

Abstract
A novel fluorine-18 prosthetic ligand, 5-(1,3-dioxolan-2-yl)-2-(2-(2-(2- fluoroethoxy)ethoxy)ethoxy)pyridine [18F]2, has been synthesized. The prosthetic ligand is formed in high radiochemical yield (rcy = 71 ± 2 %, n = 3) with excellent radiochemical purity (rcp = 99 ± 1 %, n = 3) in a short reaction time (10 min). [18F]2 is a small, neutral, organic complex, easily synthesized in four steps from a readily available starting material. It can be anchored onto a target molecule containing an aminooxy functional group under acidic conditions by way of an oxime bond. We report herein two examples [18F]23 and [18F]24, potential imaging agents for β-amyloid plaques, which were labeled with this prosthetic group. This approach could be used for labeling proteins and peptides containing an aminooxy group. Biodistribution in male ICR mice for both oxime labeled complexes [18F]23 and [18F]24 were compared to that of the known β-amyloid plaque indicator, [18F]-AV-45, florbetapir 1. Oximes [18F]23 and [18F]24 are larger in size and therefore should reduce the blood-brain barrier (BBB) penetration. The brain uptake for oxime [18F]23 appeared to be reduced, but still retained some capability to cross the BBB. Oxime [18F]24 showed promising results after 2 min post injection (0.48 % dose/gram), however the uptake increased after 30 min post injection (0.92 % dose/gram) suggesting an in-vivo decomposition/metabolism of compound [18F]24. We have demonstrated a general protocol for the fluoride-18 labeling with a new prosthetic ligand [18F]2 that is tolerant towards several functional groups and is formed via chemoselective oxime coupling.
Keywords: fluorine-18, prosthetic ligand, aldehyde, aminooxy, oxime, chemoselective
Introduction
The use of molecular imaging agents play a significant role in the advancement of medical procedures and is an important part of ongoing research (1). In the forefront of molecular imaging is the use of positron emission tomography (PET), which relies on a biomarker that is labeled with short-lived positron emitting radionuclides. These PET imaging agents produce ‘hot’ spots, where a large abundance of radioactivity congregates, due to the radioactive molecular probes. Fluorine-18 is one isotope that is widely used for PET imaging due to its unique physical and nuclear characteristics (ease of formation, half-life of 109.7 min, high resolution, and relative minor radiation dose to patients) (2).
Two methods are commonly used to incorporate fluorine-18 into a molecule, either by an electrophilic substitution or through nucleophilic displacement (1–5). Electrophilic fluorination reactions are typically used in the carrier added (ca) form of [18F]F+ or [18F]F2, but have the drawbacks of low specific activity (theoretical achievable maximum radiochemical yield is limited to 50 %) (6) and difficulty in controlled labeling (1). An alternative option is the use of [18F]F− for nucleophilic substitution. High specific activity (theoretically 1.71 × 103 Ci/μmol) (3, 4) is one advantage nucleophilic displacement posses over electrophilic substitution, which is achieved when [18F]F− is produced by the proton irradiation of enriched [18O] water via the reaction 18O(p, n)18F. Typical SN2 reactions dominate this avenue of chemistry. Due to the fact that the fluoride anion is highly electronegative and it is produced in an aqueous media, it is strongly hydrated and hence is not sufficiently reactive for nucleophilic substitution reactions. The fluoride anion therefore needs to be activated in the form of a ‘naked’ anion for nucleophilic substitution, usually with “crown” ether such as K[222]. The K[222] contains two highly basic tertiary nitrogen atoms. The strong basic conditions are not suitable for compounds containing acidic protons, and thus may lead to their decomposition. To circumvent this problem, pre-existing fluorine-18 labeled molecules, known as prosthetic ligands, or biofunctional-labeling agents, are used to indirectly attach the fluorine-18 radionuclide onto a desired ligand, providing the fluorine-18 radiolabeled molecule (1, 5).
Several prosthetic ligands have been used with compounds that otherwise could not be labeled via a direct fluoride-18 nucleophilic substitution. The incorporation of these groups onto a target molecule can be achieved by methods that include the formation of an acyl (7–12), sulfide (13–16), alkyl (17–20), hydrozone (21) or oxime (9, 22–31) bond formation, or with the use of photochemistry (32) and click chemistry (33–36). Most of these prosthetic ligands are not ideal for routine applications because they require time consuming and labor intense procedures with tedious purification processes. In general, prosthetic ligands may also be restricted to the type of substrate used when coupled with the target ligand due to interfering nucleophilic groups. This may lead to cross-linked labeled products (37). Therefore, site-specific linkage of a prosthetic ligand is necessary when attaching it to a target molecule.
The oxime bond formation is highly chemoselective (38, 39) and takes place under acidic conditions between aminooxy groups and aldehydes or ketones. Since the reaction is chemoselective, the use of protecting groups are unnecessary and the oxime coupling reaction is functional group tolerant.
The approach in designing a new prosthetic ligand for labeling compounds that pose a problem to direct fluoride-18 nucleophilic substitution, such as biologically active peptides, will need to follow four criteria. First, the prosthetic group is labeled with a [18F]fluoride anion via SN2 displacement, forming a stable C-F bond on an sp3 hybridized carbon atom. The prosthetic ligand containing an end-capped pegylated fluorine atom is similar to [18F]-AV-45 (1) (40, 41), which has been well tested and prepared at multi-curie levels; therefore we expect the fluorination reaction will proceed smoothly.
[18F]-AV-45, (E)-4-(2-(6-(2-(2-(2-18F-fluoroethoxy)ethoxy)ethoxy)pyridin-3-yl)vinyl)-N-methyl benzenamine, florbetapir (1) (Figure 1), is a new imaging agent targeting β-amyloid plaques in the brain (40, 41). This agent is currently under a phase III clinical study. It is a small, neutral, and moderately lipophilic agent (measured log D = 1.70, calculated log P = 2.85). We reasoned that using compounds with similar structural features as 1 were good candidates to test the coupling reaction of the prosthetic group, validating the chemistry prior to its application to more complicated compounds, such as peptides and proteins.
Figure 1.
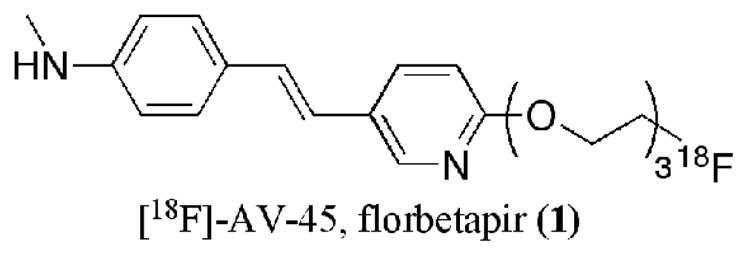
Structure of [18F]-AV-45, florbetapir (1).
A second criterion for the prosthetic ligand is the compound to be a neutral molecule, soluble in organic media, to aid in the purification process (solid phase extraction). Third, the site at which the prosthetic group attaches to the target molecule (i.e. via an aldehyde) would need to form chemoselectively an oxime bond with an aminooxy functional group. Finally, it would be ideal for the coupling between both the prosthetic ligand and the aminooxy compound to occur quickly, in an environment suitable to tolerate many different functional groups. The use of an oxime coupling reaction with aldehydes, for the attachment of prosthetic groups, has been successfully reported (22–27, 29–31, 42).
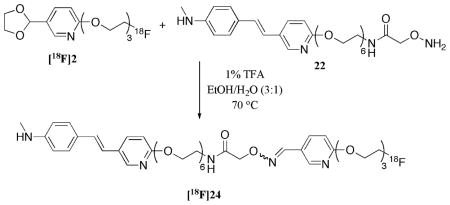
Herein we report the synthesis and characterization of a novel radiolabeled ligand, [18F]2, which meet all four criteria listed above as a suitable prosthetic group (Figure 2). To test and optimize the oxime coupling reaction, we synthesized two aminooxy compounds, 21 and 22, and used them as model complexes for the labeling with our prosthetic ligand, [18F]2. Biodistribution studies were performed for these two radiolabeled new oximes.
Figure 2.

Structure of the prosthetic group [18F]2 and its transformation to fluorine-18 labeled oxime.
Experimental Procedures
General information
All reagents used were commercial products purchased from Sigma- Aldrich and were used without further purification unless otherwise indicated. Fluoride-18 (nca) was purchased from IBA Molecular North America, Inc. All anhydrous reactions were run under positive pressure of argon, ultra high purity. All syringes and reaction flasks required for anhydrous reactions were flame dried and then cooled under argon atmosphere. All anhydrous solvents were dried by passage through an alumina column using Innovation Technology, Inc. system. Flash chromatography was carried out with silica gel 60 (230–400 mesh, Sigma-Aldrich). Analytical thin-layer chromatography (TLC) was performed on pre-coated aluminum backed plates (silica gel 60 F254). 1H and 13C NMR spectra were recorded on 200 MHz/50 MHz (Bruker DPX spectrometer), in deuterated chloroform (CDCl3) with residual chloroform protons (δ 7.26 ppm for 1H NMR and δ 77.23 ppm for 13C NMR) as the internal reference. Chemical shifts are reported in parts per million (ppm). Coupling constants are reported in hertz (Hz). The multiplicity is defined by s (singlet), d (doublet), t (triplet), dd (doublet of doublets), ddd (doublet of doublets of doublets), br (broad), and m (multiplet). High-Resolution Mass Spectrometry experiments were performed at University of Pennsylvania, Department of Radiology, Radiopharmaceutical Chemistry Section, using an Agilent Technologies LC/MSD TOF Mass Spectrometer.
2-Bromo-5-(1,3-dioxolan-2-yl)pyridine (6)
Ethylene glycol (0.54 mL, 601 mg, 9.7 mmol) was added to a suspension of 6-bromo-3-pyridine carbaldehyde (5) (1.0 g, 5.4 mmol) and p-toluene sulfonic acid, monohydride (102 mg, 0.54 mmol) in 27 mL of benzene. The flask was then fitted with a reflux condenser and placed in an oil bath set to 92° C. The reaction mixture was refluxed for 19 h, upon which time it was removed from the oil bath and allowed to cool to room temperature. The reaction was quenched with dilute K2CO3 (aq) solution to adjust the pH ~ 11. The reaction media was then partitioned between ethyl acetate and H2O. The organic phase was separated from the aqueous phase with the use of ethyl acetate (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, filtered, and concentrated to afford 1.21 g (98 %) of acetal 6, a yellow oil. 1H NMR was consistent with the reported literature (43).
