUC0–8 h, ng· h/mL | 3868 (2563, 6399) | 8332 (6508, 24 145) | 4986 (2557, 20 490) | 5299 (2249, 18 844) | 2540 (723, 22 053) | 643 (132, 3662) |
| Cmax, ng/mL | 842 (399, 1500) | 2090 (975, 4020) | 1370 (388, 3150) | 1380 (314, 2760) | 802 (149, 3060) | 198 (27, 590) | |
| t1/2, h | 4.7 (3.2, 7.0) | 4.1 (2.8, 6.8) | 4.4 (2.5, 7.9) | 5.7 (2.4, 11.1) | 3.1 (2.8, 11.1) | 3.1 (2.4, 5.1) | |
| 700 mg | AUC0–8 h, ng· h/mL | 10 527 (2732, 12 798) | 22 217 (10 646, 45 355) | 10 123 (3954, 20 490) | 7384 (3573, 15 413) | 6448 (2502, 8085) | 1404 (920, 1841) |
| Cmax, ng/mL | 2325 (616, 3190) | 4260 (1870, 8170) | 1950 (769, 3350) | 1450 (754, 2720) | 1052 (413, 1280) | 279 (184, 386) |
Data are presented as medians (minimum, maximum) for n = 6 participants, except for the 560-mg cohort, where n = 5. One participant was dropped from the pharmacokinetic analysis for the 560-mg cohort due to incorrect dosing for pharmacokinetic sampling on day 8. Superscript letters denote the number of participants in each cohort with quantifiable plasma concentrations:
5 of 6 participants,
4 of 6 participants, and
3 of 6 participants. Not detectable (ND) indicates plasma concentrations were below the limit of quantitation or where only 1 or 2 of 6 participants in a cohort had quantifiable plasma concentrations.
Median half-life could not be calculated due to extensive enterohepatic cycling after tmax in >4 participants. SA, silybin A; SB, silybin B; ISA, isosilybin A; ISB, isosilybin B; SC, silychristin; SD, silydianin.
Steady-State Pharmacokinetics of Sum Parent and Sum Total Silymarin Flavonolignans
The 6 major silymarin flavonolignans have the same molecular weight and similar chemical structures, and it is not known whether one or more of the flavonolignans contribute to silymarin’s hepatoprotective or antiviral properties. In addition, flavonolignan conjugates, which comprise the majority of total flavonolignan concentrations in plasma, may also contribute to the pharmacologic activity of silymarin. Therefore, the sum concentration for all parent silymarin flavonolignans and the sum concentration for all total silymarin flavonolignans in plasma were used to examine and define the dose-exposure relationships.
Figure 1A depicts the steady-state plasma concentration versus time profiles over the 8-hour dosing interval for the sum of all parent flavonolignans. For all doses, peak concentrations were achieved at 1.5 hours followed by approximately parallel elimination phases with half-lives between 0.7 and 1.4 hours. Similarly, Figure 1B depicts the steady-state plasma concentration versus time profiles for the sum of all total flavonolignan concentrations. For all doses, time to peak concentrations ranged between 1 and 2 hours followed by parallel elimination half-lives between 4.4 and 6.2 hours.
Figure 1.

Steady-state (A) sum parent and (B) sum total flavonolignan concentration versus time profiles at doses of 140, 280, 560, and 700 mg. Legend for Figure 1 is presented in panel A. Bars representing the first and third quartile ranges are presented along with the median values for the 140-mg and 700-mg dose groups to provide a representative example of the variability in the raw plasma concentration versus time data that were used to derive the pharmacokinetic data presented in Table IV.
Pharmacokinetic parameters obtained from the data depicted in Figures 1A,B are presented in Table IV. To evaluate dose-exposure proportionality, we normalized the AUC0–8 h to account for differences in body weight and administered dose between patients. Although the dose weight–normalized AUC0–8 h for the sum of all parent flavonolignans was approximately 2-fold higher at the 700-mg dose compared to the other dose groups, differences between dose groups, which ranged from 1.5 to 3.8, did not reach significance (P = .054). In contrast, more convincing evidence of good dose-exposure proportionality was observed for the sum of total flavonolignan concentrations, as suggested by the approximately constant dose-normalized AUC0–8 h across all dose groups (P = .42).
Table IV.
