

Abstract
Prenatal exposure to infection and subsequent inflammatory responses have been implicated in the etiology of schizophrenia and autism. In this review, we summarize current evidence from human and animal studies supporting the hypothesis that the pathogenesis of these two disorders is linked via exposure to inflammation at early stages of development. Moreover, we propose a hypothetical model in which inflammatory mechanisms may account for multiple shared and disorder-specific pathological characteristics of both entities. In essence, our model suggests that acute neuroinflammation during early fetal development may be relevant for the induction of psychopathological and neuropathological features shared by schizophrenia and autism, while post-acute latent and persistent inflammation may contribute to schizophrenia- and autism-specific phenotypes, respectively.
Neuro-immune factors are now being recognized to play important roles in the etiology of neurological and neuropsychiatric disorders, including those with an onset of pathogenesis during early brain development. Indeed, the brain is highly vulnerable to environmental insults during early development (1). Therefore, immunological processes targeting the developing brain can be expected to have long-lasting impact on brain and behavioral functions throughout the life span (2,3).
Over the last three decades, one line of research has focused on the impact of prenatal maternal infection on the development of childhood and adult neuropsychiatric disorders, particularly schizophrenia and autism (4,5). In addition to their suggested etiopathogenetic contribution to chronic mental illness, intrauterine infection and subsequent maternal/fetal inflammatory responses have also been associated with neonatal white matter damage and cerebral palsy (6-9). In particular, the fetal inflammatory response to intrauterine infection seems to contribute to neonatal brain injury and subsequent neurological disability (6-9). In this article, we focus on the link between prenatal infection and neuropsychiatric disease, especially schizophrenia and autism.
Schizophrenia is a chronic psychotic disorder that affects approximately 1% of the population worldwide (10). The disease onset is typically in late adolescence or early adulthood and includes psychotic symptoms which are commonly referred to as positive, negative and cognitive symptoms (10). Positive symptoms are features that are not present in healthy individuals but appear as a result of the disease process. These include visual and/or auditory hallucinations, delusions, paranoia and major thought disorders. Negative symptoms refer to features that are normally present, but are reduced or absent as a result of the disease process, including social withdrawal, apathy, anhedonia, alogia, and behavioral perseveration. Cognitive symptoms of schizophrenia involve disturbances in executive functions, working memory impairment, and inability to sustain attention.
The main psychopathological features of autism are severe deficiencies in social interaction and communication, and restricted and repetitive behaviors (11). Autism is one of three disorders collectively called autism spectrum disorders (ASDs), the other two being Asperger syndrome and Pervasive Developmental Disorder-Not Otherwise Specified (PDD-NOS). In contrast to autism, individuals with Asperger syndrome generally lack delays in cognitive development and communication, while PDD-NOS is diagnosed when the full set of criteria for autism or Asperger syndrome are not met (11). Overt symptoms of autism often begin by the age of six months, become established by two or three years, and tend to persist throughout life (11). The current prevalence of ASD is estimated to be approximately 0.5-1%, and this rate seems to increase by approximately 15% annually (12). It remains a matter of debate whether this is a genuine increase in disease incidence or related to changes in diagnostic practice (12).
In what follows, we first outline some of the major commonalities between schizophrenia and autism, followed by a summary of epidemiologic and laboratory evidence supporting a causal relationship between prenatal infection and both schizophrenia and autism. We focus on key neuroimmunological factors that seem to shape the “neuropsychiatric vulnerability” of infants born to mothers with intrauterine infection. We then propose and discuss a hypothetical model in which inflammatory processes may simultaneously account for both disorder-specific and shared pathological features of schizophrenia and autism.
Schizophrenia and autism: Separate entities, or pieces of the same puzzle?
Schizophrenia and autism seem to overlap at multiple levels. It is therefore not surprising that autism was initially believed to be an early manifestation of schizophrenia, and as such it was often referred to as “childhood psychosis” or “schizophrenic syndrome of childhood” (13,14). In 1971, ASD was finally separated conceptually from schizophrenia based on a delineation of symptomatic differences (including age of onset), family histories, and differential treatment responses in individuals with suspected adult schizophrenia versus infantile autism (13,14).
At the behavioral level, deficits in social interaction and cognition, disruption of emotional processing and sensorimotor gating, and impairments in executive functions, are psychopathological features commonly observed in both disorders (11,15). Moreover, at least a subset of individuals with autism frequently experience psychotic phases involving auditory and visual hallucinations, similarly to acutely ill patients with schizophrenia (16). Based on this, it has been suggested that individuals with autism are at increased risk of developing psychosis, and that the presence of neurodevelopmental deficits typically associated with autism may represent an alternative “entry-point” into a common final pathway of psychosis (16).
Schizophrenia and autism also appear to share multiple features at the brain morphological level. For example, structural and functional abnormalities in the cerebellum, insular cortex and fusiform gyrus are present in both disorders (15-18). Patients with schizophrenia and autism also display a similar pattern of deficient neuronal activation during a social cognition task, with both patient groups showingreduced activation in the right amygdala, fusiform gyrus, and ventrolateral prefrontal cortex (19). The impression of overlapping brain abnormalities between schizophrenia and autism has also been supported by a recent meta-analytic anatomical likelihood estimation consolidating multiple imaging datasets, revealing appreciable brain structural concordance between the two disorders, especially in the right parahippocampal gyrus, posterior cingulate, putamen, claustrum and left thalamus (15).