2-(2-(2-(5-(1,3-dioxolan-2-yl)pyridin-2-yloxy)ethoxy)ethoxy)ethanol (7)
Acetal 6 (302 mg, 1.31 mmol) was dissolved in 13 mL of anhydrous N,N-dimethyl formamide and added to a microwave reaction vessel containing Cs2CO3 (1.28 g, 3.93 mmol) and triethylene glycol (0.43 mL, 475 mg, 3.16 mmol). The reaction vessel was sealed and placed in a microwave reactor (Biotage Initiator) set at 150 °C for 50 min. The reaction was allowed to cool to room temperature and then vented with a needle prior to breaking the seal. The reaction mixture was partitioned between dichloromethane and H2O. The organic phase was extracted from the aqueous phase with the use of dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (25 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (100 % ethyl acetate) afforded 240 mg (61 %) of alcohol 7, a viscous pale yellow oil. Rf = 0.16 (100 % ethyl acetate); 1H NMR (200 MHz, CDCl3): δ 8.20 (d, J = 2.3 Hz, 1H), 7.68 (dd, J = 8.6, 2.4 Hz, 1H), 6.80 (d, J = 8.6 Hz, 1H), 5.60 (s, 1H), 4.52–4.47 (m, 2H), 4.16–3.97 (m, 4H), 3.87–3.82 (m, 2H), 3.71–3.58 (m, 8H), 2.49 (br, s, 1H, OH); 13C NMR (50 MHz, CDCl3): δ 164.7, 146.0, 137.5, 126.9, 111.6, 102.5, 72.7, 70.9, 70.6, 69.9, 65.5, 65.4, 62.0; HRMS (ESI) m/z calcd for C14H22NO6 [M+H]+ 300.1447, found 300.1440.
2-(2-(2-(5-(1,3-dioxolan-2-yl)pyridin-2-yloxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (8)
Alcohol 7 (50 mg, 0.17 mmol) was added to a flame dried round bottom flask and dissolved in 1 mL anhydrous dichloromethane. Triethyl amine (30 μL, 25 mg, 0.25 mmol) was added and the reaction mixture was cooled to 0 °C. p-Toluenesulfonyl chloride (35 mg, 0.18 mmol) followed by N,N-4- dimethylaminopyridine (4.0 mg, 0.03 mmol) were quickly added and the reaction was placed in an ice/water bath at 0 °C. The reaction was allowed to warm to room temperature over the course of 5 h at which it was partitioned between ethyl acetate and H2O. The organic layer was then extracted from the aqueous layer with the use of ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (15 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (45–70 % ethyl acetate/hexanes) provided 62 mg (82 %) of tosylate 8, a viscous cloudy oil. Rf = 0.27 (60 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 8.20 (d, J = 2.4 Hz, 1H). 7.80 (d, J = 8.2 Hz, 2H), 7.68 (dd, J = 8.6, 2.4 Hz, 1H), 7.33 (dd, J = 8.6, 0.6 Hz, 2H), 6.78 (d, J = 8.6 Hz, 1H), 5.77 (s, 1H), 4.49–4.44 (m, 2H), 4.18–4.09 (m, 4H), 4.06–3.98 (m, 2H), 3.82–3.78 (m, 2H), 3.71–3.60 (m, 6H), 2.43 (s, 3H); 13C NMR (50 MHz, CDCl3): δ 168.5, 164.6, 146.0, 137.4, 133.4, 130.1, 128.2, 126.8, 111.5, 102.4, 73.1, 71.0, 70.8, 69.9, 69.4, 68.9, 65.5, 65.4, 21.8; HRMS (ESI) m/z calcd for C21H28NO8S [M+H]+ 454.1536, found 454.1524.
5-(1,3-dioxolan-2-yl)-2-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)pyridine (2)
Tosylate 8 (53 mg, 0.117 mmol) was dissolved in 2 mL of tetrahydrofuran and added to a 10 mL round bottomed flask containing tetra-n-butylammonium fluoride hydrate (80 mg, 0.30 mmol). The color of the solution changes instantly upon addition from clear solution to a black solution. The 10 mL round bottomed flask was then fitted with a reflux condenser and set in an oil bath at 76 °C. After 15 h, the reaction was removed from the heat, allowed to cool to room temperature, diluted with 15 mL ethyl acetate and concentrated. Purification by flash chromatography (45–60 % ethyl acetate/hexanes) provided 20 mg (57 %) of a clear oil, compound 2. Rf = 0.38 (60 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 8.20 (d, J = 2.3 Hz, 1H), 7.68 (dd, J = 8.6, 2.4 Hz, 1H), 6.79 (d, J = 8.9 Hz, 1H), 5.77 (s, 1H), 4.70–4.65 (m, 1H), 4.52–4.42 (m, 3H), 4.17–3.98 (m, 4H), 3.88–3.80 (m, 3H), 3.74–3.65 (m, 5H); 13C NMR (50 MHz, CDCl3): δ 164.7, 145.9, 137.3, 126.9, 111.5, 102.4, 85.1, 81.8, 71.1, 71.0, 70.9, 70.5, 70.0, 65.5; HRMS (ESI) m/z calcd for C14H11FNO5 [M+H]+ 302.1404, found 302.1398; HPLC conditions: Gemini 5μ C18 110A column, 250 × 4.60 mm, 5 micron; rt = 4.79 min (280 nm) in acetonitrile/10 mM ammonium formate buffer 60/40 with a flow rate of 1.0 mL/min.
6-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)nicotinaldehyde (9)
Pyridinium p-toluenesulfonate (38.0 mg, 0.150 mmol) was added to a solution of acetal 7 (150 mg, 0.50 mmol) in 5 mL acetone and 9 μL H2O. The flask was then fitted with a reflux condenser and placed in an oil bath set to 70 °C. After 3 h, the reaction was removed from the oil bath, allowed to cool to room temperature and concentrated. The reaction mixture was then partitioned between dichloromethane and NaHCO3 (aq, sat). The organic layer was separated from the aqueous layer with the use of 5 % methanol/dichloromethane (5 × 10 mL). The combined organic layers were then washed with brine (20 mL), dried over MgSO4, filtered, and concentrated to provide 114 mg (89 %) of a thick pale yellow oil, aldehyde 9. Rf = 0.08 (100 % ethyl acetate); 1H NMR (200 MHz, CDCl3): δ 9.96 (d, J = 0.6 Hz, 1H), 8.62–8.61 (m, 1H), 8.08 (dd, J = 8.7, 2.4 Hz, 1H), 6.91 (ddd, J = 8.6, 0.7, 0.6 Hz, 1H), 4.63–4.58 (m, 2H), 3.92–3.87 (m, 2H), 3.76–3.68 (m, 6H), 3.65–3.60 (m, 2H), 2.34 (br, s, 1H, OH); 13C NMR (50 MHz, CDCl3): δ 189.6, 167.3, 152.7, 137.7, 126.9, 112.5, 72.6, 70.8, 70.5, 69.5, 66.2, 61.8; HRMS (ESI) m/z calcd for C12H17NO5Na [M+Na]+ 278.1004, found 278.1005.
2-(2-(2-(5-formylpyridin-2-yloxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (10)
Alcohol 9 (52 mg, 0.20 mmol) was added to a flame dried round bottom flask, dissolved in 1 mL of anhydrous dichloromethane and cooled to 0 °C. Anhydrous pyridine (50 μL, 48 mg, 0.61 mmol) was added to the reaction mixture at 0 °C. After allowing the reaction mixture to stir at 0 °C for 5 min, p-toluenesulfonyl chloride (58 mg, 0.31 mmol) was quickly added to the solution. After 10 min, the reaction mixture was removed from the ice/water bath and allowed to warm to room temperature overnight. After 19 h, the reaction mixture was diluted with ethyl acetate and partitioned between H2O. The organic phase was extracted from the aqueous phase with the use of ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated. Purification by flash chromatography (50–60 % ethyl acetate/hexanes) provided 70 mg (84 %) of tosylate 10, a thick milky oil. Rf = 0.32 (60 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 9.95 (d, J = 0.5 Hz, 1H), 8.60 (dd, J = 2.3, 0.5 Hz, 1H), 8.06 (dd, J = 8.7, 2.4 Hz, 1H), 7.82–7.76 (m, 2H), 7.35–7.31 (m, 2H), 6.87 (ddd, J = 8.7, 0.6, 0.6 Hz, 1H), 4.58–4.54 (m, 2H), 4.18–4.13 (m, 2H), 3.86–3.81 (m, 2H), 3.71–3.60 (m, 6H), 2.43 (s, 3H); 13C NMR (50 MHz, CDCl3): δ 189.7, 167.5, 152.8, 145.0, 137.9, 130.1, 128.4, 127.1, 112.6, 71.0, 70.9, 69.7, 69.4, 69.0, 66.3, 21.8; HRMS (ESI) m/z calcd for C19H24NO7S [M+H]+ 410.1273, found 410.1277.
6-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)nicotinaldehyde (3)
Tosylate 10 (40 mg, 0.10 mmol) was dissolved in tetrahydrofuran (1 mL) and added to a round bottom flask containing tetra-nbutylammonium fluoride hydrate (61 mg, 0.23 mmol). The flask was fitted with a reflux condenser and heated to 76 °C under an inert atmosphere of argon. After 18 h, the reaction was removed from the oil bath and let cool to room temperature. Note: most of the solvent had evaporated over the course of the reaction; the reflux condensers had a leak. The reaction media was then condensed and purified by flash chromatography (50 % ethyl acetate/hexanes) to afford 10 mg (40 %) of fluoride 3, a thin clear oil. Rf = 0.35 (60 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 9.95 (s, 1H), 8.60 (d, J = 2.3 Hz, 1H), 8.06 (dd, J = 8.9, 2.3 Hz, 1H), 6.88 (dd, J = 8.7, 0.5 Hz, 1H), 4.69–4.65 (m, 1H), 4.61–4.57 (m, 2H), 4.45–4.41 (m, 1H), 3.90–3.80 (m, 3H), 3.72–3.65 (m, 5H); 13C NMR (50 MHz, CDCl3): δ 189.7, 167.5, 152.9, 137.8, 127.1, 112.6, 85.1, 81.7, 71.1, 71.0, 70.9, 70.5, 69.8, 66.4; HRMS (ESI) m/z calcd for C12H17FNO4 [M+H]+ 258.1142, found 258.1142.