Steady-State Pharmacokinetics of Sum Parent and Sum Total Silymarin Flavonolignans
| Pharmacokinetic Parameters | 140 mg | 280 mg | 560 mg | 700 mg | P Value* | |
|---|---|---|---|---|---|---|
| Sum Parent | AUC0–8 h, ng· h/mL | 190 (0, 449)a,α | 559 (363, 744)α | 698 (548, 1519)α | 2802 (1132, 5023)α | <.001 |
| Cmax, ng/mL | 54 (0, 459)a,α | 301 (94, 465)β | 496 (178, 903) | 1015 (424, 2707)α,β | .001 | |
| t1/2, h | 0.7 (0.6, 2)a | 1.2 (0.6, 1.5) | 1.4 (1, 2) | 1.4 (0.5, 2.4) | .515 | |
| AUC/dose per 70 kg | 1.5 (0, 3.9)α | 2.3 (1.8, 3.5) | 2.2 (1.4, 3.7) | 3.8 (1.7, 6.4) | .054 | |
| Sum Total | AUC0–8 h, ng· h/mL | 6862 (2655, 12 432)α,β,γ | 19 258 (11 261, 24 198)α,δ | 35 676 (17 068, 95 675)β,ε | 56 727 (25 665, 103 809)γ,δ,ε | .002 |
| Cmax, ng/mL | 1568 (427, 2013)α,β,γ | 3018 (2521, 4208)α,β | 7965 (2455, 14 133)β | 10 652 (4132, 18 923)β,γ | .003 | |
| t1/2, h | 4.4 (2.1, 5.2) | 5.2 (2.5, 8.6) | 6.2 (3.3, 8.4) | 5.5 (3.4, 7.3)c | .677 | |
| AUC/dose per 70 kg | 60.8 (23.2, 76.7) | 90.0 (40.7, 105.6) | 78.5 (46.8, 233.1) | 77.4 (37.7, 187.7) | .415 |
Data are presented as medians (minimum, maximum). Superscript letters denote the number of participants in each cohort with quantifiable plasma concentrations:
5 of 6 participants,
4 of 6 participants, and
3 of 6 participants.
From 5000 permutations based on the Kruskal-Wallis statistic to test the equality of distributions across the dose groups. Statistically significant (P < .05, based on 2-sample Wilcoxon statistic adjusted for multiple comparison using Holm’s step-down method). Pairwise comparisons are indicated by the same Greek symbols. For example, Sum Parent AUC0–8 h is significantly different for any pair of dose groups, but Sum Parent Cmax is only significantly different between the 140- and 700-mg and between the 280- and 700-mg dose groups.
Enterohepatic Cycling of Silymarin Flavonolignans
To explore potential mechanisms for the nonlinear pharmacokinetics observed for some flavonolignans, especially for silybin B and silychristin, we evaluated the steady-state pharmacokinetics over a 48-hour period. Figure 2A depicts the concentration versus time profiles for each of the 6 flavonolignans following a dose of 700 mg. Plasma concentrations of parent flavonolignans peaked by 2 hours postdose but then declined with different elimination phases. For isosilybin A and isosilybin B, elimination was monophasic with half-lives of 1.5 hours. In contrast, the elimination of silybin A and silybin B appeared biphasic with half-lives of 1.1 hours for the initial phase and between 5 and 6 hours for the second phase, which occurred approximately 6 hours post-dose. Although silychristin’s elimination was monophasic, its half-life of 6.5 hours approximated that of silybin B’s second elimination phase (data not shown).
Figure 2.

The 48-hour pharmacokinetic profiles for (A) parent and (B) total flavonolignan plasma concentrations of 6 silymarin flavonolignans at 700 mg silymarin. Legend for Figure 2 is presented in panel A. SC, silychristin; SD, silydianin; SA, silybin A; SB, silybin B; ISA, isosilybin A; ISB, isosilybin B.
Figure 2B depicts the concentration versus time profiles for the total concentration for each flavonolignan at a dose of 700 mg. All flavonolignans reached peak plasma concentrations by 4 hours postdose, whereas their elimination phases were characterized by varying evidence of a second elimination phase. All flavonolignans demonstrated 1 or more minor peaks or “shoulders” at 12, 18, or 24 hours, suggestive of a 6-hour enterohepatic cycling period. These secondary peaks were more apparent in the profiles for individual participants (data not shown) than in the median data depicted in Figure 2B. A second elimination phase was most notable for silybin B and silychristin and appeared to plateau around 18 hours. As seen in Table V, which presents the 48-hour steady-state exposures for parent and total flavonolignans, silybin B and silychristin achieved the highest exposures as a result of their prolonged second phase of elimination.