At the cellular level, one of the best established neuronal markers known to be altered in both schizophrenia and autism is Reelin (20). Since Reelin has crucial neurodevelopmental functions such as providing a detachment signal for migrating cells, it has been suggested that reduced Reelin signaling may contribute the early neurodevelopmental pathology involved in schizophrenia and autism (20).
It appears that schizophrenia and autism also share specific abnormalities at the neurochemical level, which seem to relate primarily to abnormalities in the serotonergic system. For example, similar to a subset of schizophrenia patients, some individuals with autism display abnormal serotonin synthesis capacity and serotonin receptor dysfunctions (21,22).
Finally, schizophrenia and autism seem to share several genetic and environmental risk factors 15), with prenatal exposure to infection being one of them. The role of prenatal infection in alerting neurodevelopmental trajectories relevant to schizophrenia and autism is discussed in the subsequent section.
Maternal infection and the “cytokine hypothesis” of neurodevelopmental disorders
Epidemiologic studies suggest that the risk of schizophrenia is increased followingh prenatal maternal viral infections such as influenza, rubella, measles, and polio, as well as infections with bacterial pathogens and genital and/or reproductive infections (4). Thus, the association between maternal infection during pregnancy and schizophrenia in the offspring does not appear to be pathogen-specific. Similarly, prenatal/perinatal exposure to numerous pathogens including rubella, measles, and cytomegalovirus has been implicated in the etiology of autism (5,23), suggesting that the infection-associated risk of autism may also not be pathogen-specific. This notion finds support from a recent nationwide study in Denmark on more than 20,000 children born to mothers who required hospitalization because of infection during pregnancy (24). Hospital admission due to maternal exposure to various viral or bacterial infections significantly increased the offspring's risk of ASD, an effect that seemed unrelated to hospitalization per se (24).
These findings suggest that factors common to the immune response to a variety of pathogens are candidate mediators of the association between prenatal/perinatal infection and neurodevelopmental disorders. One hypothesis suggests that enhanced expression of pro-inflammatory cytokines and other mediators of inflammation in the maternal, fetal and neonatal compartments may interfere with brain development, thereby increasing the risk for long-term brain dysfunction later in life (25,26). Cytokines are low-molecular weight proteins secreted by immune cells and other cell types in response to a number of environmental stimuli, particularly infections (27). They have wide-ranging roles in the innate and adaptive immune systems, where they help regulate the recruitment and activation of lymphocytes as well as immune cell differentiation and homeostasis. In addition, some cytokines possess direct effector mechanisms, including induction of cell apoptosis and inhibittion of protein synthesis. Furthermore, pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α are essential to the inflammatory response by mediating febrile reactions, activating phagocytotic cells such as macrophages or dendritic cells, facilitating vascular permeability, and promoting the release of plasma-derived inflammatory mediators such as bradykinin and components of the complement system (27). In the adult central nervous system (CNS), cytokines and their receptors are expressed by glial and neuronal cell types and are important mediators of the neuro-immune crosstalk in various brain and behavioral functions (28). In addition, many cytokines and cytokine receptors are constitutively expressed during fetal brain development (29,30), implying essential roles for these molecules in the regulation and modulation of normal brain development. Indeed, cytokines in general, and pro-inflammatory cytokines in particular, appear to have appreciable effects on various neurodevelopmental processes, neuronal plasticity and neuronal survival (31). It can therefore be expected that abnormal levels of these molecules during critical periods of early brain development may adversely affect neurodevelopmental processes, thereby contributing to an increased susceptibility for complex brain disorders of developmental origin such as schizophrenia and autism.
Two major lines of evidence support the hypothesis that cytokine-mediated aberrance of early brain development might be relevant for autism and schizophrenia. First, epidemiological studies demonstrate a significant association between high maternal levels of the pro-inflammatory cytokines TNF-α (32) and IL-8 (33) during pregnancy and elevated risk of schizophrenia spectrum disorder (SSD) in the offspring. Second, experimental animal work shows that prenatal exposure to pro-inflammatory agents in the absence of viral or bacterial infection is sufficient to cause long-lasting brain and behavioral dysfunction relevant to schizophrenia and autism (26,34,35). Subsequent to the establishment of a mouse model of prenatal human influenza infection, which mimics various brain and behavioral abnormalities with potential relevance to both schizophrenia and autism, several research groups have used cytokine-releasing agents or specific pro-inflammatory cytokines when modeling the neurodevelopmental consequences of prenatal immune challenge (26,34,35). This includes maternal exposure to the bacterial endotoxin, lipopolysaccharide (LPS), or the synthetic double-stranded RNA analogue, polyriboinosinic-polyribocytidilic acid (PolyI:C). Both immunogens trigger the production and release of pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α (36). PolyI:C is also a potent inducer of the type I interferons IFN-α and IFN-β (36). In essence, LPS administration mimics infection with gram-negative bacteria, while administration of PolyI:C mimics the acute-phase inflammatory response to viral infection (36). Importantly, maternal exposure to LPS or PolyI:C during pregnancy is capable of enhancing pro-inflammatory cytokine levels in the three maternal-fetal compartments, namely the placenta, the amniotic fluid and the fetus, including the fetal brain (37,38). The efficacy of prenatal exposure to cytokine-releasing agents to induce fetal brain inflammation, together with its long-term impact on brain and behavioral abnormalities relevant to schizophrenia and autism, underscores the essential role of prenatal cytokine-associated inflammation in mediating the effects of maternal infection on the offspring. The concept is further supported by the findings that blocking the actions of the pro-inflammatory cytokine IL-6 in the pregnant maternal host by genetic or pharmacological interventions prevents the long-term brain and behavioral consequences of prenatal PolyI:C treatment (39); and that over-expression of the anti-inflammatory cytokine IL-10 prevents the emergence of multiple behavioral and pharmacological abnormalities typically seen following prenatal PolyI:C-induced immune challenge (40).