(E)-17-(5-(4-(tert-butoxycarbonyl(methyl)amino)styryl)pyridin-2-yloxy)-3,6,9,12,15- pentaoxaheptadecyl 4-methylbenzenesulfonate (12)
Sodium hydride (60 %, 22 mg, 0.54 mmol) was added to a flame dried round bottom flask. Anhydrous N,N-dimethyl formamide (0.5 mL) was added and the suspension was cooled to 0 °C. A solution of alcohol 11 (69 mg, 0.13 mmol), dissolved in 0.5 mL of anhydrous N,N-dimethyl formamide, was added to the reaction mixture at 0 °C. Diethylene glycol bis(p-toluenesulfonate) was then added to the reaction mixture at 0 °C, which was allowed to warm up to room temperature over the course of the night. After 24 h, the reaction was quenched with NaHCO3 (sat. aq.) (10 mL). The organic phase was separated from the aqueous phase with the use of ethyl acetate (4 × 10 mL). The combined organic layers were then washed with brine (1 × 20 mL), dried over MgSO4, filtered and concentrated. Purification by flash chromatography (2–4 % methanol/dichloromethane) afforded 73 mg (73 %) of a yellow oil, tosylate 12. Rf = 0.19 (5 % methanol/dichloromethane); 1H NMR (200 MHz, CDCl3): δ 8.16 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 8.4, 2H), 7.77 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.34–7.30 (m, 2H), 7.21 (d, J = 8.6 Hz, 2H), 6.95 (s, 2H), 6.77 (d, J = 8.8 Hz, 1H), 4.50–4.45 (m, 2H), 4.17–4.12 (m, 2H), 3.87–3.82 (m, 2H), 3.72–3.53 (m, 18 H), 3.26 (s, 3H), 2.42 (s, 3H), 1.45 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 163.3, 154.8, 145.8, 144.9, 143.4, 135.5, 134.3, 129.9, 128.1, 127.5, 126.9, 126.6, 125.7, 124.7, 111.6, 80.6, 70.9, 70.8, 70.7, 70.7, 69.9, 69.4, 68.8, 65.5, 37.3, 28.5, 21.8; HRMS (ESI) m/z calcd for C38H53N2O11S [M+H]+ 745.3370, found 745.3384.
(E)-tert-butyl 4-(2-(6-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)pyridin-3- yl)vinyl)phenyl(methyl)carbamate (14)
Tosylate 12 (380 mg, 0.62 mmol) and sodium azide (121 mg, 1.86 mmol) were added to a flame dried round bottom flask. Anhydrous N,N-dimethyl formamide (6 ml) was added to the reaction mixture and placed in an oil bath set to 60 °C. After 4 h, the reaction mixture was removed from the heat, allowed to cool to room temperature, and then partitioned between ethyl acetate and H2O. The organic phase was separated from the aqueous phase with the use of ethyl acetate (3 × 20 mL). The combined organic layers were then washed with brine (2 × 20 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (35–50 % ethyl acetate/hexanes) afforded 300 mg (quantitative) of a clear oil, azide 14. Rf = 0.48 (50 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 8.19 (d, J = 2.4 Hz, 1H), 7.79 (dd, J = 8.8, 2.6 Hz, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 6.97 (s, 2H), 6.80 (d, J = 8.6 Hz, 1H), 4.53–4.48 (m, 2H), 3.90–3.85 (m, 2H), 3.77–3.66 (m, 6H), 3.39 (t, J = 5.2 Hz, 2H), 3.27 (s, 3H), 1.46 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 163.4, 154.9, 145.9, 143.5, 135.6, 134.4, 127.5, 127.0, 126.7, 125.8, 124.8, 111.7, 80.6, 71.0, 70.3, 65.5, 50.9, 37.3, 28.6; HRMS (ESI) m/z calcd for C25H34N5O5 [M+H]+ 484.2650, found 484.2546.
(E)-tert-butyl 4-(2-(6-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)pyridin-3- yl)vinyl)phenyl(methyl)carbamate (16)
Triphenyl phosphine (271 mg, 1.03 mmol) was added to a solution of azide 14 (200 mg, 0.41 mmol) in 12.5 mL of tetrahydrofuran. After 1 h, water (1.5 mL) was added to the reaction mixture. The reaction mixture was heated to 60 °C for 18 h. The reaction was then removed from the heat and allowed to cool to room temperature and concentrated. Purification by flash chromatography (96:4:1 – 85:15:1 dichloromethane:methanol:ammonium hydroxide) provided 133 mg (70 %) amine 16, a white waxy material. Rf = 0.19 (8.5:1.5:0.1 dichloromethane:methanol:ammonium hydroxide); 1H NMR (200 MHz, CDCl3): δ 8.14 (d, J = 2.3 Hz, 1H), 7.74 (dd, J = 8.6, 2.4 Hz, 1H), 7.40 (d, J = 8.6 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 6.92 (s, 2H), 6.76 (d, J = 8.7 Hz, 1H), 4.48–4.43 (m, 2H), 3.85–3.80 (m, 2H), 3.71–3.58 (m, 4H), 3.49 (t, J = 5.2 Hz, 2H), 3.22 (s, 3H), 2.84 (t, J = 5.2 Hz, 2H), 2.30 (br, s, 2H, NH2), 1.42 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 163.2, 154.7, 145.7, 143.3, 135.5, 134.2, 127.4, 126.9, 126.5, 125.6, 124.6, 111.5, 80.5, 73.0, 70.7, 70.4, 69.8, 65.3, 41.7, 37.3, 28.4; HRMS (ESI) m/z calcd for C25H36N3O5 [M+H]+ 458.2655, found 458.2642.
Dicarbamate (19)
Amine 16 (132 mg, 0.29 mmol) and succinimide 18 (108 mg, 0.38 mmol) were dissolved in 6 mL of anhydrous N,N-dimethyl formamide and to this solution was added N,Ndiisopropylethylamine (1.0 mL, 744 mg, 5.76 mmol). The reaction was allowed to stir at room temperature. After 1 h, the reaction was concentrated (23 mbar at 60 °C, to facilitate the removal of N,Ndimethyl formamide) and purified by flash chromatography (2–6 % methanol/dichloromethane) to provide 81 mg (45 %) of dicarbamate 19, a clear gummy material. Rf = 0.29 (5 % methanol/dichloromethane); 1H NMR (200 MHz, CDCl3): δ 8.18 (d, J = 2.2 Hz, 1H), 7.98 (br, s, 1H, NH), 7.80 (dd, J = 8.7, 2.3 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.6 Hz, 2H), 6.96 (s, 2H), 6.80 (d, J = 8.7 Hz, 1H), 4.52–4.47 (m, 2H), 4.34 (s, 2H), 3.90–3.85 (m, 2H), 3.76–3.47 (m, 8H), 3.27 (s, 3H), 1.46 (s, 9H), 1.46 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 169.1, 163.2, 157.6, 154.8, 145.7, 143.3, 135.6, 134.3, 127.5, 126.9, 126.6, 125.6, 124.6, 111.6, 82.6, 80.6, 76.0, 70.7, 70.5, 69.8, 69.7, 65.4, 39.0, 37.3, 28.5, 28.3; HRMS (ESI) m/z calcd for C32H47N4O9 [M+H]+ 631.3343, found 631.3345.
(E)-2-(aminooxy)-N-(2-(2-(2-(5-(4-(methylamino)styryl)pyridin-2- yloxy)ethoxy)ethoxy)ethyl)acetamide (21)
Trifluoroacetic acid (3 mL) was added to a solution of carbamate 19 (394 mg, 0.63 mmol) in dichloromethane (3 mL). The reaction was allowed to stir at room temperature for 3 h after which time the reaction mixture was concentrated down from 1N HCl (2 × 3 mL) and placed under high vacuum for 30 min. Purification by flash chromatography (97:3:1 – 90:10:1 dichloromethane:methanol:ammonium hydroxide) provided 158 mg (59 %) of aminooxy 21 a yellow, waxy solid. Due to its instability, full characterization was achieved from the subsequent step of the formation of oxime 23; aminooxy 21 was determined by HRMS. Rf = 0.49 (9.0:1.0:0.1 dichloromethane:methanol:ammonium hydroxide); HRMS (ESI) m/z calcd for C22H31N4O5 [M+H]+ 431.2294, found 431.2300.
2-((E)-(6-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)pyridin-3-yl)methyleneaminooxy)-N-(2-(2-(2-(5- ((E)-4-(methylamino)styryl)pyridin-2-yloxy)ethoxy)ethoxy)ethyl)acetamide (23)
A solution of aldehyde 3 (7 mg, 0.03 mmol) dissolved in 1.4 mL of ethanol, 0.1 mL of H2O, and 20 μL of trifluoroacetic acid was added to hydroxylamine 21 (12 mg, 0.03 mmol). The reaction mixture was placed in an oil bath set to 70 °C. After 2 h, the reaction mixture was removed from the heat and allowed to cool to room temperature. The reaction was then quenched with Na2CO3 (aq) and partitioned between 10 % methanol/dichloromethane. The organic layer was separated from the aqueous layer with the use of 10 % methanol/dichloromethane (4 × 10 mL). The combined organic layers were washed with brine (15 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (97:3:0.5 – 93:7:0.5 dichloromethane:methanol:ammonium hydroxide) provided 9 mg (50 %) of oxime 23, a viscous yellow oil. Rf = 0.33 (9.5:0.5:0.1 dichloromethane:methanol:ammonium hydroxide); 1H NMR (200 MHz, CDCl3): δ 8.28 (s, 1H), 8.28–8.14 (m, 2H), 7.95 (ddd, J = 8.9, 8.7, 2.2 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 6.96–6.78 (m, 4H), 6.64 (d, J = 8.6 Hz, 2H), 4.67–4.62 (m, 3H), 4.49–4.39 (m, 6H), 3.87–3.79 (m, 5H), 3.69–3.58 (m, 12H), 3.50–3.46 (m, 2H), 2.83 (s, 3H); 13C NMR (50 MHz, CDCl3): δ 172.5, 166.6, 164.0, 151.5, 149.6, 148.5, 146.3, 146.0, 137.7, 137.0, 130.3, 129.8, 128.8, 127.5, 123.2, 120.6, 113.6, 112.8, 112.2, 85.9, 82.6, 74.1, 72.0, 71.9, 71.5, 70.9, 70.7, 67.0, 66.7, 40.3, 30.8; HRMS (ESI) m/z calcd for C34H45FN5O8 [M+H]+ 670.3252, found 670.3236; HPLC conditions: Gemini 5μ C18 110A column, 250 × 4.60 mm, 5 micron; rt = 6.83 min (280 nm) in acetonitrile/10 mM ammonium formate buffer 60/40 at a flow rate of 1.0 mL/min.