Table V.
AUC0–48 h (ng·h/mL) for Parent Flavonolignans and Total Silymarin Flavonolignans
| Dose Group | SA | SB | ISA | ISB | SC | SD |
|---|---|---|---|---|---|---|
| Parent | ||||||
| flavonolignans | ||||||
| 140 mg | 213 (0, 678)a | 16 (0, 187)c | ND | ND | ND | ND |
| 280 mg | 363 (331, 611) | 87 (58, 122) | ND | ND | ND | ND |
| 560 mg | 671 (493, 1275) | 135 (76, 317) | ND | ND | ND | ND |
| 700 mg | 1783 (808, 2739) | 647 (313, 1245) | 123 (18, 233) | 222 (72, 423) | 308 (104, 1555) | 71 (0, 103)a |
| Total | ||||||
| flavonolignans | ||||||
| 140 mg | 2751 (879, 11 774) | 5190 (1660, 25 590) | 3238 (900, 15 425) | 2120 (825, 11 748) | 2654 (815, 5139) | 378 (0, 11 133)a |
| 280 mg | 6506 (2989, 7874) | 18 943 (12 592, 22 218) | 4563 (2747, 6170) | 4276 (1892, 6040) | 12 909 (7205, 23 957) | 1537 (756, 2591) |
| 560 mg | 7721 (5268, 12 701) | 19 106 (13 832, 51 927) | 12 218 (6050, 39 459) | 7703 (4105, 37 909) | 10 004 (4019, 88 697) | 1289 (273, 7257) |
| 700 mg | 21 436 (4955, 29 009) | 40 618 (22 385, 94 789) | 17 709 (6649, 40 026) | 14 331 (6037, 34 239) | 24 918 (9663, 37 013) | 2929 (1785, 5209) |
Data are presented as medians (minimum, maximum) for n = 6 participants, except for the 560-mg cohort, where n = 5. One participant was dropped from the pharmacokinetic analysis for the 560-mg cohort due to incorrect dosing for pharmacokinetic sampling on day 8. Superscript letters denote the number of participants in each cohort with quantifiable plasma concentrations:
5 of 6 participants,
4 of 6 participants, and
3 of 6 participants. Not detectable (ND) indicates plasma concentrations were below the limit of quantitation or where only 1 or 2 of 6 participants in a cohort had quantifiable plasma concentrations. SA, silybin A; SB, silybin B; ISA, isosilybin A; ISB, isosilybin B; SC, silychristin; SD, silydianin.
Steady-state Peak and Trough Concentrations
To verify that plasma trough concentrations achieved steady state, we determined silymarin flavonolignan concentrations in blood samples obtained predose on days 4, 6, and 8 and also at 8 hours postdose on day 8. Trough concentrations were only consistently detected at the 700-mg dose. At 8 hours postdose, mean ± SD plasma concentrations for silybin A and silybin B ranged from troughs of 26 ± 8 and 12 ± 7 ng/mL to peaks of 703 ± 459 and 274 ± 200 ng/mL, respectively, and for the sum of all parent flavonolignans from a trough of 59 ± 13 ng/mL to a peak of 1235 ± 322 ng/mL (data not shown). For the sum of total flavonolignan concentrations, the 8-hour post-dose troughs were 620, 1660, 2610, and 3890 ng/mL at doses of 140, 280, 560, and 700 mg, respectively. At the 700-mg dose, flavonolignan steady-state trough concentrations on day 6, day 8, and 8 hours postdose on day 8 ranged between 77% and 95% of the plasma trough concentration obtained on day 4. These data suggest that flavonolignans did not accumulate in plasma with a chronic dosing interval of 8 hours and that parent flavonolignans achieved steady-state levels that were only approximately 1.5% of the steady-state trough levels achieved for total flavonolignan concentrations.
Renal Elimination of Silymarin Flavonolignans
Only flavonolignan conjugates were detected in urines and equaled approximately 4.2, 9.6, and 15.8 mg of parent flavonolignan at doses of 140, 280, and 560 mg, respectively. These amounts represented 3.1% ± 0.3% (mean ± SD) of the doses administered and approximately 93% of the total amounts excreted over 48 hours. The apparent renal clearance (CLR(0–8 h)) for flavonolignan conjugates ranged from 530 to 608 mL/h for all doses (data not shown).