Schizophrenia and autism: Shared pathogenesis via prenatal inflammation?
In view of the emerging role of prenatal infection and associated inflammatory processes in the etiology of both schizophrenia and autism, we speculate that the pathogenesis of both disorders might have prenatal inflammation as a common pathway. In fact, this proposition might help explain why the two disorders share several phenotypic characteristics as discussed above. Numerous findings from pertinent rodent models suggest that this is indeed the case. Prenatal immune challenge in mice and rats leads to deficiency in social interaction and sensorimotor gating, the latter being assessed using the paradigm of prepulse inhibition (PPI) of the acoustic startle reflex (26,34,35). Furthermore, offspring born to animals who sustained an immune challenge during gestation display deficits in working memory and abnormal executive functions such impaired discrimination reversal learning, as well as deficits in spatial and non-spatial information processing (26,34,35). These prenatal inflammation-induced behavioral manifestations are accompanied by neuroanatomical and neurochemical abnormalities seen in both schizophrenia and autism, including neuromorphological alterations in cerebellum, basal ganglia and amygdala, deficient Reelin expression in hippocampus and prefrontal cortex, and alterations in serotonin synthesis and metabolism (26,34,35).
It needs to be emphasized that despite the apparent similarities between schizophrenic and autistic disease, a plethora of distinctive neuropathological and psychopathological characteristics allow to distinguish the two disorders. One example is that autism has generally been associated with excessive brain growth (macrocephaly) involving thickening of cortex, increased gray and white matter, and lack of gray matter loss (41), while schizophrenia seems to be associated with microcephaly involving thinner cortical areas, reduced gray and white matter, and accelerated gray matter loss (18).
Although it is beyond the scope of this article to summarize all characteristics unique to either schizophrenia or autism, we would like to emphasize that contrary to our hypothesis, it has recently been postulated that schizophrenia and autism are inter-related but diametrical both in terms of phenotypic expression as well as underlying etiopathology (42). Hence, although the two disorders may be characterized by dysfunctions in the same set of traits, these dysfunctions and the resulting phenotypes may be diametrically opposite (42). In view of the two apparently mutually exclusive positions that schizophrenia and autism are either related (15,16) or remote (42), our proposal that the two are fundamentally linked via prenatal inflammation may seem counter-intuitive. However, as discussed in more detail below, we suggest that the nature and developmental character of perinatal inflammation can provide a satisfactory explanation for the induction of phenotypic features in schizophrenia and autism that are either shared by both, or unique to either one.
A model of inflammation leading to both shared and disorder-specific features of schizophrenia and autism
Under normal conditions, inflammation is controlled by various homeostatic processes that limit or counteract inflammation once it has been induced by an environmental stimulus, e.g. infection (43). Such control mechanisms ensure that inflammatory processes efficiently remove invading pathogens and contribute to tissue repair and wound healing without placing non-infected, healthy and unwounded tissue in jeopardy. Dysfunction of such surveillance mechanisms may lead to chronic inflammation, known from numerous pathological conditions such as rheumatoid arthritis, atherosclerosis, obesity, and diabetes (43).
Immunomodulatory control systems exist both in the periphery and the CNS (43,44). In the CNS, microglia and astrocytes are the major immunocompetent cells, which drive both the induction as well as limitation of inflammatory processes (45). This is achieved through the synthesis of pro- and anti-inflammatory cytokines, up- or down-regulation of various cell surface receptors such as pathogen recognition receptors, cytokine receptors, and numerous receptors crucial for antigen presentation. Microglia appear to play crucial roles in both neuronal protection and pathology, and are often referred to as a “double-edged sword” (3,45,46). On the one hand, they secrete neurotrophic factors pivotal for cellular repair, and recruit immune cells into the brain for clearance of infection or cellular debris. On the other hand, chronic or exaggerated microglial activation has been linked to multiple neuroinflammatory and neurodegenerative diseases, including Parkinson's disease, Multiple Sclerosis, Alzheimer's, and Huntington's disease (46).
We propose the following theoretical model in which perinatal inflammation may simultaneously account for both shared and unique pathological features of schizophrenia and autism (Figure 1):
Figure 1. A model of inflammation leading to both shared and disorder-specific features of schizophrenia and autism.