(E)-17-(5-(4(tert-butoxycarbonyl(methyl)amino)styryl)pyridin-2-yloxy)-3,6,9,12,15- pentaoxaheptadecyl 4-methylbenzenesulfonate (13)
Sodium hydride (60 %, 22 mg, 0.54 mmol) was dissolved in 0.5 mL of N,N-dimethyl formamide in a flame-dried round bottom flask. This solution was cooled to 0 °C. Alcohol 11 (69 mg, 0.13 mmol), dissolved in 0.5 mL of N,N-dimethyl formamide, was added to the reaction mixture at 0 °C. After 3 min of stirring at 0 °C, diethylene glycol bis(p-toluenesulfonate) (337 mg, 0.80 mmol) was added to the reaction. The ice/water bath was removed and the reaction was allowed to warm to room temperature. After 24 h, the reaction media was buffered with NaHCO3 (aq, sat) and partitioned between ethyl acetate. The organic layer was extracted from the aqueous layer with the use of ethyl acetate (4 × 20 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (2–3 % methanol/dichloromethane) provided 73 mg (73%) of tosylate 13, a yellow oil. Rf = 0.19 (5 % methanol/dichloromethane); 1H NMR (200 MHz, CDCl3): δ 8.16 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.34–7.30 (m, 2H), 7.21 (d, J = 8.6 Hz, 2H), 6.95 (s, 2H), 6.77 (d, J = 8.8 Hz, 1H), 4.50–4.45 (m, 2H), 4.17–4.12 (m, 2H), 3.87–3.82 (m, 2H), 3.72–3.53 (m, 18H), 3.26 (s, 3H), 2.42 (s, 3H), 1.45 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 163.3, 154.8, 145.8, 144.9, 143.4, 135.5, 134.3, 129.9, 128.1, 127.5, 126.9, 125.7, 126.9, 126.6, 125.7, 124.7, 111.6, 80.6, 70.9, 70.8, 70.7, 70.7, 69.9, 69.4, 68.8, 65.5, 37.3, 28.5, 21.8; HRMS (ESI) m/z calcd for C38H53N2O11S [M+H]+ 745.3370, found 745.3384.
(E)-tert-butyl 4-(2-(6-(17-azido-3,6,9,12,15-pentaoxaheptadecyloxy)pyridin-3- yl)vinyl)phenyl(methyl)carbamate (15)
Tosylate 13 (300 mg, 0.40 mmol) was added to a flame-dried flask containing a solution of sodium azide (79 mg, 1.21 mmol) in 4 mL of anhydrous N,N-dimethyl formamide. The reaction mixture was heated to 60 °C. After 3 h, the reaction mixture was removed from the heat and allowed to cool to room temperature. It was then partitioned between ethyl acetate and H2O. The organic phase was separated from the aqueous phase with the use of ethyl acetate (3 × 40 mL). The combined organic layers were washed with brine (2 × 20 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (80–85 % ethyl acetate/hexanes) gave 150 mg (60 %) of a viscous yellow oil, azide 15. Rf = 0.24 (85 % ethyl acetate/hexanes); 1H NMR (200 MHz, CDCl3): δ 8.18 (d, J = 2.2 Hz, 1H), 7.79 (dd, J = 9.0, 2.6 Hz, 1H), 7.45 (d, J = 8.9 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 6.97 (s, 1H), 6.79 (d, J = 8.6 Hz, 1H), 4.52–4.47 (m, 2H), 3.89–3.84 (m, 2H), 3.71–3.65 (m, 18H), 3.41–3.36 (m, 2H), 3.27 (s, 3H), 1.46 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 163.3, 154.8, 145.8, 143.3, 135.3, 134.3, 127.5, 126.9, 126.6, 125.7, 124.8, 111.6, 80.6, 70.8, 70.8, 70.2, 69.9, 65.5, 50.8, 37.3, 28.5; HRMS (ESI) m/z calcd for C31H45N5O8Na [M+Na]+ 638.3166, found 638.3157.
(E)-tert-butyl 4-(2-(6-(17-amino-3,6,9,12,15-pentaoxaheptadecyloxy)pyridin-3- yl)vinyl)phenyl(methyl)carbamate (17)
Triphenyl phosphine (51 mg, 0.20 mmol) was added to a solution of azide 15 (48 mg, 0.08 mmol) in 2.7 mL of tetrahydrofuran. After 1 h, water (0.3 mL) was added to the reaction and heated to 60 °C for 17 h. The reaction was then removed from the heat and allowed to cool to room temperature and concentrated. Purification by flash chromatography (95:5:1 – 85:15:1 dichloromethane:methanol:ammonium hydroxide) provided 37 mg (80 %) of amine 17, a white wax. Rf = 0.28 (8.5:1.5:0.1 dichloromethane:methanol:ammonium hydroxide); 1H NMR (200 MHz, CDCl3): δ 8.23 (d, J = 2.3 Hz, 1H), 7.99 (dd, J = 8.8, 2.5 Hz, 1H), 7.55 (d, J = 8.6 Hz, 2H), 7.25 (d, J = 8.6 Hz, 2H), 7.12 (s, 2H), 6.85 (d, J = 8.7 Hz, 1H), 4.46–4.44 (m, 2H), 3.89–3.84 (m, 2H), 3.71–3.53 (m, 20H), 3.26 (s, 3H), 2.86 (t, J = 5.4 Hz, 2H), 1.47 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 164.6, 156.6, 146.9, 144.4, 137.2, 136.5, 128.8, 128.8, 127.9, 127.2, 125.8, 112.3, 82.0, 72.3, 71.7, 71.6, 71.4, 71.3, 70.8, 66.8, 41.9, 38.0, 28.7; HRMS (ESI) m/z calcd for C31H48N3O8 [M+H]+ 590.3441, found 590.3442.
Dicarbamate (20)
Amine 17 (37 mg, 0.06 mmol) dissolved in 2 mL of anhydrous N,N-dimethyl formamide was added to succinimide 18 (24 mg, 0.08 mmol) in a flame-dried 10 ml round bottom flask. N,N-Diethyl iso-propyl amine (0.22 mL, 163 mg, 1.26 mmol) was then added to the reaction mixture and allowed to stir at room temperature. After 1 h, the reaction was concentrated (23 mbar at 60 °C, to facilitate the removal of N,N-dimethyl formamide) and purified by flash chromatography (2–4 % methanol/dichloromethane) to provide 29 mg (60 %) of dicarbamate (20), a clear, gummy compound. Rf = 0.31 (5 % methanol/dichloromethane); 1H NMR (200 MHz, CDCl3): δ 8.18–8.17 (m, 2H), 7.87 (br, s, 1H, NH), 7.79 (dd, J = 8.6, 2.5 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 6.96 (s, 2H), 6.78 (d, J = 8.6 Hz, 1H), 4.50–4.45 (m, 2H), 4.33 (s, 2H), 3.87–3.82 (m, 2H), 3.78–3.45 (m, 20H), 3.26 (s, 3H), 1.46 (s, 9H), 1.45 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 169.3, 163.3, 157.7, 155.0, 145.8, 143.5, 135.6, 134.4, 127.5, 127.0, 126.7, 125.8, 124.7, 111.6, 82.7, 80.7, 76.1, 70.8, 70.7, 70.5, 69.9, 69.8, 65.6, 53.3, 39.2, 37.5, 28.5, 28.4; HRMS (ESI) m/z calcd for C38H59N4O12 [M+H]+ 763.4129, found 763.4130.
(E)-2-(aminooxy)-N-(17-(5-(4-(methylamino)styryl)pyridine-2-yloxy)-3,6,9,12,15- pentaoxaheptadecyl)acetamide (22)
Carbamate 20 (29 mg, 0.04 mmol) was dissolved in 1 mL dichloromethane and to it was added 1 mL of trifluoroacetic acid. The reaction was allowed to stir at room temperature for 5 h after which time the reaction mixture was concentrated down from 1N HCl (2 × 3 mL) and placed under high vacuum to obtain 137 mg of a pale, yellow viscous oil of the aminooxy·xHCl salt 22. Due to its instability, full characterization was achieved from the subsequent step of formation of oxime 24.
2-((E)-(6-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)pyridin-3-yl)methyleneaminooxy)-N-(17-(5-((E)- 4-(methylamino)styryl)pyridin-2-yloxy)-3,6,9,12,15-pentaoxaheptadecyl)acetamide (24)
Trifluoroacetic acid (10 μl) was added to a solution of acetal 2 (10 mg, 0.03 mmol) dissolved in 1.0 mL ethanol and 0.5 mL H2O to lower the pH ~ 0. This was then placed in an oil bath and heated to 60 °C. After 5 min, a solution of diamine 22 (21 mg, 0.04 mmol) in 0.7 mL ethanol and 0.1 mL H2O was added to the reaction mixture at 60 °C. After 10 min, the reaction mixture was removed from the heat, allowed to cool to room temperature, and concentrated down from 1N HCl (2 × 1 mL). The crude reaction mixture was then partitioned between 50 % NaHCO3 (aq, sat)/ice and 5 % methanol/ethyl acetate. The organic layer was separated form the aqueous layer with the use of 5 % methanol/ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered, and concentrated. Purification by flash chromatography (5–10 % methanol/dichloromethane) provided 18 mg (67 %) of oxime 24, a pale yellow material. Rf = 0.28 (5 % methanol/dichloromethane); 1H NMR (200 MHz, CDCl3): δ 8.26–8.24 (m, 2H), 8.11 (d, J = 2.5 Hz, 1H), 7.93 (ddd, J = 16.6, 8.9, 2.3 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 7.02–6.75 (m, 4H), 6.60 (d, J = 8.6 Hz, 2H), 4.64–4.57 (m, 3H), 4.47–4.36 (m, 6H), 3.85–3.78 (m, 6H), 3.67–3.40 (m, 24H), 2.79 (s, 3H); 13C NMR (50 MHz, CDCl3): δ 172.5, 166.5, 164.1, 151.5, 149.6, 148.5, 145.9, 137.8, 136.8, 130.3, 129.7, 128.8, 127.5, 123.3, 120.6, 113.6, 112.9, 112.3, 85.9, 82.6, 74.1, 71.9, 71.8, 71.7, 71.7, 71.6, 71.5, 70.9, 70.7, 70.6, 67.0, 66.7, 40.2, 30.7; HRMS (ESI) m/z calcd for C40H57N5O11 [M+H]+ 802.4039, found 802.4016; HPLC conditions: Gemini 5μ C18 110A column, 250 × 4.60 mm, 5 micron; rt = 6.40 min (280 nm) in acetonitrile/10 mM ammonium formate buffer 60/40 with a flow rate of 1.0 mL/min.