Effect of Silymarin on Serum Transaminases and HCV RNA Titer
Figure 3 depicts baseline and end-of-study HCV RNA titers for the 6 active and 2 placebo patients enrolled into the 700-mg dose cohort. When compared to screening baseline values, no reductions in serum transaminases or in HCV RNA titer were observed at the end of the 7-day treatment period for any dose cohort (other data not shown).
Figure 3.
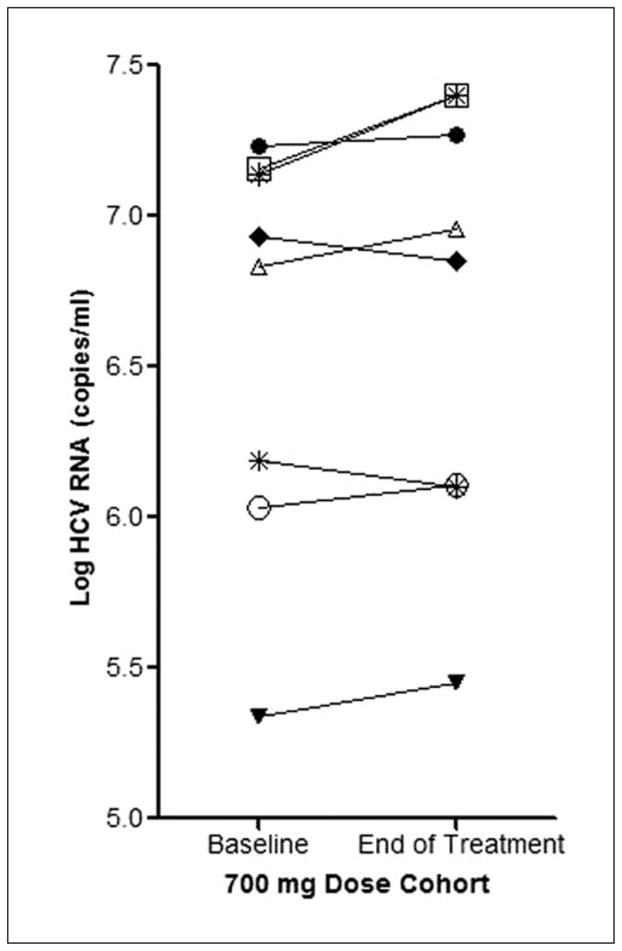
Effect of silymarin on hepatitis C virus (HCV) RNA titer. Baseline and end-of-treatment HCV RNA titers are depicted for the 8 patients enrolled in the 700-mg dose cohort who achieved the highest flavonolignan concentrations. * denotes the 2 placebo patients.
DISCUSSION
The therapeutic benefits of silymarin at customary doses ranging between 210 and 800 mg per day have not been consistently demonstrated in various liver disease populations.29–33 For example, changes in standard surrogate clinical endpoints, such as serum ALTs, have not been observed in patients with chronic HCV and early stage of the disease.22,34 In contrast, silymarin has been shown to decrease the complications of liver disease and mortality in patients with cirrhotic disease.32,35,36
High concentrations of silymarin in the range of 10 to 40 μg/mL have been shown to inhibit viral replication in a human HCV replicon system.23,24 We have recently observed low systemic exposures and short 1- to 2-hour half-lives for all 6 major silymarin flavonolignans using single, higher than customary oral doses of silymarin.37 These data suggest that multiple daily oral doses must be administered at least 3 times per day if steady-state concentrations and antiviral effects are to be achieved. The design of the current study used oral doses of silymarin that were 3- to 5-fold higher than customary doses and were administered daily as 3 divided doses of 5 capsules to achieve steady-state blood concentrations. At the highest dose of 700 mg, which may represent a reasonable pill burden limit for patients, the steady-state peak plasma concentrations of silybin A and silybin B achieved were still 1 to 2 orders of magnitude below those concentrations associated with antiviral effects in vitro, and trough concentrations were approximately 25-fold lower than peak concentrations. These steady-state concentrations provide an important benchmark for any clinical investigation evaluating the HCV antiviral activity or the disease-modifying effects of oral28 or intravenous25 formulations of silymarin flavonolignans as single therapy or as an adjuvant to the standard of care. For example, conflicting results from early clinical trials evaluating the use of silymarin flavonolignans in combination with peginterferon and ribavirin on sustained virologic response rates in HCV patients may reflect unrecognized and variable pharmacokinetic interactions influencing steady-state concentrations of the active flavonolignans.25,38
In this study, nonlinear increases in plasma concentration were observed for silybin B, which increased more rapidly than silybin A as the dose of silymarin was increased. In addition, at the highest dose of 700 mg, silybin A only represented 54% of the flavonolignans detected in plasma due to the appearance of other flavonolignans, especially silychristin. In contrast, the relative proportions of the total concentration for each flavonolignan remained constant as the dose increased with the total concentration of silybin B, accounting for the greatest proportion at 38%. Differences in the extent in which flavonolignans are conjugated and undergo biliary excretion presumably account for some of these observations. As discussed above, the amounts of silybin A (23.2 mg), silybin B (32.0 mg), and silychristin (24.9 mg) in each silymarin capsule are approximately similar. However, at the lower customary doses, the plasma concentrations of silybin A are 4- to 8-fold higher than those for silybin B, whereas concentrations of silychristin are not detectable. At higher doses, the saturation of conjugating enzymes may be responsible for the greater increases in the plasma concentrations of flavonolignans, which appear to be more extensively conjugated, such as silybin B39 and silychristin, compared to silybin A. In a recent study using isolated perfused rat livers, the percentage of the flavonolignan dose excreted into bile as conjugates varied for each flavonolignan and was least for silybin A and greatest for silychristin and silydianin.19 Although silybin B and silychristin exhibited nonlinear pharmacokinetics, nonlinear pharmacokinetics was not observed for the sum of all parent flavonolignans possibly because silybin A, which accounts for the majority of parent flavonolignans detected in plasma, would be less influenced by the saturation of conjugation pathways. However, the results from the current study may not be generalizable to other liver disease patient populations because silymarin’s pharmacokinetics appears to vary according to type and grade/stage of liver disease.26