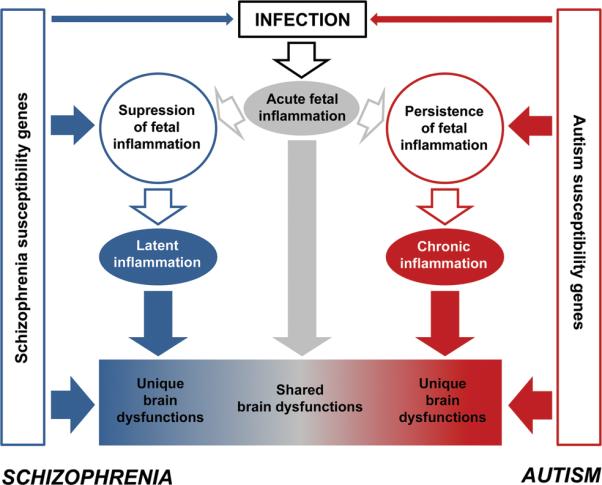
Maternal infection during critical stages of pregnancy leads to acute fetal inflammation. The consequences of acute fetal neuroinflammation on early neurodevelopmental consequences may facilitate the development of psychopathological and neuropathological phenotypes shared between schizophrenia and autism. Upon the initiation of acute fetal inflammation, the maternal and/or fetal system may then either be driven into persistent inflammation, or alternatively may gain control over the ongoing inflammatory process. The genetic background may critically influence the transition of acute fetal inflammation into either persistence or suppression of inflammation. The occurrence of persistent inflammation may be related to a failure in the negative feedback control of inflammation, and may eventually result in chronic inflammation persisting into the perinatal period and beyond. By contrast, mounting sufficient anti-inflammatory and/or immunosuppressive activity in the maternal and/or fetal systems may counteract the acute fetal inflammatory response on the one hand, but on the other hand, may prime the early life organisms to latent immune abnormalities persisting into the postnatal period. Latent inflammation may then be unmasked in postnatal life by exposure to specific environmental stimuli such as stress or immune re-exposure. Persistent inflammation may be more relevant for the etiopathogenesis of autism, and thus may contribute to phenotypic abnormalities specifically seen in autism. On the other hand, latent immune abnormalities may be essential to the pathogenesis of schizophrenia-specific brain abnormalities. The genetic background further contributes to the emergence of unique brain dysfunctions independently of or in interaction with the outlined inflammatory pathways.
Acute inflammation and characteristics shared by schizophrenia and autism
In line with previous theoretical accounts (25,26), maternal infection during critical stages of pregnancy leads to acute cytokine-associated inflammation in the fetal system, including the fetal brain. Acute neuroinflammation during early fetal brain development may negatively affect ongoing neurodevelopmental processes such as neuronal/glial cell differentiation, proliferation, migration, and survival. We propose that acute fetal neuroinflammation, together with its effects on early neurodevelopmental processes, may facilitate the development of psychopathological and neuropathological phenotypes shared by schizophrenia and autism. Hence, acute fetal neuroinflammation may be responsible for aberrant development of neural substrates of social behavior and cognition, emotional processing, sensorimotor gating, and certain executive functions, the disruption of which has been similarly implicated in schizophrenia and autism.
Latent versus persisting inflammation and characteristics unique to schizophrenia or autism
Upon the initiation of acute prenatal inflammation, the maternal and/or fetal system may then either be driven into persistent inflammation or gain control over the inflammatory processes. The former may be related to a failure in the homeostatic control of inflammation, and may eventually result in chronic inflammation persisting into late fetal and neonatal periods. By contrast, mounting sufficient anti-inflammatory and/or immunosuppressive activity in the maternal and/or fetal system may dampen the acute fetal inflammatory response. Still, the developing organism might sustain latent immune abnormalities that may not become relevant until exposure to specific environmental stimuli over the postnatal life span. According to our model, the induction of persistent inflammation may be more relevant for the etiopathogenesis of autism by contributing to phenotypic abnormalities specifically seen in this disorder. By contrast, latent immune inflammation may be essential to the pathogenesis of schizophrenia-specific brain and behavioral abnormalities.
Several lines of evidence support our suggested model. First, autism but not schizophrenia seems to be characterized by relatively severe chronic inflammation, both in the periphery and in the CNS, although altered immune regulations have been implicated in both disorders. For example, a nearly 50-fold increase in TNF-α level was found in the cerebrospinal fluid (CSF) of autistic children (47), and severe inflammation is present in the brains of autistic patients from a broad range of ages (48), characterized by prominent activation of microglia and astroglia cells and enhanced pro-inflammatory cytokine and chemokine expression in multiple brain areas, including the cerebellum, cortex, and white matter tracts. Several recent studies corroborate the presence of ongoing systemic inflammation and neuroinflammation in autistic individuals (49,50).
Experimental work in rodents provides further evidence that prenatal immune challenge can lead to long-lasting inflammatory changes. Of special interest are the findings of enhanced microglial densities and elevation of peripheral pro-inflammatory cytokine levels in offspring of mothers chronically exposed to LPS during pregnancy (51,52). Such prenatal LPS-induced inflammatory changes can persist even until adulthood (51,52), suggesting that immune challenge early in life can permanently alter postnatal immune functions. This idea is further supported by the finding that sub-chronic maternal treatment with IL-2 on gestational days 12 to 17 leads to elevated B and T cell counts in response to antigenic stimulation in the juvenile offspring (53). It is important to note that postnatal immune changes are primarily seen following prenatal exposure to relatively severe forms of maternal immune challenge, i.e., following chronic (51,52) or sub-chronic (53) maternal inflammation. By contrast, acute prenatal inflammation (e.g., by single PolyI:C administration) appears to be largely devoid of long-term (neuro)inflammatory effects in the adolescent or adult offspring (40,54).
In contrast to autism, chronic (neuro)inflammation seems less prominent in schizophrenia. Indeed, even though peripheral pro-inflammatory markers such as IL-6 appear to be elevated in at least a subgroup of schizophrenic patients, the reported increases are relatively modest (55). Furthermore, despite considerable research efforts, imaging and post-mortem immunohistochemical studies provide very little evidence for over-activation of microglia and astrocytes in the brains of schizophrenic patients (56). These human data are consistent with the experimental results discussed above suggesting that schizophrenia-related brain and behavioral abnormalities induced by acute prenatal immune challenge can occur in the absence of overt neuroinflammation and/or astrogliosis (40,54).
