

Abstract
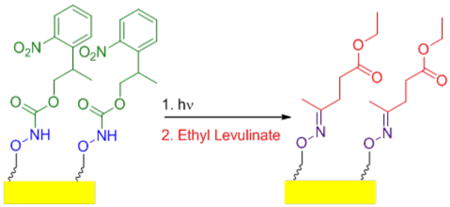
A photo-caged aminooxyalkanethiol synthesized in 7 steps and 15% overall yield was used to form a self-assembled monolayer (SAM). Photo-deprotection on the surface was confirmed by FT-IR spectroscopy and contact angle goniometry. Conjugation of a small molecule ketone, ethyl levulinate, further confirmed the presence of aminooxy groups on the surface.
Introduction
Improved efficiency of biomarker discovery has resulted in a demand for high-throughput protein screening methods to identify disease at early onset, as well as to monitor the progression of treatments through biosignature detection.1-4 Self-assembled monolayers (SAMs) are a viable option for the fabrication of bioactive arrays required for high-throughput screening. This is because SAMs present well defined surfaces for protein immobilization and subsequent investigation of biomolecules at biologically relevant concentrations.5 The ability to pattern bioactive arrays of proteins at the micron or nanoscale has been comprehensively examined.6, 7 Surface immobilization at this scale allows for detection of multiple biomarkers on the same surface, resulting in a more resolved biosignature and a more robust protein array.7, 8 As such, we sought to provide a method for fabrication of a photo-activated surface for site-specific conjugation of molecules of biological interest via oxime bonds. In particular we report the synthesis of a 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) protected aminooxy alkane thiol with subsequent conjugation of ethyl levulinate to photo-deprotected surfaces (Scheme 1).
Scheme 1.
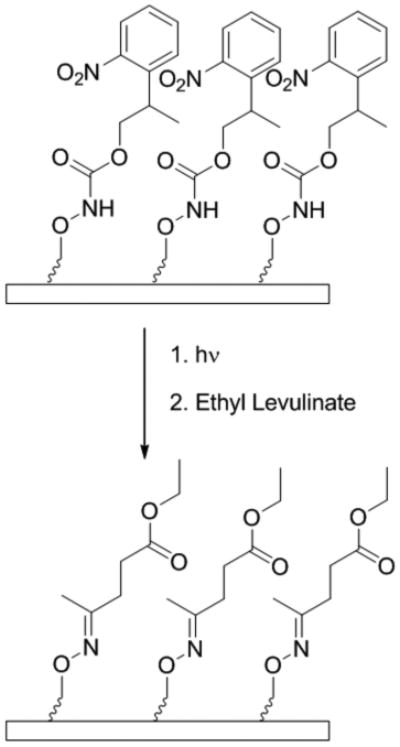
Deprotection and conjugation of a NPPOC aminooxy SAM
Several methods of immobilization have been utilized to attach biomolecules to surfaces. One method involves nonspecific adsorption onto a surface; however this often results in major conformational changes.9-11 Alternatively, biomolecules may be covalently linked to the surface. Early routes included carbodiimide coupling of the free amine groups of the protein to a surface bearing terminal carboxylic acid moietiesor reversible coupling with surface carbonyl groups via imine chemistry.12, 13 These methods however, are not site-selective and often result in reduction of protein bioactivity.13
To achieve site-specific coupling, free cysteines have been targeted. However, while effective, free cysteines are rare in natural proteins and additional steps of adding a surface reactive amino acid through techniques such as mutagenesis may be required.14,15 Alternatively, Huisgen cycloaddition or “click” chemistry has been used by modifying the biomolecule to contain an azide or alkyne moiety with an alkane thiol of complementary reactivity patterned on the surface.16-18 Phosphonate protein interactions as well as Diels-Alder reactions have also been exploited.19,20 Another approach involved reaction of an aminooxy moiety with a protein containing a ketone or aldehyde to form oxime bonds.21-23
Surface immobilization of proteins via oxime chemistry was first demonstrated by Boncheva in 1999,24 and we have used light and a photo-acid generator to liberate surface aminooxy groups on polymer films.25 We have also directly generated nanofeatures of aminooxy groups with electron beam lithography.26 Chan and coworkers were able to immoblize ketone-decorated gold colloids onto aminooxy SAMs on a gold surface.27 Yousaf has produced a hydroquinone SAM that once oxidized to the corresponding quinone reacts to immobilize aminooxy RGD peptides for cell patterning.28 The same group has also produced a photo-caged aminooxy SAM that has been shown to immobilize ligands for cell adhesion after removal of the nitroveratryloxycarbonyl (NVOC) group by ultra-violet (UV) exposure; in this case a semicarbazide solution was required to remove the aldehyde generated upon deprotection to prevent oxime bond formation with the photobyproduct.29 We present an alternate stratagey involving the NPPOC moiety that does not require a scavenger because an aldehyde photobyproduct is not produced upon photo-deprotection30, 31. SAMs of a NPPOC protected aminooxy alkane thiol were fabricated. Subsequent deprotection and conjugation to a ketone-containing biologically relevant small molecule were demonstrated.