In addition to the saturation of conjugating enzymes, the nonlinear pharmacokinetics for some parent flavonolignans may reflect extensive enterohepatic cycling, which can result in their delayed elimination because conjugates can be cleaved in the gut and returned to systemic circulation as unchanged parent. The pharmacokinetics for the silymarin formulation used in this study is consistent with that reported by other laboratories,40 and the secondary peaks observed in the plasma concentration versus time profile at 12 to 24 hours postdose appear well past the normal time window associated with gastrointestinal (GI) emptying, dissolution time, and small intestinal transit for this conventional capsule preparation. Therefore, enterohepatic cycling rather than formulation problems or delayed GI emptying is the most likely source for the observed secondary peaks. Enterohepatic cycling was most apparent for the total concentrations of silybin A and silychristin, as evidenced by a second elimination phase that appeared to plateau around 18 hours and by secondary peaks that were observed at a recycling time of about every 6 hours. Therefore, nonlinear pharmacokinetics most likely reflects saturation of flavonolignan conjugation as well as the enterohepatic cycling of flavonolignan conjugates.
Although the small number of participants in each dose cohort represents a potential limitation to the study findings, these data suggest that higher than customary oral doses of silymarin up to 2.1 g/d appear safe and well tolerated. However, the steady-state plasma concentrations of parent flavonolignans achieved in this study were an order of magnitude too low to cause reductions in serum transaminases or HCV titers after 7 days of chronic dosing. In contrast, the total flavonolignan concentration in plasma, which primarily reflects flavonolignan conjugates, ranged from a trough of 3.9 μg/mL to a peak of 10.6 μg/mL at a dose of 700 mg and approached the range of silymarin concentrations shown to inhibit viral replication in vitro.23 The absence of effects on HCV RNA titers after 7 days of treatment suggests that flavonolignan conjugates may not possess antiviral activity. Potent antiviral effects have been observed in patients treated with a similar dose (10–20 mg/kg) and duration (7 days) with an intravenous formulation of the sodium succinate salt of silibinin (silybin A + silybin B).25 Therefore, oral doses of silymarin beyond 2.1 g/d may be necessary to achieve target plasma concentrations of parent flavonolignans for antiviral effects, although silymarin’s high first-pass metabolism represents a significant hurdle for developing any oral dose regimen unless approaches for slowing the rate of elimination can be developed. Recent in vitro investigations on the mechanisms of silymarin’s anti-inflammatory, antiviral, or immunomodulatory effects have used cell culture systems that lack the capacity of liver cells to metabolize and excrete silymarin.23,24,41 Because such in vitro studies routinely employ silymarin concentrations 1 to 2 orders of magnitude higher than those achieved in this study, the concentrations of unconjugated parent flavonolignans associated with pharmacologic effects in these culture systems are most likely several orders of magnitude greater than those achieved in vivo. However, portal blood concentrations reaching the liver after an oral dose may be several orders of magnitude higher than peripheral blood concentrations due to the rapid conjugation and biliary excretion of silymarin flavonolignans by hepatocytes, which limits peripheral blood concentrations. Enterohepatic cycling of silymarin conjugates would contribute to both free and conjugate concentrations reaching the liver by portal blood even hours after an oral dose. Therefore, potential immunomodulatory and anti-inflammatory effects of silymarin might be localized to the nonparenchymal cell types that reside in the liver (ie, Kupffer cells, sinusoidal endothelial cells, stellate cells, infiltrated lymphocytes), which may represent the cellular targets for any silymarin-based regimen. The relationship between steady-state plasma concentrations and the intrahepatic concentrations of flavonolignans is not known. Therefore, it is possible that a more prolonged treatment period with doses up to 2.1 g/d may result in HCV antiviral activity or effects on disease progression, which are being evaluated in the ongoing SyNCH phase II trials.