What switches between latent versus persistent inflammation trajectories?
The induction of latent versus persistent neuroinflammatory trajectories subsequent to acute prenatal inflammation is likely to be controlled and modified by multiple mechanisms. First and foremost is the organism's capability to mount appropriate anti-inflammatory, immunomodulatory, and/or immunosuppressive responses to acute inflammation, which might be smaller in individuals with autism as in those with schizophrenia. Indeed, autism appears to be associated with cellular and molecular processes that facilitate the persistence of inflammation. For example, autistic patients have reduced serum levels of the immunosuppressive cytokine TGF-β1 (58) and increased serum levels of the high mobility group box 1 (HMGB1) protein (59). HMGB1 is a highly conserved, ubiquitous protein that activates inflammation-associated immune responses (59). The persistence of neuroinflammation in autism may also be related to alterations in glutamate-immune interactions (60). Glutamate is a potent activator of microglia cells (45,61), and the presence of increased CNS glutamate levels (21) together with up-regulation of specific glutamate receptor subtypes (62) on microglia cells may facilitate the persistence of neuroinflammation subsequent to the induction of acute fetal inflammation (Figure 1, right).
In schizophrenia, on the other hand, enhanced anti-inflammatory and/or immunosuppressive actions appear to concur with (modest) pro-inflammatory alterations. For example, multiple molecules with potent anti-inflammatory and immunosuppressive properties seem to be increased in subjects diagnosed with schizophrenia, including IL-10, TGF-β, soluble IL-1 receptor antagonist (sIL-1RA), and soluble TNF receptor (sTNF-R) (55,63,64). A common feature of these molecules is that they can effectively antagonize the biological activities and/or production of multiple pro-inflammatory molecules, thereby regulating inflammation in both acute and chronic conditions (27). The presence of altered peripheral levels of IL-10 and related cytokines has often been explained by an imbalance between T help (65). Interestingly, there seems to be a genetic basis for the reported alterations in peripheral anti-inflammatory and immunosuppressive cytokine levels in schizophrenic patients. For example, IL-10 promoter variants that lead to high IL-10 production are more common among individuals with schizophrenia (66). Likewise, converging evidence suggests a role for the interleukin-1 gene complex (involving sIL-1RA) in schizophrenia (67). Taken together, enhanced potential to mount anti-inflammatory, immunomodulatory, and/or immunosuppressive responses may explain at least in part why acute fetal inflammation may lead only to minimal or absent chronic neuroinflammation in the postnatal period (Figure 1, left).
A second mechanism that might switch between the autistic and schizophrenic phenotypes is sensitization for a second hit. In our model, this concept would be accounted for by the possibility that exposure to acute fetal inflammation may induce latent immune abnormalities that can be unmasked by exposure to certain environmental stimuli throughout postnatal life. Such exposure to additional immunological or non-immunological environmental factors, together with maturational processes occurring during peri-adolescent development, may thus be needed to disclose the latent impact of acute fetal neuroinflammation on schizophrenia. This idea of multiple hits with either sensitizing or preconditioning effects is also central to several theories of prenatal immune priming which have been put forward by others in the context of peripheral immunity, CNS inflammation, perinatal brain damage, retinopathy and various forms of learning and memory (68-71).
As reviewed in detail elsewhere (3), numerous experimental studies in rodents show that immunological exposure in early (prenatal or neonatal) life can cause the organism to respond differently (and often more vigorously) to subsequent immunological or non-immunological challenges. Such early immune priming may provide an environmental basis for the findings thatproduction of pro-inflammatory cytokines such as IL-6 and TNF-α following stimulation with phytohemagglutinin and LPS is significantly higher in schizophrenic patients relative to controls (55,72). In keeping with these ideas, we propose that priming of immune dysfunctions, rather than presence of chronic inflammation, may be relevant for infection-associated pathogenetic mechanisms in schizophrenia. In particular, we suggest that exposure to additional postnatal environmental factors such as stress or immune re-challenge in high-risk individuals (particularly in those with a history of prenatal infection) may lead to altered inflammatory responses, which in turn may further negatively affect brain maturation, thereby facilitating transition into chronic schizophrenic disease. Consistent with this suggestion, exposure to environmental insults such as stress and drugs of abuse, which are believed to facilitate or even trigger the onset of full-blown psychosis in individuals at high-risk for schizophrenia (73), are also known to exert direct influences on the immune system (74,75). Our model would thus predict that latent inflammation may also be relevant for the delayed onset of full-blown schizophrenic disease. Against this background, it is highly interesting to note that the addition of anti-inflammatory treatment to standard antipsychotic medication for schizophrenia seems to be superior to antipsychotic treatment alone especially when such anti-inflammatory therapy is initiated in the early phases of the disease (76).
However, it needs to be pointed out that sensitized inflammatory responses resulting from early immune priming may seem paradoxical in view of enhanced anti-inflammatory/immunosuppressive potency in schizophrenia discussed above. Indeed, enhanced anti-inflammatory/immunosuppressive actions may be expected to attenuate rather than facilitate the development of inflammatory responses to distinct environmental stimuli such as immunological challenge or stress. We must admit that we do not have a parsimonious explanation for this at the present stage. However, we would like to emphasize enhanced anti-inflammatory and immunosuppressive actions may increase the sensitivity to infections (77), and this may act in concert with the presence of latent immune abnormalities (induced by perinatal immune priming) in the development of altered inflammatory responses in psychosis-prone subjects.