Radiochemistry
A typical procedure for the formation of radioactive intermediate [18F]2 is as follows: A Sep-Pak® light Accell Plus QMA (Waters Corporation) cartridge was activated with 1N NaHCO3 (10 mL), H2O (10 mL), and dried with argon. [18O]-Enriched aqueous solution of fluoride-18 (~ 2 mCi) was taken up in 1 mL of H2O (diluted) and passed through the activated QMA (light) cartridge. A solution of 1 mL (0.93 mL acetonitrile, 0.07 mL H2O) containing Kryptofix®[K222] (11 mg 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane) and 2 mg K2CO3, was then passed through the QMA cartridge into a reaction vessel to collect the K[222]18F complex. This complex was dried at 110 °C with a flow of argon until most of the solvent was evaporated. The solution was further azeotroped with the use of acetonitrile (2 × 1 mL) at 110 °C with a stream of argon gas. A solution of tosylate 8 (1 mg) in 1 mL of anhydrous acetonitrile was added to the ‘dry’ K[222]18F complex at 110 °C. After 10 min, the reaction vessel was removed from the oil bath, cooled in an ice/water bath and quenched with 8 mL of H2O. The reaction medium was then passed through an OASIS HLB 3cc cartridge (activated by flushing with 3 mL ethanol followed by 10 mL H2O). The OASIS cartridge was subsequently washed with H2O (2 × 3 mL) and the desired radioactive ligand was eluted with ethanol (0.75 mL). The activity was measured from each washing to obtain the decay corrected radio chemical yield (rcy). The eluted ligand was used without any further purification. Radiochemical purity (rcp) of the prosthetic ligand [18F]2 was determined by HPLC. HPLC conditions: Gemini 5μ C18 110A column, 250 × 4.60 mm, 5 micron; acetonitrile/10mM ammonium formate buffer solution 60/40 with a flow rate of 1.0 mL/min; rt = 4.79 min.
A typical procedure of the formation of oxime [18F]23 and [18F]24 is as follows: Water (0.2 mL) followed by trifluoroacetic acid (~ 15 μL) was added to compound [18F]2. The reaction vessel was then placed in an oil bath set to 70 °C. After 2 min at 70 °C, a solution of aminooxy 21 or 22 (~ 4 mg, >1.1 equiv of tosylate 8) in 0.3 mL of ethanol and 0.1 mL of H2O was added to the reaction mixture. After an additional 25 min, the reaction vessel was removed from the oil bath, cooled in an ice/water bath, and was quenched with 0.2 mL of 1N NaHCO3. This crude solution was then purified by HPLC (Gemini 5μ C18 110A column, 250 × 10.00 mm, 5 micron; acetonitrile/10 mM ammonium formate buffer solution 60/40 as mobile phase with a flow rate of 2.0 mL/min; fractions were collected from 14.56 to 16.17 min). The fractions were then pooled together and diluted with 3 mL H2O. The reaction media was then passed through an OASIS HLB 3cc cartridge (activated by flushing with 3 mL ethanol followed by 10 mL H2O). The OASIS cartridge was washed with an organic solvent (ethanol or acetonitrile) (0.75 mL) to elute the desired radioactive oxime. The radiochemical purity and the specific activity for compounds [18F]23 and [18F]24 were determined by HPLC (Gemini 5μ C18 110A column, 250 × 4.60 mm, 5 micron; acetonitrile/10 mM ammonium formate buffer 60/40 with a flow rate of 1.0 mL/min; retention time for [18F]23 rt = 6.83 min; retention time for [18F]24 rt = 7.37 min).
Biodistribution
In-vivo biodistribution study in mice
Biodistribution studies were performed on ligands [18F]23 and [18F]24, to evaluate their biological properties. Each of these radiotracers were tested in normal male ICR mice weighing 20 – 25 grams. Each of the biodistribution studies used 6 mice per group with two time points taken at 2 min and 30 min post-injection. The mice were put under isoflurane anesthesia (1 – 2 %, 1 L oxygen/minute). A saline solution (0.15 mL) containing 25 μCi of each isomer, [18F]23 or [18F]24, was injected via the lateral tail vein. The mice were sacrificed at 2 and 30 minutes post-injection by cardiac excision while under isoflurane anesthesia. The organs of interest were removed, weighed, and the radioactivity was counted with a gamma counter (Packard Cobra). The percent dose per gram was calculated by a comparison of the tissue activity counts to counts of 1.0 % of the initial dose. The initial dose consisted of 100 times diluted aliquots of the injected material measured at the same rate.
Results and Discussion
Chemical Synthesis
The formation of the ‘cold’ prosthetic ligand 2 is depicted in Scheme 1. The synthesis began with the formation of acetal 6 (43) through an acid catalyzed protection of 6-bromo-3- pyridine carbaldehyde 5. The triethylene glycol chain was tethered onto the molecule under basic conditions (cesium carbonate) with the aid of a microwave reactor. Alcohol 7 was converted into tosylate 8, which was subsequently refluxed in the presence of tetra-n-butylammonium fluoride hydrate to provide the desired acetal, compound 2.
Scheme 1a. Synthetic Pathway for the Preparation of Fluorides 2 and 3.

aReagents and conditions: (i) ethylene glycol, p-TsOH, benzene, reflux (98 %); (ii) triethylene glycol, Cs2CO3, DMF, microwave, 150 °C, 50 min (61 %); (iii) p-TsCl, Et3N, DMAP, CH2Cl2 (82 %); (iv) TBAF·H2O, THF, reflux (57 %); (v) PPTS, acetone, H2O, 70 °C (89 %); (vi) p-TsCl, pyridine, CH2Cl2, 0 °C to rt (84 %); (vii) TBAF·H2O, THF, reflux (40 %).
The formation of compound 3 is depicted in Scheme 1. The acetal functional group of intermediate 7 was removed under acidic conditions to provide aldehyde 9. It should be noted that direct addition of triethylene glycol to 6-bromo-3-pyridine carbaldehyde 5, the same conditions used to form intermediate 7, proved to be incompatible, providing only 2 % of the desired aldehyde 9. Therefore, two steps were necessary to synthesize aldehyde 9; nucleophilic substitution of triethylene glycol followed by the removal of the acetal protecting group. The alcohol functional group of compound 9 was converted into the tosylate to provide intermediate 10. Compound 10 was transformed into aldehyde 3 with the use of tetra-n-butylammonium fluoride hydrate in refluxing tetrahydrofuran.
The syntheses of ‘cold’ oximes 23 and 24 are illustrated in Scheme 2. Compound 13 was prepared from alcohol 11 (n = 4) with the use of sodium hydride and diethylene glycol bis(p-toluenesulfonate). Each of the tosylate groups in species 12 and 13 (44) were displaced with sodium azide at elevated temperatures. The azide functionality was then reduced to an amine, which lead to the formation of compounds 14 and 15, by way of a Staudinger reaction (45, 46). It was crucial that the correct reaction conditions were used to reduce only the azide functionality without saturating the carbon-carbon double bond. Succinimide 18 (47) was attached with 14 and 15 with excess Hünig’s base to provide the dicarbamates 19 and 20, respectively. Removal of both t-butoxycarbonyl (Boc) protecting groups was achieved using a 1:1 mixture of dichloromethane:trifluoroacetic acid. Subsequent addition of the free aminooxy compound into aldehyde 3 lead to the formation of the desired oxime 23 (n = 3). Likewise, aminooxy 22 was added to acetal 2 under acidic conditions to provide the desired oxime 24 (n = 6). The acetal group was removed in situ to form the aldehyde, which under an acidic environment has a high affinity to react with aminooxy groups (38, 39). The one pot deprotection/addition sequence to form the desired oxime proved to be essential for the radiosynthesis of compounds [18F]23 and [18F]24. Although two isomers (cis- and trans-) are potentially formed, it appears only one isomer was observed in the formation of both oximes 23 and 24 (confirmed by HPLC). It is hypothesized that this isomer is the more stable trans-complex. Work is currently underway to elucidate their structures by attempts to obtain crystal structures with the use of forming the salts with several different counter ions. Oximes 23 and 24 are the ‘cold’ standards for our labeling studies.
Scheme 2a. Synthetic Pathway for the Formation of Oximes 23 (n = 3) and 24 (n = 6).

aReagents and conditions: (i) diethylene glycol bis(p-toluenesulfonate), NaH, DMF, 0 °C to rt (73 %); (ii) NaN3, DMF, 60 °C 3 h (n = 3: 100 %; n = 6: 60 %); (iii) PPh3, THF/H2O (9:1), 60 °C (n = 3: 70 %; n = 6: 80 %); (iv) 18, i-Pr2NEt, DMF (n = 3: 45 %; n = 6: 60 %); (v) TFA/CH2Cl2, (1:1) (n = 3: 73 %; n = 6: 100 %); (vi) 3, TFA, EtOH/H2O, 70 °C (n = 3: 50 %); (vii) 2, TFA, EtOH/H2O, 70 °C (n = 6: 67 %).