Acknowledgments
The authors thank the patients who volunteered for this trial, as well as Joseph Colagreco, Mary Hammond, and Deborah Moretti, who served as the study coordinators, and Sharon Lawlor, who was the DCC coordinator, for their invaluable assistance in the conduct of this trial. The authors also thank Roche Molecular Diagnostics for providing the HCV RNA testing and Craig W. Hendrix, MD, who graciously agreed to serve as the independent safety monitor.
This research was supported from the following sources: National Institutes of Health Grants jointly funded by NCCAM and NIDDK; UO1 AT003571-01 (Beth Israel Deaconess Medical Center), UO1 AT003560-01 (University of North Carolina at Chapel Hill), UO1 AT003573-01 (University of Pennsylvania), UO1 AT003566-01 (University of Pittsburgh), and UO1 AT003574-01 (Thomas Jefferson University); and RR00046 from the General Clinical Research Centers program of the Division of Research Resources. In addition, Rottapharm|Madaus, Italy, provided silymarin and placebo and partly funded the trial.
Footnotes
Financial disclosure: This was an investigator-initiated trial, and Rottapharm|Madaus had no direct or indirect involvement in the design of the trial, data collection, or preparation or submission of the manuscript for this registered (http://clinicaltrials.gov/ct2/show/NCT00389376) investigator-initiated trial. None of the authors has a personal conflict of interest with the manufacturer of any of the marketed silymarin formulations.
No conflicts of interest exist.
References
- 1.Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 2.Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 3.Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med. 2006;355:2444–2451. doi: 10.1056/NEJMct061675. [DOI] [PubMed] [Google Scholar]
- 4.Cornberg M, Wedemeyer H, Manns MP. Treatment of chronic hepatitis C with PEGylated interferon and ribavirin. Curr Gastroenterol Rep. 2002;4:23–30. doi: 10.1007/s11894-002-0034-y. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990–1997: results of a follow-up national survey. JAMA. 1998;280:1569–1575. doi: 10.1001/jama.280.18.1569. [DOI] [PubMed] [Google Scholar]
- 6.Strader DB, Bacon BR, Lindsay KL, et al. Use of complementary and alternative medicine in patients with liver disease. Am J Gastroenterol. 2002;97:2391–2397. doi: 10.1111/j.1572-0241.2002.05993.x. [DOI] [PubMed] [Google Scholar]
- 7.Flora K, Hahn M, Rosen H, Benner K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am J Gastroenterol. 1998;93:139–143. doi: 10.1111/j.1572-0241.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- 8.Seeff LB, Curto TM, Szabo G, et al. HALT-C Trial Group. Herbal product use by persons enrolled in the hepatitis C Antiviral Long-Term Treatment Against Cirrhosis (HALT-C) trial. Hepatology. 2008;47:605–612. doi: 10.1002/hep.22044. [DOI] [PubMed] [Google Scholar]
- 9.Kim NC, Graf TN, Sparacino CM, Wani MC, Wall ME. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum) Org Biomol Chem. 2003;1:1684–1689. doi: 10.1039/b300099k. [DOI] [PubMed] [Google Scholar]
- 10.Psotova J, Chlopcikova S, Grambal F, Simánek V, Ulrichová J. Influence of silymarin and its flavonolignans on doxorubicin-iron induced lipid peroxidation in rat heart microsomes and mitochondria in comparison with quercetin. Phytother Res. 2002;16(suppl 1):S63–S67. doi: 10.1002/ptr.811. [DOI] [PubMed] [Google Scholar]
- 11.Kren V, Ulrichova J, Kosina P, et al. Chemoenzymatic preparation of silybin beta-glucuronides and their biological evaluation. Drug Metab Dispos. 2000;28:1513–1517. [PubMed] [Google Scholar]