In contrast to the delayed onset of schizophrenia, overt symptoms of autism often begin as early as by the age of six months and become established by age two or three years (11,12). Notably, this early disease onset would coincide with the presence of persistent neuroinflammation in the neonatal period, so that enhanced expression of pro-inflammatory markers may exert a direct influence on postnatal brain functions relevant to the expression of autistic behavior. In support of this, several cytokines seem to modulate processes of learning and memory, anxiety-related behaviors, and social interaction (3,78,79).
Third, abnormal inflammatory processes are likely to have a differential impact on brain structures and functions depending on the developmental/maturational stage at which they occur. Age-dependent effects may further modulate the impact of inflammation on pathological features unique to either schizophrenia or autism. For example, in contrast to other brain structures, the development of the cerebellum reaches peak levels only in late fetal and neonatal periods (80). Therefore, we would expect persistent neuroinflammation to a have more prominent impact on cerebellar development compared to acute fetal neuroinflammation at earlier time points in (81). The fact that cerebellar pathology seems to be more pronounced in autism than in schizophrenia (81,82) lends further support to our model of persistent neuroinflammation in autism versus latent neuroinflammation in schizophrenia.
Fourth, the presence of distinct genetic factors may play additional critical roles in regulating the switch between latent versus persistent inflammation trajectories, and may therefore critically determine the impact of inflammation on pathological features unique to either schizophrenia or autism (Figure 1). Indeed, multiple genetic factors are likely to interact with perinatal and/or postnatal inflammatory processes in shaping the shared or disorder-specific pathogenesis of schizophrenia and autism. Such genetic components seem highly essential since both autism and schizophrenia exhibit strong patterns of heritability (83,84).
Conclusions
Prenatal infection and inflammatory responses appear to play a significant role in the etiology of both schizophrenia and autism. This suggests that the pathogenesis of schizophrenia and autism may be fundamentally linked via prenatal inflammation. According to our proposed model, acute neuroinflammation during early fetal development may be relevant for the induction of psychopathological and neuropathological phenotypes shared by schizophrenia and autism, while subsequent latent versus persistent inflammation may lead to phenotypic characteristics of schizophrenia versus autism, respectively. The precise mechanisms whereby latent or persistent inflammation negatively affects neurobiological and neurochemical abnormalities relevant to schizophrenia and/or autism are currently the subject of extensive experimental studies. The further elucidation of such mechanisms might lead to the establishment of novel immunomodulatory interventions that could help prevent abnormal brain development and long-term mental illness in subjects with prenatal infectious/inflammatory histories. The identification of preventive strategies is particularly important for chronic mental illnesses such as schizophrenia and autism with little hope for complete functional recovery once the disorder has developed. We hope that the model presented here will help achieving this goal.
Acknowledgments
Statements of financial support: The studies performed at the Laboratory of Behavioural Neurobiology, ETH Zurich, were supported by the Swiss National Science Foundation (grant 3100AO-100309 and grant 3100AO-116719) and ETH Zurich (grant – 11 07/03). J.F. received additional support from a 2009 NARSAD Distinguished Investigator Award. O.D. is supported by the European Commission (NEOBRAIN, LSHM-CT-2006-036534; NEUROBID, HEALTH-F2-2009-241778), the NIH (1R21EY019253-01), and the Richard Saltonstall Charitable Foundation.
Abbreviations
- ASD
Autism spectrum disorders
- CNS
Central nervous system
- LPS
Lipopolysaccharide
- PolyI:C
Polyriboinosinic-polyribocytidilic acid
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: All authors declare that they have no conflicts of interest to disclose.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Rees S, Inder T. Fetal and neonatal origins of altered brain development. Early Hum Dev. 2005;81:753–761. doi: 10.1016/j.earlhumdev.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libbey JE, Sweeten TL, McMahon WM, Fujinami RS. Autistic disorder and viral infections. J Neurovirol. 2005;11:1–10. doi: 10.1080/13550280590900553. [DOI] [PubMed] [Google Scholar]
- 6.Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Dammann O, Leviton A. Role of the fetus in perinatal infection and neonatal brain damage. Curr Opin Pediatr. 2000;12:99–104. doi: 10.1097/00008480-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Shatrov JG, Birch SC, Lam LT, Quinlivan JA, McIntyre S, Mendz GL. Chorioamnionitis and cerebral palsy: a meta-analysis. Obstet Gynecol. 2010;116:387–392. doi: 10.1097/AOG.0b013e3181e90046. [DOI] [PubMed] [Google Scholar]
- 9.Leviton A, Allred EN, Kuban KC, Hecht JL, Onderdonk AB, O'shea TM, Paneth N. Microbiologic and histologic characteristics of the extremely preterm infant's placenta predict white matter damage and later cerebral palsy. The ELGAN study. Pediatr Res. 2010;67:95–101. doi: 10.1203/PDR.0b013e3181bf5fab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110:1–23. doi: 10.1016/j.schres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Rapin I, Tuchman RF. Autism: definition, neurobiology, screening, diagnosis. Pediatr Clin North Am. 2008;55:1129–1146. doi: 10.1016/j.pcl.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res. 2009;65:591–598. doi: 10.1203/PDR.0b013e31819e7203. [DOI] [PubMed] [Google Scholar]