Radiolabeling
Our initial goal was to synthesize and evaluate a new prosthetic ligand with a future goal of radiolabel biologically active peptides and proteins by way of an oxime coupling reaction. Preliminary studies were done on two model aminooxy species, compounds 23 and 24. We first fully characterized the ‘cold’ oximes, and then radiolabeled each aminooxy compound with our prosthetic ligand. These small molecules were used as models, instead of the protein or peptide oxime adducts, due to their compatibility with more practical and manageable reagents and characterization methods (i.e. cost effectiveness and characterization without the use of MALDI). The radiolabeling of complexes 21 and 22 were used to demonstrate the capability of this methodology while attempting to optimize the reaction conditions to form the oxime bond.
The first attempt at the formation of a prosthetic ligand was through the direct fluoride-18 (nca) labeling of aldehyde 10 (Scheme 3). The formation of an oxime with the use of an aminooxy group and an aldehyde under acidic conditions can be achieved. The fluoride-18 labeling of aldehyde 10 afforded compound [18F]3. The formation of compound [18F]3 provided a reasonable radiochemical yield; however, with low radiochemical purity (42%). Several unidentified peaks were observed in the HPLC radioactive trace.
Scheme 3a. Direct Fluoride-18 (nca) Labeling of Aldehyde 10.

aReagents and conditions: (i) K[222]18F, K2CO3, DMSO, 100 °C, 10 min (45 % rcy; 42 % rcp).
To circumvent the low radiochemical purity, the aldehyde group was protected prior to the fluoride-18 radiolabeling. Therefore, acetal 8 was used in place of aldehyde 10. The formation of compound [18F]2 was achieved through a direct SN2 displacement of a tosylate group (see Figure 3 for HPLC traces for cold and hot complexes). The acetal functionality protected the compound from the harsh environment present for the nucleophilic attack of a fluoride anion. The acetal, [18F]2, is a stable, neutral molecule soluble in organic media. After successful fluoride-18 labeling, the acetal group was deprotected under an aqueous acidic media and converted to the aldehyde; aldehydes will chemoselectively react with aminooxy functional groups under these same conditions to provide an irreversible oxime bond (38, 39). This allows for both the de-blocking of the acetal functionality and the formation of the oxime bond, to bestow the radiolabeled tracer.
Figure 3.

(A) HPLC profile of radio-trace [18F]2. (B) HPLC profile of UV-spectra for compound 2 at 277 nm. Both samples were run on Gemini 5μ C18 110A 250 × 4.60 mm column; ACN/10 mM AFB 60/40 with a flow rate of 1.0 mL/min; rt = 4.10 min.
Several variables were studied (i.e. solvent, temperature, and time) to determine the most efficient and suitable conditions for radiosynthesis of acetal [18F]2. A solvent screening was performed at a constant temperature (80 °C) and time (15 min) with the use of 4,7,13,16,21,24-hexaoxa-1,10- diazabicyclo[8.8.8]-hexacosane (K[222]) and potassium carbonate as the catalysts for the preparation of ‘naked’ fluoride (see Table 1). The radiochemical yields (rcy) were determined from the isolation of the product after solid phase extraction (OASIS HLB 3cc cartridge), and the radiochemical purities (rcp) were determined by HPLC (for conditions see experimental section). These results suggested that dimethyl sulfoxide and acetonitrile are the solvents of choice. Due to the higher observed radiochemical purity (rcp) achieved with acetonitrile as the solvent (91 ± 4 %, n = 3, see Table 1), all further experiments were performed with the use of acetonitrile.
Table 1.
Use of Varying the Solvent for the Radiolabeling of Tosylate 8 (n = 3)
 | |||
|---|---|---|---|
| entry | solvent | % rcy (isolated) | % rcp (HPLC) |
| 1 | DMSO | 32 (± 2) | 84 (± 15) |
| 2 | DMF | 14 (± 6) | 78 (± 11) |
| 3 | t-BuOH | 7 (± 2) | 53 (± 12) |
| 4 | ACN | 35 (± 2) | 91 (± 4) |
A time study was performed for the radiolabeling of acetal [18F]2 in the presence of ‘dry’ K[222]18F catalyst (Table 2). Reproducibility and greatest radiochemical yields were obtained when compound 8 was heated in an open test tube at 110 °C for 10 min (rcy = 71 ± 2 %; rcp > 99 ± 1 %, n = 3, Table 2, entry 2). The radiochemical yield was lower for shorter time spans (5 min). Noticeably lower radiochemical yields were routinely observed when the reaction was allowed to proceed for more than 15 min. This suggests that decomposition of the desired product at elevated temperatures over extended time periods (i.e. greater than 15 min). When the reaction was run in a sealed tube for 15 min at 110 °C, lower radiochemical yields and lower radiochemical purities were observed (Table 2, entry 4). The lower radiochemical yields observed in the sealed tube experiments may be its concentration; when the reaction was performed in an open test tube, solvent was able to evaporate (acetonitrile’s bp = 82 °C). It is unclear yet as to why the radiochemical purities would decrease in a sealed environment.
Table 2.
Time Study Results on the Radiochemical Synthesis of Compound [18F]2
 | |||
|---|---|---|---|
| entry | time (min) | % rcy (isolated) | % rcp (HPLC) |
| 1 | 5 | 51 | 93 |
| *2 | 10 | 71 (± 2) | 99 (± 1) |
| 3 | 15 | 65 | 96 |
| D4 | 15 | 49 | 94 |
| 5 | 20 | 61 | >99 |
reaction was run in a sealed tube
n = 3
As mentioned earlier, aminooxy groups readily form oximes with aldehydes under acid conditions. An acetal can be converted to an aldehyde, in-situ, under aqueous acidic conditions. If the insitu formed aldehyde were then subjected to the aminooxy under these same conditions, labeling via the oxime bond would be accomplished in one pot. To test our theory, aminooxy 22 was added to an HCl acidic aqueous/ethanol solution with acetal [18F]2 at 70 °C (Table 3). The pH of the solution was estimated at 0. At 5 min time intervals, an aliquot (5 μL) was removed from the reaction vessel, neutralized with 1N NaHCO3 (10 μL, pH ~ 9), and diluted with 100 μL of acetonitrile. The relative rates of the formation of product, which was equal to the disappearance of the starting material, were analyzed by HPLC. The reaction appeared to be complete after 35 min, with no starting material observed by HPLC analysis.
Table 3.
Formation of F-18 Labeled Oxime [18F]24 Over 5 min Increments
 | ||
|---|---|---|
| entry | time (min) | % rcy (= %rcp) |
| 1 | 5 | 30 |
| 2 | 10 | 65 |
| 3 | 15 | 80 |
| 4 | 20 | 89 |
| 5 | 25 | 93 |
| 6 | 30 | 94 |
| 7 | 35 | 98 |
Species [18F]24 could be purified and isolated by semi-preparative HPLC with a reversed phase column (rt ~ 15 min) in 41% decay corrected radiochemical yield end of synthesis (eos) with a radiochemical purity > 99%. The total preparation time for [18F]24 was 100 min (over two steps). The specific activity at eos was ~ 100 Ci/mmol. The radiolabeling of oxime [18F]23 provided similar results with a decay corrected radiochemical yield (eos) of 38 % (over two steps) and a radiochemical purity of 94 %. The specific activity was not determined for oxime [18F]23.
In Vitro Binding Assays
Human brain sections containing Alzheimer Disease (AD) plaque were obtained. The gray matter was separated from the white matter. The gray matter containing the AD plaques were dissected and processed. The brain tissue homogenates were prepared in phosphate buffered solution (PBS), pH = 7.2. The specific binding of [18F]-AV-45 1 was carried out in 12 × 75 mm borosilicate glass tubes. The reaction mixture contained 50 μL of brain homogenates (20–50 μg), 50 μL of ligand diluted in PBS and 50 μL of inhibitors (10−5 – 10−10 M diluted serially in PBS containing 0.1% bovine serum albumin in a final volume of 1 mL. Nonspecific binding was determined in the presence of IMPY (600 nM) within the same assay tubes. In competition experiments, compounds at concentrations up to 10−5 M were examined for their abilities to compete for the binding of [18F]-AV-45 1. Incubations were carried out routinely at 37 °C for 120 min, and the bound and free radioactivity were separated by vacuum filtration. The samples were then filtered through glass fiber filters soaked in PBS (GF/B Whatman) using a Brandel M-24R cell harvester followed by 2 × 3-mL washes of PBS at room temperature. The filters containing bound [18F] ligand were immediately counted with a gamma counter (Cobra II, Perkin Elmer) with 70% counting efficiency. The results of the inhibition experiments were subjected to nonlinear regression analysis using equilibrium binding data analysis by which Ki values were calculated as [18F]-AV-45 1 Ki = 3.80 ± 1.82 nM (n = 5), [18F]23 Ki = 11.78 ± 3.06 nM (n = 5), [18F]24 Ki = 25.2 ± 0.9 nM (n = 6).
Biodistribution
Biodistribution studies of [18F]23 and [18F]24 were conducted in male ICR mice. The % dose/gram was compared to that of [18F]-AV-45 (1) (Table 4). [18F]-AV-45 (1) (40, 41) is used as a PET imaging agent targeting β-amyloid plaques within the brain. Both oximes contain the core backbone structure found in [18F]-AV-45 (1). The use of the neutral (calculated log P = 1.00) and hydrophilic spacer (triethylene glycol) on the prosthetic group was thought to increase the overall hydrophilicity and therefore deter the compound from passing the blood-brain barrier (BBB). Compounds [18F]23 and [18F]24 may be useful to image β-amyloid plaques located on the cerebral blood vessels outside the brain.
Table 4.