Results and discussion
Synthesis of photo-caged aminooxy alkane thiol
The synthetic strategy employed for the formation of the photolabile alkanethiol is outlined in Scheme 2. The triethylene glycol alkene was synthesized from triethylene glycol and 11-bromo-1-undecene following a literature procedure.32 After purification, product 2 was isolated in 67% yield. The yield was lowered by formation of dialkylated triethylene glycol byproduct. Compound 2 was subjected to Mitsunobu conditions with N-hydroxyphthalimide.33 Using triphenylphosphine and diisopropyl azodicarboxylate (DIAD), the terminal alcohol of 2 was substituted. The triphenylphosphine oxide byproduct was precipitated from the reaction mixture with hexanes. This method removed the side product, enabling efficient chromatography and isolation of the desired product 3 in quantitative yield.
Scheme 2.
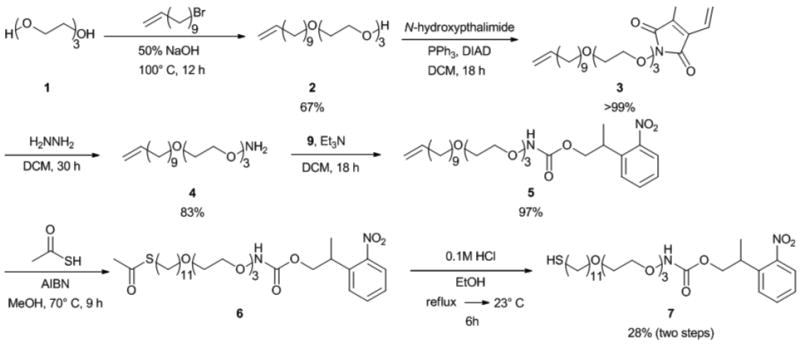
Synthesis of photo-caged aminooxyalkane thiol 7
The phthalimide protecting group of 3 was removed with hydrazine.33 The reaction was monitored by TLC to ensure complete consumption of the starting material. Additional hydrazine was added over the course of the reaction to complete the deprotection. Purification of 4 by flash column chromatography resulted in a yield of 83%.
To install the thiol end group, we first attempted to modify 4 with thioacetic acid using 2,2′-azobisisobutyronitrile (AIBN) as the radical initiator and light. While this thioene reaction typically proceeds with good yields,34 the isolation of the thioacteate alkanethiol bearing the aminooxy end group proved to be challenging. To circumvent this problem, we opted to install the NPPOC group first followed by the formation of thioacetate.
The activated photolabile group was synthesized from 2-(2-nitrophenyl)propanol (Scheme 3). Typical conditions to activate this group involve the use of phosgene.31 Instead, disuccinimidyl carbonate (DSC) was coupled to the alcohol of 8 to form 9 in 69% yield, to avoid use of this toxic compound. The carbonate was then added to 4 to give 5 in 97% yield.
Scheme 3.

Synthesis of activated carbonate 9
Compound 5 was then subjected to thioacetic acid and AIBN using thermal activation. Chromatography was employed to remove starting material and byproducts. Again, separation of the alkene starting material 5 and the resulting thioacetate 6 proved difficult. Therefore 6 was not isolated prior to removal of the acetate group.
The final step required deprotection of the thioacteate to reveal the thiol. Hydrochloric acid in ethanol was used, and the product was purified. The yield was 28% for the two step conversion of 5 to 7. Overall, the desired NPPOC-protected aminooxy product was synthesized in seven convergent steps, with a yield of 15%.
SAM formation deprotection and conjugation
Upon obtaining 7, SAM formation, deprotection, and subsequent surface conjugation were attempted. A piranha cleaned gold wafer was incubated in a 5 mM ethanolic solution of 7 for 24 h. The resulting SAM 7 was evaluated by contact angle (76±2°) and IR (Figure 1a). In particular, the peak at 1130 cm-1 confirmed the presence of PEG (C-O stretching) while the signals at 1260(N-CO-O), 1530(NO2) and 1720 cm-1(C=O) showed that the NPPOC protecting group was present on the surface. Ellipsometry also indicated successful formation of the SAM, giving a surface thickness of 2.4±0.3 nm.
Fig. 1.