- 13.Kolvin I. Studies in the childhood psychoses. I. Diagnostic criteria and classification. Br J Psychiatry. 1971;118:381–384. doi: 10.1192/bjp.118.545.381. [DOI] [PubMed] [Google Scholar]
- 14.Kolvin I, Ounsted C, Humphrey M, McNay A. Studies in the childhood psychoses. II. The phenomenology of childhood psychoses. Br J Psychiatry. 1971;118:385–395. doi: 10.1192/bjp.118.545.385. [DOI] [PubMed] [Google Scholar]
- 15.Cheung C, Yu K, Fung G, Leung M, Wong C, Li Q, Sham P, Chua S, McAlonan G. Autistic disorders and schizophrenia: related or remote? An anatomical likelihood estimation. PLoS ONE. 2010;5:e12233. doi: 10.1371/journal.pone.0012233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toal F, Bloemen OJ, Deeley Q, Tunstall N, Daly EM, Page L, Brammer MJ, Murphy KC, Murphy DG. Psychosis and autism: magnetic resonance imaging study of brain anatomy. Br J Psychiatry. 2009;194:418–425. doi: 10.1192/bjp.bp.107.049007. [DOI] [PubMed] [Google Scholar]
- 17.McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS, Yip L, Murphy DG, Chua SE. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain. 2005;128:268–276. doi: 10.1093/brain/awh332. [DOI] [PubMed] [Google Scholar]
- 18.Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinkham AE, Hopfinger JB, Pelphrey KA, Piven J, Penn DL. Neural bases for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr Res. 2008;99:164–175. doi: 10.1016/j.schres.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fatemi SH. Reelin glycoprotein: structure, biology and roles in health and disease. Mol Psychiatry. 2005;10:251–257. doi: 10.1038/sj.mp.4001613. [DOI] [PubMed] [Google Scholar]
- 21.Lam KS, Aman MG, Arnold LE. Neurochemical correlates of autistic disorder: a review of the literature. Res Dev Disabil. 2006;27:254–289. doi: 10.1016/j.ridd.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Abi-Dargham A, Laruelle M, Aghajanian GK, Charney D, Krystal J. The role of serotonin in the pathophysiology and treatment of schizophrenia. J Neuropsychiatry Clin Neurosci. 1997;9:1–17. doi: 10.1176/jnp.9.1.1. [DOI] [PubMed] [Google Scholar]
- 23.Chess S. Autism in children with congenital rubella. J Autism Child Schizophr. 1971;1:33–47. doi: 10.1007/BF01537741. [DOI] [PubMed] [Google Scholar]
- 24.Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- 25.Gilmore JH, Jarskog LF. Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res. 1997;24:365–367. doi: 10.1016/s0920-9964(96)00123-5. [DOI] [PubMed] [Google Scholar]
- 26.Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009;35:959–972. doi: 10.1093/schbul/sbn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curfs JH, Meis JF, Hoogkamp-Korstanje JA. A primer on cytokines: sources, receptors, effects, and inducers. Clin Microbiol Rev. 1997;10:742–780. doi: 10.1128/cmr.10.4.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998;9:259–275. doi: 10.1016/s1359-6101(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 29.Burns TM, Clough JA, Klein RM, Wood GW, Berman NE. Developmental regulation of cytokine expression in the mouse brain. Growth Factors. 1993;9:253–258. doi: 10.3109/08977199308991585. [DOI] [PubMed] [Google Scholar]
- 30.Mehler MF, Kessler JA. Hematolymphopoietic and inflammatory cytokines in neural development. Trends Neurosci. 1997;20:357–365. doi: 10.1016/s0166-2236(96)01045-4. [DOI] [PubMed] [Google Scholar]
- 31.Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- 32.Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun. 2001;15:411–420. doi: 10.1006/brbi.2001.0644. [DOI] [PubMed] [Google Scholar]
- 33.Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004;161:889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- 34.Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Kimura M, Toth LA, Agostini H, Cady AB, Majde JA, Krueger JM. Comparison of acute phase responses induced in rabbits by lipopolysaccharide and double-stranded RNA. Am J Physiol. 1994;267:R1596–R1605. doi: 10.1152/ajpregu.1994.267.6.R1596. [DOI] [PubMed] [Google Scholar]
- 37.Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry. 2006;11:47–55. doi: 10.1038/sj.mp.4001748. [DOI] [PubMed] [Google Scholar]
- 39.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- 41.Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 42.Crespi B, Badcock C. Psychosis and autism as diametrical disorders of the social brain. Behav Brain Sci. 2008;31:241–261. doi: 10.1017/S0140525X08004214. [DOI] [PubMed] [Google Scholar]
- 43.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 44.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 46.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 47.Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36:361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 49.Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, Altaye M, Wills-Karp M. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2006;172:198–205. doi: 10.1016/j.jneuroim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 50.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2010;25:40–45. doi: 10.1016/j.bbi.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borrell J, Vela JM, Arévalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- 52.Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry. 2010;15:372–383. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- 53.Ponzio NM, Servatius R, Beck K, Marzouk A, Kreider T. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann N Y Acad Sci. 2007;1107:118–128. doi: 10.1196/annals.1381.013. [DOI] [PubMed] [Google Scholar]