Biodistribution study % dose/gram of [18F]-AV-45 (1), [18F]23, and [18F]24 in male ICR mice
| [18F]-AV-45 (1)* | [18F]23 | [18F]24 | ||||
|---|---|---|---|---|---|---|
| Organ | 2 min n=3 | 30 min n=3 | 2 min n=3 | 30 min n=3 | 2 min n=3 | 30 min n=3 |
| Blood | 3.14 ± 0.69 | 2.80 ± 0.44 | 9.34 ± 1.37 | 4.94 ± 0.91 | 4.19 ± 1.04 | 2.81 ± 0.22 |
| Heart | 6.25 ± 1.79 | 2.18 ± 0.32 | 13.1 ± 3.08 | 3.72 ± 0.84 | 12.3 ± 1.79 | 4.17 ± 0.61 |
| Muscle | 1.06 ± 0.39 | 1.78 ± 0.34 | 1.32 ± 0.50 | 1.56 ± 0.27 | 1.42 ± 0.46 | 2.01 ± 0.21 |
| Lung | 6.87 ± 1.36 | 3.20 ± 0.54 | 7.09 ± 1.37 | 3.53 ± 0.38 | 3.70 ± 1.06 | 2.43 ± 0.33 |
| Kidney | 10.9 ± 2.63 | 6.31 ± 0.58 | 14.1 ± 2.85 | 6.83 ± 1.93 | 15.4 ± 3.54 | 7.78 ± 0.85 |
| Pancreas | -- | - | 5.13 ± 1.08 | 2.28 ± 0.48 | 3.32 ± 0.58 | 2.22 ± 0.23 |
| Spleen | 4.57 ± 1.07 | 1.81 ± 0.24 | 4.81 ± 1.18 | 3.57 ± 0.68 | 3.11 ± 0.45 | 1.63 ± 0.19 |
| Liver | 21.5± 1.07 | 12.9 ± 0.72 | 22.2 ± 3.99 | 19.1 ± 3.65 | 29.4 ± 5.09 | 10.9 ± 1.17 |
| Skin | 1.18 ± 0.23 | 2.36 ± 0.29 | 0.79 ± 0.20 | 1.50 ± 0.25 | 1.14 ± 0.44 | 1.78 ± 0.13 |
| Brain | 7.77 ± 1.70 | 1.59 ± 0.22 | 1.69 ± 0.44 | 0.96 ± 0.16 | 0.48 ± 0.12 | 0.92 ± 0.11 |
| Bone | 1.43 ± 0.09 | 1.22 ± 0.17 | 2.00 ± 0.33 | 2.27 ± 0.41 | 1.76 ± 0.31 | 1.62 ± 0.10 |
Data taken from Zhang, W., et. al. (2005) F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Aβ aggregates in the brain. Nucl. Med. Biol. 32, 799–809.
Table 4 depicts a comparison of the biodistribution for [18F]-AV-45 (1), [18F]23, and [18F]24 in normal mice. We were particularly interested in comparing the brain uptakes of [18F]-AV-45 (1) and the two new derivatives, [18F]23 and [18F]24. The addition of the prosthetic group led to AV-45 derivatives with a much higher molecular weight, exceeding the limit for penetrating the intact BBB (48, 49). As expected, [18F]-AV-45 (1) (molecular weight = 359.43 g/mol) has a high initial brain uptake after 2 min post injection (7.77 % dose/gram). The tracer then proceeds to gradually wash out after 30 min post injection. Initial brain uptake of oxime [18F]23 (molecular weight = 668.74 g/mol) was rather respectable (1.69 % dose/gram). On the other hand, [18F]24 (molecular weight = 799.91 g/mol) showed significantly lower uptake (0.48 % dose/gram) at the 2 min post injection mark, indicting a far less efficient passage of the tracer through the intact BBB. To our surprise an increase of uptake in the brain at 30 min post injection (0.92 % dose/gram) was observed. We are suggesting the in vivo metabolites of [18F]24 may have contributed to the increase in brain uptake.
Compounds [18F]23 and [18F]24 display high initial uptake and slow retention, remaining rather consistent throughout the 30 minutes, in most of the organs tested. Heart uptake and retention drop fairly fast with time, showing low heart activity at 30 min post injection. There was no significant uptake within the lung, along with no significant retention within the first arteriovenous capillary bed. Rapid uptake is observed within the kidneys but is soon excreted to the urinary bladder. It should be noted that each labeled oxime showed the washing out of activity through the liver. During the time frame of the studies performed, there is very little de-fluorination occurring as depicted from the values in the bone (femur).
Conclusions
A new prosthetic radiolabeled ligand, [18F]2, was successfully synthesized in a total of four steps, starting with readily available 6-bromo-3-pyridine carbaldehyde 5. The fluoride-18 labeling of this compound was achieved in high radiochemical yields (71 ± 2 %, n = 3) with excellent radiochemical purity (> 99 %, based on HPLC analyses) in short reaction time (10 min). The species could be easily purified through a solid phase extraction (OASIS HLB 3cc cartridge), eluted with a small volume of ethanol, and used directly in the next step of the synthetic sequence without any further purification. The coupling with species [18F]2 was demonstrated in two model complexes, oximes [18F]23 and [18F]24.
Biodistribution studies were performed on each of the labeled oximes [18F]23 and [18F]24, and compared to that of [18F]-AV-45 (1). The major differences between [18F]-AV-45 (1), [18F]23, and [18F]24 are their size (molecular weight) and the hydrophilic spacer (triethylene glycol) from the prosthetic ligand. [18F]-AV-45 (1) is a small and neutral molecule designed to serve as a brain-imaging agent to target Aβ-plaques inside the brain. Compounds [18F]23 and [18F]24 are larger in size (PEG, n = 3 and n = 6 respectively); therefore they should reduce the BBB penetration. The use of the hydrophilic spacer (triethylene glycol) on the prosthetic group was thought to increase the overall hydrophilicity of the molecule. The brain uptake for oxime [18F]23 appeared to be reduced, but it retained some capability to cross the BBB. Oxime [18F]24 showed promising results after 2 min post injection (0.48 % dose/gram), however the uptake increased after 30 min post injection (0.92 % dose/gram) suggesting an in-vivo decomposition/metabolism of compound [18F]24.
The prosthetic ligand, [18F]2, has several advantages over other known prosthetic ligand in the literature: high radiochemical yield, ease of formation, the ability to label base sensitive compounds, and the chemoselective formation of the oxime bond. Future work will focus on using this prosthetic ligand to label larger molecules that are incompatible with direct labeling with fluoride-18 (nca) anion. We intend to use this methodology to label biologically active peptides and proteins modified with an aminooxy functional group.
Supplementary Material
Acknowledgments
This work was supported by the grants from NIH (AG-022559) and a grant from Avid Radiopharmaceuticals.
Footnotes
Supporting Information Spectroscopic data of key compounds 8, 2, 23, and 24 are provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Tolmachev V, Stone-Elander S. Radiolabelled proteins for positron emission tomography: Pros and cons of labelling methods. Biochim Biophys Acta, Gen Subj. 2010;1800:487–510. doi: 10.1016/j.bbagen.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Cherry SR. Fundamentals of positron emission tomography and applications in preclinical drug development. J Clin Pharmacol. 2001;41:482–491. doi: 10.1177/00912700122010357. [DOI] [PubMed] [Google Scholar]
- 3.Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 4.Mach RH, Schwarz SW. Challenges for developing PET tracers: isotopes, chemistry, and regulatory aspects. PET Clinics. 2010;5:131–153. doi: 10.1016/j.cpet.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Kuhnast B, Dolle F. The challenge of labeling macromolecules with fluorine-18: three decades of research. Curr Radiopharm. 2010;3:174–201. [Google Scholar]
- 6.Schubiger PA, Lehmann L, Friebe M. PET Chemistry. Vol. 62. Springer; New York: 2006. [Google Scholar]
- 7.Hultsch C, Berndt M, Bergmann R, Wuest F. Radiolabeling of multimeric neurotensin(8–13) analogs with short-lived positron emitter fluorine-18. Appl Radiat Isot. 2007;65:818–826. doi: 10.1016/j.apradiso.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Fredriksson A, Johnstrom P, Stone-Elander S, Jonasson P, Nygren PA, Ekberg K, Johansson BL, Wahren J. Labeling of human C-peptide by conjugation with Nsuccinimidyl- 4-[18F]fluorobenzoate. J Labelled Compd Radiopharm. 2001;44:509–519. [Google Scholar]
- 9.Schottelius M, Poethko T, Herz M, Reubi JC, Kessler H, Schwaiger M, Wester HJ. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin Cancer Res. 2004;10:3593–3606. doi: 10.1158/1078-0432.CCR-03-0359. [DOI] [PubMed] [Google Scholar]
- 10.Guhlke S, Wester HJ, Bruns C, Stoecklin G. (2-[18F]fluoropropionyl-(D)phe1)- octreotide, a potential radiopharmaceutical for quantitative somatostatin receptor imaging with PET: Synthesis, radiolabeling, in vitro validation and biodistribution in mice. Nucl Med Biol. 1994;21:819–25. doi: 10.1016/0969-8051(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 11.Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM, Cuthbertson A. One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6- tetrafluorophenyl ester ([18F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J Med Chem. 2010;53:1732–1740. doi: 10.1021/jm9015813. [DOI] [PubMed] [Google Scholar]
- 12.Wust F, Hultsch C, Bergmann R, Johannsen B, Henle T. Radiolabeling of isopeptide Ne-(gamma -glutamyl)-L-lysine by conjugation with N-succinimidyl 4- [18F]fluorobenzoate. Appl Radiat Isot. 2003;59:43–48. doi: 10.1016/s0969-8043(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 13.De Bruin B, Kuhnast B, Hinnen F, Yaouancq L, Amessou M, Johannes L, Samson A, Boisgard R, Tavitian B, Dolle F. 1-[3-(2-[18F]Fluoropyridin-3- yloxy)propyl]pyrrole-2,5-dione: Design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjugate Chem. 2005;16:406–420. doi: 10.1021/bc0497463. [DOI] [PubMed] [Google Scholar]
- 14.Prante O, Einsiedel J, Haubner R, Gmeiner P, Wester HJ, Kuwert T, Maschauer S. 3,4,6-Tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl phenylthiosulfonate: a thiolreactive agent for the chemoselective 18F-glycosylation of peptides. Bioconjugate Chem. 2007;18:254–262. doi: 10.1021/bc060340v. [DOI] [PubMed] [Google Scholar]