- 12.Manna SK, Mukhopadhyay A, Van NT, Aggarwall BB. Silymarin suppresses TNF-induced activation of NF-kappa B, c-Jun N-terminal kinase, and apoptosis. J Immunol. 1999;163:6800–6809. [PubMed] [Google Scholar]
- 13.Schumann J, Prockl J, Kiemer AK, Vollmar AM, Bang R, Tiegs G. Silibinin protects mice from T cell-dependent liver injury. J Hepatol. 2003;39:333–340. doi: 10.1016/s0168-8278(03)00239-3. [DOI] [PubMed] [Google Scholar]
- 14.Tsai JH, Liu JY, Wu TT, et al. Effects of silymarin on the resolution of liver fibrosis induced by carbon tetrachloride in rats. J Viral Hepatol. 2008;15:508–514. doi: 10.1111/j.1365-2893.2008.00971.x. [DOI] [PubMed] [Google Scholar]
- 15.Jia JD, Bauer M, Cho JJ, et al. Antifibrotic effect of silymarin in rat secondary biliary fibrosis is mediated by downregulation of procollagen alpha1(I) and TIMP-1. J Hepatol. 2001;35:392–398. doi: 10.1016/s0168-8278(01)00148-9. [DOI] [PubMed] [Google Scholar]
- 16.Mayer KE, Myers RP, Lee SS. Silymarin treatment of viral hepatitis: a systematic review. J Viral Hepatol. 2005;12:559–567. doi: 10.1111/j.1365-2893.2005.00636.x. [DOI] [PubMed] [Google Scholar]
- 17.Wen Z, Dumas TE, Schrieber SJ, Hawke RL, Fried MW, Smith PC. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab Dispos. 2008;36:65–72. doi: 10.1124/dmd.107.017566. [DOI] [PubMed] [Google Scholar]
- 18.Schandalik R, Gatti G, Perucca E. Pharmacokinetics of silybin in bile following administration of silipide and silymarin in cholecystectomy patients. Arzneimittelforschung. 1992;42:964–968. [PubMed] [Google Scholar]
- 19.Miranda SR, Lee JK, Brouwer KL, Wen Z, Smith PC, Hawke RL. Hepatic metabolism and biliary excretion of silymarin flavonolignans in isolated perfused rat livers: role of multidrug resistance-associated protein 2 (abcc2) Drug Metab Dispos. 2008;36:2219–2226. doi: 10.1124/dmd.108.021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morazzoni P, Magistretti MJ, Giachetti C, Zanolo G. Comparative bioavailability of silipide, a new flavonolignan complex, in rats. Eur J Drug Metab Pharmacokinet. 1992;17:39–44. doi: 10.1007/BF03189986. [DOI] [PubMed] [Google Scholar]
- 21.Morazzoni P, Montalbetti A, Malandrino S, Pifferi G. Comparative pharmacokinetics of silipide and silymarin in rats. Eur J Drug Metab Pharmacokinet. 1993;18:289–297. doi: 10.1007/BF03188811. [DOI] [PubMed] [Google Scholar]
- 22.Gordon A, Hobbs DA, Bowden DS, et al. Effects of Silybum marianum on serum hepatitis C virus RNA, alanine aminotransferase levels and well-being in patients with chronic hepatitis C. J Gastroenterol Hepatol. 2006;21(pt 2):275–280. doi: 10.1111/j.1440-1746.2006.04138.x. [DOI] [PubMed] [Google Scholar]
- 23.Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DY. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappaB signaling, and HCV infection by standardized silymarin. Gastroenterology. 2007;132:1925–1936. doi: 10.1053/j.gastro.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 24.Bonifaz V, Shan Y, Lambrecht R, Donohue SE, Moschenross D, Bonkovsky HL. Effects of silymarin on hepatitis C virus and haem oxygenase-1 gene expression in human hepatoma cells. Liver Int. 2009;29:366–373. doi: 10.1111/j.1478-3231.2008.01833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferenci P, Scherzer TM, Kerschner H, et al. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology. 2008;135:1561–1567. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 26.Schrieber SJ, Wen Z, Vourvahis M, et al. The pharmacokinetics of silymarin is altered in patients with hepatitis C virus and nonalcoholic fatty liver disease and correlates with plasma caspase-3/7 activity. Drug Metab Dispos. 2008;36:1909–1916. doi: 10.1124/dmd.107.019604. [DOI] [PubMed] [Google Scholar]