- 54.Nyffeler M, Meyer U, Yee BK, Feldon J, Knuesel I. Maternal immune activation during pregnancy increases limbic GABAA receptor immunoreactivity in the adult offspring: implications for schizophrenia. Neuroscience. 2006;143:51–62. doi: 10.1016/j.neuroscience.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 55.Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. 2008;63:801–808. doi: 10.1016/j.biopsych.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 56.Bernstein HG, Steiner J, Bogerts B. Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother. 2009;9:1059–1071. doi: 10.1586/ern.09.59. [DOI] [PubMed] [Google Scholar]
- 57.Okada K, Hashimoto K, Iwata Y, Nakamura K, Tsujii M, Tsuchiya KJ, Sekine Y, Suda S, Suzuki K, Sugihara G, Matsuzaki H, Sugiyama T, Kawai M, Minabe Y, Takei N, Mori N. Decreased serum levels of transforming growth factor-beta1 in patients with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:187–190. doi: 10.1016/j.pnpbp.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 58.Emanuele E, Boso M, Brondino N, Pietra S, Barale F, Ucelli di Nemi S, Politi P. Increased serum levels of high mobility group box 1 protein in patients with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:681–683. doi: 10.1016/j.pnpbp.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 59.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 60.Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr Med Chem. 2009;16:157–170. doi: 10.2174/092986709787002745. [DOI] [PubMed] [Google Scholar]
- 61.Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 62.Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57:1618–1628. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- 63.Maes M, Bocchio Chiavetto L, Bignotti S, Battisa Tura GJ, Pioli R, Boin F, Kenis G, Bosmans E, de Jongh R, Altamura CA. Increased serum interleukin-8 and interleukin-10 in schizophrenic patients resistant to treatment with neuroleptics and the stimulatory effects of clozapine on serum leukemia inhibitory factor receptor. Schizophr Res. 2002;54:281–291. doi: 10.1016/s0920-9964(00)00094-3. [DOI] [PubMed] [Google Scholar]
- 64.Numata S, Ueno S, Iga J, Yamauchi K, Hongwei S, Hashimoto R, Takeda M, Kunugi H, Itakura M, Ohmori T. TGFBR2 gene expression and genetic association with schizophrenia. J Psychiatr Res. 2008;42:425–432. doi: 10.1016/j.jpsychires.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 65.Müller N, Schwarz MJ. Immune System and Schizophrenia. Curr Immunol Rev. 2010;6:213–220. doi: 10.2174/157339510791823673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bocchio Chiavetto L, Boin F, Zanardini R, Popoli M, Michelato A, Bignotti S, Tura GB, Gennarelli M. Association between promoter polymorphic haplotypes of interleukin-10 gene and schizophrenia. Biol Psychiatry. 2002;51:480–484. doi: 10.1016/s0006-3223(01)01324-5. [DOI] [PubMed] [Google Scholar]
- 67.Xu M, He L. Convergent evidence shows a positive association of interleukin-1 gene complex locus with susceptibility to schizophrenia in the Caucasian population. Schizophr Res. 2010;120:131–142. doi: 10.1016/j.schres.2010.02.1031. [DOI] [PubMed] [Google Scholar]
- 68.Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rousset CI, Kassem J, Olivier P, Chalon S, Gressens P, Saliba E. Antenatal bacterial endotoxin sensitizes the immature rat brain to postnatal excitotoxic injury. J Neuropathol Exp Neurol. 2008;67:994–1000. doi: 10.1097/NEN.0b013e31818894a1. [DOI] [PubMed] [Google Scholar]
- 70.Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 71.Dammann O, Brinkhaus MJ, Bartels DB, Dördelmann M, Dressler F, Kerk J, Dörk T, Dammann CE. Immaturity, perinatal inflammation, and retinopathy of prematurity: a multi-hit hypothesis. Early Hum Dev. 2009;85:325–329. doi: 10.1016/j.earlhumdev.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 72.Na KS, Kim YK. Monocytic, Th1 and th2 cytokine alterations in the pathophysiology of schizophrenia. Neuropsychobiology. 2007;56:55–63. doi: 10.1159/000111535. [DOI] [PubMed] [Google Scholar]
- 73.Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2010 doi: 10.1016/j.pneurobio.2010.09.003. (in press, doi:10.1016/j.pneurobio.2010.09.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harbuz MS, Chover-Gonzalez AJ, Jessop DS. Hypothalamo-pituitary-adrenal axis and chronic immune activation. Ann N Y Acad Sci. 2003;992:99–106. doi: 10.1111/j.1749-6632.2003.tb03141.x. [DOI] [PubMed] [Google Scholar]
- 75.Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P. Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology. 2010;215:598–605. doi: 10.1016/j.imbio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Müller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M, Möller HJ, Klauss V, Schwarz MJ, Riedel M. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res. 2010;121:118–124. doi: 10.1016/j.schres.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 77.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–3411. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 78.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev. 2009;33:355–366. doi: 10.1016/j.neubiorev.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 80.Ten Donkelaar HJ, Lammens M. Development of the human cerebellum and its disorders. Clin Perinatol. 2009;36:513–530. doi: 10.1016/j.clp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 81.Schmahmann JD, Weilburg JB, Sherman JC. The neuropsychiatry of the cerebellum - insights from the clinic. Cerebellum. 2007;6:254–267. doi: 10.1080/14734220701490995. [DOI] [PubMed] [Google Scholar]
- 82.Andreasen NC, Pierson R. The role of the cerebellum in schizophrenia. Biol Psychiatry. 2008;64:81–88. doi: 10.1016/j.biopsych.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 84.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]