- 15.Wangler B, Quandt G, Iovkova L, Schirrmacher E, Wangler C, Boening G, Hacker M, Schmoeckel M, Jurkschat K, Bartenstein P, Schirrmacher R. Kit-like 18F labeling of proteins: synthesis of 4-(di-tert-butyl[18F]fluorosilyl)benzenethiol (Si[18F]FA-SH) labeled rat serum albumin for blood pool imaging with PET. Bioconjugate Chem. 2009;20:317–321. doi: 10.1021/bc800413g. [DOI] [PubMed] [Google Scholar]
- 16.Glaser M, Karlsen H, Solbakken M, Arukwe J, Brady F, Luthra SK, Cuthbertson A. 18F-Fluorothiols: a new approach to label peptides chemoselectively as potential tracers for positron emission tomography. Bioconjugate Chem. 2004;15:1447–1453. doi: 10.1021/bc0498774. [DOI] [PubMed] [Google Scholar]
- 17.Mach RH, Leudtke RR, Unsworth CD, Boundy VA, Nowak PA, Scripko JG, Elder ST, Jackson JR, Hoffman PL, et al. Fluorine-18 labeled benzamides for studying the dopamine D2 receptor with positron emission tomography. J Med Chem. 1993;36:3707–20. doi: 10.1021/jm00075a028. [DOI] [PubMed] [Google Scholar]
- 18.Garg S, Thopate SR, Minton RC, Black KW, Lynch AJH, Garg PK. 3- Amino-4-(2-((4-[18F]fluorobenzyl)methylamino)methylphenylsulfanyl)benzonitrile, an F-18 fluorobenzyl analogue of DASB: synthesis, in vitro binding, and in vivo biodistribution studies. Bioconjugate Chem. 2007;18:1612–1618. doi: 10.1021/bc070112g. [DOI] [PubMed] [Google Scholar]
- 19.Olberg DE, Hjelstuen OK, Solbakken M, Arukwe J, Karlsen H, Cuthbertson A. A novel prosthetic group for site-selective labeling of peptides for positron emission tomography. Bioconjugate Chem. 2008;19:1301–1308. doi: 10.1021/bc800007h. [DOI] [PubMed] [Google Scholar]
- 20.Olberg DE, Hjelstuen OK, Solbakken M, Arukwe JM, Dyrstad K, Cuthbertson A. Site-specific addition of an 18F-N-methylaminooxy-containing prosthetic group to a vinylsulfone modified peptide. J Labelled Compd Radiopharm. 2009;52:571–575. [Google Scholar]
- 21.Bruus-Jensen K, Poethko T, Schottelius M, Hauser A, Schwaiger M, Wester HJ. Chemoselective hydrazone formation between HYNIC-functionalized peptides and 18Ffluorinated aldehydes. Nucl Med Biol. 2006;33:173–183. doi: 10.1016/j.nucmedbio.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Toyokuni T, Walsh JC, Dominguez A, Phelps ME, Barrio JR, Gambhir SS, Satyamurthy N. Synthesis of a new heterobifunctional linker, N-[4- (aminooxy)butyl]maleimide, for facile access to a thiol-reactive 18F-labeling agent. Bioconjugate Chem. 2003;14:1253–1259. doi: 10.1021/bc034107y. [DOI] [PubMed] [Google Scholar]
- 23.Berndt M, Pietzsch J, Wuest F. Labeling of low-density lipoproteins using the 18F-labeled thiol-reactive reagent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide. Nucl Med Biol. 2007;34:5–15. doi: 10.1016/j.nucmedbio.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Namavari M, Padilla De Jesus O, Cheng Z, De A, Kovacs E, Levi J, Zhang R, Hoerner Joshua K, Grade H, Syud Faisal A, Gambhir Sanjiv S. Direct site-specific radiolabeling of an affibody protein with 4-[18F]fluorobenzaldehyde via oxime chemistry. Mol Imaging Biol. 2008;10:177–81. doi: 10.1007/s11307-008-0142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poethko T, Schottelius M, Thumshirn G, Hersel U, Herz M, Henriksen G, Kessler H, Schwaiger M, Wester HJ. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and ocreotide analogs. J Nucl Med. 2004;45:892–902. [PubMed] [Google Scholar]
- 26.Namavari M, Cheng Z, Zhang R, De A, Levi J, Hoerner JK, Yaghoubi SS, Syud FA, Gambhir SS. A novel method for direct site-specific radiolabeling of peptides using [18F]FDG. Bioconjugate Chem. 2009;20:432–436. doi: 10.1021/bc800422b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Link JM, Stekhova S, Yagle KJ, Smith C, Krohn KA, Tait JF. Site-specific labeling of annexin V with F-18 for apoptosis imaging. Bioconjugate Chem. 2008;19:1684–1688. doi: 10.1021/bc800164d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hultsch C, Schottelius M, Auernheimer J, Alke A, Wester HJ. 18FFluoroglucosylation of peptides, exemplified on cyclo(RGDfK) Eur J Nucl Med Mol. 2009;36:1469–1474. doi: 10.1007/s00259-009-1122-0. [DOI] [PubMed] [Google Scholar]
- 29.Wuest F, Hultsch C, Berndt M, Bergmann R. Direct labelling of peptides with 2- [18F]fluoro-2-deoxy-D-glucose ([18F]FDG) Bioorg Med Chem Lett. 2009;19:5426–5428. doi: 10.1016/j.bmcl.2009.07.108. [DOI] [PubMed] [Google Scholar]
- 30.Abdel-Jalil RJ, Aqarbeh M, Loeffler D, Shen B, Orabi SA, Voelter W, Machulla HJ. Synthesis and radiolabeling of new N-[(4-[18F]fluorobenzylidene) aminooxy) alkyl]-2-nitroimidazoles as possible hypoxia imaging pharmaceuticals. J Radioanal Nucl Chem. 2010;283:239–243. [Google Scholar]
- 31.Glaser M, Morrison M, Solbakken M, Arukwe J, Karlsen H, Wiggen U, Champion S, Kindberg GM, Cuthbertson A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjugate Chem. 2008;19:951–957. doi: 10.1021/bc700472w. [DOI] [PubMed] [Google Scholar]
- 32.Wester HJ, Hamacher K, Stoecklin G. A comparative study of n.c.a fluorine-18 labeling of proteins via acylation and photochemical conjugation. Nucl Med Biol. 1996;23:365–372. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]
- 33.Thonon D, Kech C, Paris J, Lemaire C, Luxen A. New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[18F]-fluorobenzene for PET. Bioconjugate Chem. 2009;20:817–823. doi: 10.1021/bc800544p. [DOI] [PubMed] [Google Scholar]
- 34.Marik J, Sutcliffe JL. Click for PET: rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6681–6684. [Google Scholar]
- 35.Inkster JAH, Adam MJ, Storr T, Ruth TJ. Labeling of an antisense oligonucleotide with [18F]FPy5yne. Nucleosides, Nucleotides Nucleic Acids. 2009;28:1131–1143. doi: 10.1080/15257770903400691. [DOI] [PubMed] [Google Scholar]
- 36.Inkster JAH, Guerin B, Ruth TJ, Adam MJ. Radiosynthesis and bioconjugation of [18F]FPy5yne, a prosthetic group for the 18F labeling of bioactive peptides. J Labelled Compd Radiopharm. 2008;51:444–452. [Google Scholar]
- 37.Okarvi SM. Recent progress in fluorine-18 labelled peptide radiopharmaceuticals. Eur J Nucl Med. 2001;28:929–938. doi: 10.1007/s002590100508. [DOI] [PubMed] [Google Scholar]
- 38.Adams JP. Imines, enamines and oximes. Perkin. 2000;1:125–139. [Google Scholar]
- 39.Singh Y, Edupuganti OP, Villien M, Defrancq E, Dumy P. The oxime bond formation as a useful tool for the preparation of oligonucleotide conjugates. C R Chim. 2005;8:789–796. [Google Scholar]
- 40.Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F, Benedum TE, Kilbourn MR, Skovronsky D, Kung HF. Preclinical properties of 18F-AV-45: a PET agent for Aβ plaques in the brain. J Nucl Med. 2009;50:1887–1894. doi: 10.2967/jnumed.109.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin KJ, Hsu WC, Hsiao IT, Wey SP, Jin LW, Skovronsky D, Wai YY, Chang HP, Lo CW, Yao CH, Yen TC, Kung MP. Whole-body biodistribution and brain PET imaging with [18F]AV-45, a novel amyloid imaging agent - a pilot study. Nucl Med Biol. 2010;37:497–508. doi: 10.1016/j.nucmedbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Poethko T, Schottelius M, Thumshirn G, Herz M, Haubner R, Henriksen G, Kessler H, Schwaiger M, Wester HJ. Chemoselective pre-conjugate radiohalogenation of unprotected mono- and multimeric peptides via oxime formation. Radiochim Acta. 2004;92:317–327. [Google Scholar]
- 43.Zoppellaro G, Ivanova A, Enkelmann V, Geies A, Baumgarten M. Synthesis, magnetic properties and theoretical calculations of novel nitronyl nitroxide and imino nitroxide diradicals grafted on terpyridine moiety. Polyhedron. 2003;22:2099–2110. [Google Scholar]
- 44.Zhang W, Oya S, Kung MP, Hou C, Maier DL, Kung HF. F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Aβ aggregates in the brain. Nucl Med Biol. 2005;32:799–809. doi: 10.1016/j.nucmedbio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Mormeneo D, Casas J, Llebaria A, Delgado A. Synthesis and preliminary antifungal evaluation of a library of phytosphingolipid analogues. Org Biomol Chem. 2007;5:3769–3777. doi: 10.1039/b709421c. [DOI] [PubMed] [Google Scholar]
- 46.Wascholowski V, Giannis A. Sphingolactones: selective and irreversible inhibitors of neutral sphingomyelinase. Angew Chem Int Ed Engl. 2006;45:827–30. doi: 10.1002/anie.200501983. [DOI] [PubMed] [Google Scholar]
- 47.Deroo S, Defrancq E, Moucheron C, Kirsch-De Mesmaeker A, Dumy P. Synthesis of an oxyamino-containing phenanthroline derivative for the efficient preparation of phenanthroline oligonucleotide oxime conjugates. Tetrahedron Lett. 2003;44:8379–8382. [Google Scholar]
- 48.Kung HF, Choi SR, Qu W, Zhang W, Skovronsky D. 18F stilbenes and styrylpyridines for PET imaging of Aβ plaques in alzheimer’s disease: a miniperspective. J Med Chem. 2010;53:933–941. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dischino DD, Welch MJ, Kilbourn MR, Raichle ME. Relationship between lipophilicity and brain extraction of carbon-11-labeled radiopharmaceuticals. J Nucl Med. 1983;24:1030–1038. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.