- 27.Flaig TW, Gustafson DL, Su LJ, et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007;25:139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]
- 28.Lang I. Phase I/II clinical trial to explore silymarin in chronic live diseases. Gastroenterology. 2006;131:990. [Google Scholar]
- 29.Salmi HA, Sarna S. Effect of silymarin on chemical, functional, and morphological alterations of the liver: a double-blind controlled study. Scand J Gastroenterol. 1982;17:517–521. doi: 10.3109/00365528209182242. [DOI] [PubMed] [Google Scholar]
- 30.Lang I, Nekam K, Deak G, et al. Immunomodulatory and hepatoprotective effects of in vivo treatment with free radical scavengers. Ital J Gastroenterol. 1990;22:283–287. [PubMed] [Google Scholar]
- 31.Feher J, Deak G, Muzes G, et al. Liver-protective action of silymarin therapy in chronic alcoholic liver diseases. Orv Hetil. 1989;130:2723–2727. [PubMed] [Google Scholar]
- 32.Pares A, Planas R, Torres M, et al. Effects of silymarin in alcoholic patients with cirrhosis of the liver: results of a controlled, double-blind, randomized and multicenter trial. J Hepatol. 1998;28:615–621. doi: 10.1016/s0168-8278(98)80285-7. [DOI] [PubMed] [Google Scholar]
- 33.Tanasescu C, Petrea S, Baldescu R, Macarie E, Chiriloiu C, Purice S. Use of the Romanian product Silimarina in the treatment of chronic liver diseases. Med Interne. 1988;26:311–322. [PubMed] [Google Scholar]
- 34.Tanamly MD, Tadros F, Labeeb S, et al. Randomised double-blinded trial evaluating silymarin for chronic hepatitis C in an Egyptian village: study description and 12-month results. Dig Liver Dis. 2004;36:752–759. doi: 10.1016/j.dld.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 35.Ferenci P, Dragosics B, Dittrich H, et al. Randomized controlled trial of silymarin treatment in patients with cirrhosis of the liver. J Hepatol. 1989;9:105–113. doi: 10.1016/0168-8278(89)90083-4. [DOI] [PubMed] [Google Scholar]
- 36.Lucena MI, Andrade RJ, de la Cruz JP, Rodriguez-Mendizabal M, Blanco E, Sánchez de la Cuesta F. Effects of silymarin MZ-80 on oxidative stress in patients with alcoholic cirrhosis: results of a randomized, double-blind, placebo-controlled clinical study. Int J Clin Pharmacol Ther. 2002;40:2–8. doi: 10.5414/cpp40002. [DOI] [PubMed] [Google Scholar]
- 37.Soule TA, Schrieber SJ, Wen Z, et al. Single dose escalation phase I study to evaluate the pharmacokinetics (PK) of silymarin (Legalon®) and the effect of food in patients with chronic hepatitis C (HCV) Clin Pharmacol Expt Ther. 2008;83(suppl 1s):PIII–18. [Google Scholar]
- 38.Par A, Roth E, Miseta A, et al. Effects of silymarin supplementation in chronic hepatitis C patients treated with peg-interferon + ribavirin: a placebo-controlled double blind study. Orv Hetil. 2009;150(2):73–79. doi: 10.1556/OH.2009.28517. [DOI] [PubMed] [Google Scholar]
- 39.Han YH, Lou HX, Ren DM, Sun LR, Ma B, Ji M. Stereoselective metabolism of silybin diastereoisomers in the glucuronidation process. J Pharm Biomed Anal. 2004;34:1071–1078. doi: 10.1016/j.jpba.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Kim YC, Kim EJ, Lee ED, et al. Comparative bioavailability of silibinin in healthy male volunteers. Int J Clin Pharmacol Ther. 2003;41:593–596. doi: 10.5414/cpp41593. [DOI] [PubMed] [Google Scholar]
- 41.Gharagozloo M, Khoshdel Z, Amirghofran Z. The effect of an iron (III) chelator, silybin, on the proliferation and cell cycle of Jurkat cells: a comparison with desferrioxamine. Eur J Pharmacol. 2008;589(1–3):1–7. doi: 10.1016/j.ejphar.2008.03.059. [DOI] [PubMed] [Google Scholar]