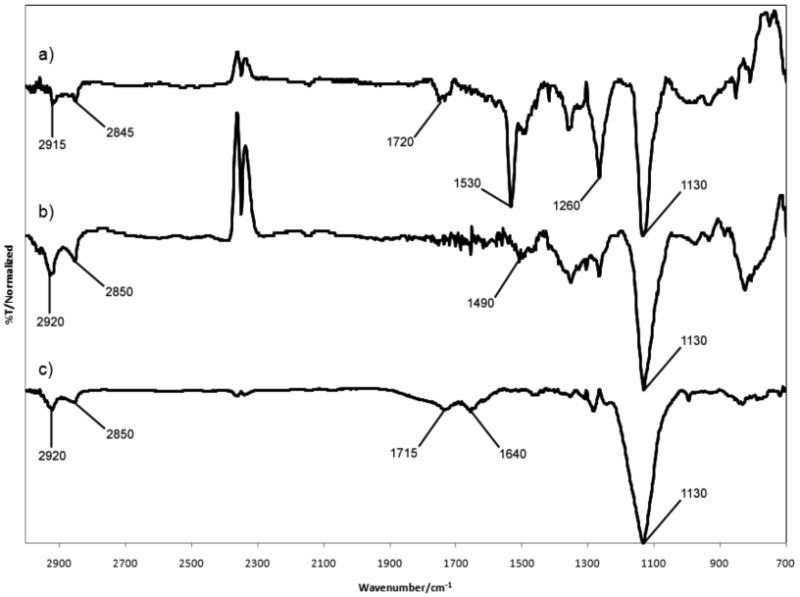
Infrared spectroscopy of a) SAM 7, b) SAM 7 after 3 h exposure to a hand held 365 nm UV light c) DP-SAM 7 after 3 h exposure to a 3 mM ethyl levulinate solution at 60°C.
Following SAM formation, photo-deprotection of SAM 7 was investigated by flood exposure to 365 nm UV light. Deprotection was monitored by IR via the disappearance of the signal at 1260 cm-1corresponding to the aminooxy carbamate moiety with approximately 50% deprotection occurring after 10 to 15 minutes. Although recent investigations report more rapid removal of the NPPOC protecting group with a UV laser at a dose of 1.2 J/cm2,35 our deprotection scheme employs a hand-held UV lamp as a less intense source of light, applied at a distance of 1.5 cm from the surface. While this is a more convenient method for UV exposure, it accounts for the increased exposure time required for deprotection. SAM 7 was exposed to a hand held UV lamp for 3 hours to ensure maximal surface deprotection. The resulting photo-deprotected surface, DP-SAM 7 was subsequently examined. The contact angle (62±7°) decreased as expected. The peaks at 1260, 1530, and 1720 cm-1 in the IR spectrum (Figure 1b) were no longer visible.
Reaction with DP-SAM 7 was subsequently examined with ethyl levulinate, a small molecule with a ketone moiety (Scheme 1). This molecule was chosen because it is widely used to modify proteins with ketone groups.36 The photo-deprotected aminooxy SAM was rinsed with 5 mL of ethanol and incubated with a 3 mM ethanolic solution of ethyl levulinate at 60 °C. Contact angle of the conjugate was measured as (64±4°). This was only a slight change compared to the deprotected aminooxy surface. Covalent conjugation of ethyl levulinate to the aminooxy surface was confirmed by observation of the oxime bond stretch in the IR spectrum (Figure 1c) at 1640 cm-1 (C=N). The ester carbonyl stretch at 1715 cm-1 was also observed. Absorption by the oxime bond is typically weak, yet it was found to be comparable to that of the carbonyl stretch; this was likely due to surface enhancement.
Taken together these data showed that sucessful deprotection and conjugation of a ketone to the SAM via oxime bond formation occurred. This suggests that the aminooxy moiety present in this photo-caged molecule may be used to immobilize proteins and other bio-molecules with ketones onto surfaces. This site selective conjugation in conjunction with standard photolithography strategies may be employed to pattern biomolecules for a range of applications, including oriented protein arrays.
Conclusions
A photo-caged aminooxy surface reactive moiety was synthesized in seven steps in 15% yield. This molecule was used to form a SAM that was subsequently shown to reveal surface aminooxy groups when exposed to 365 nm light. No scavanging additives were required. Conjugation of the resulting aminooxy terminated SAM to the biologically relevant molecule ethyl levulinate via oxime bond formation was confirmed by IR spectroscopy. Application of this alkane thiol to immobilize biomolecules on surfaces is underway.
Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. The products 232 and 831 were synthesized according to literature procedures.1H and 13C NMR spectra were obtained on either a Bruker ARX 500 MHz or AVANCE 500 MHz spectrometer. J values are given in Hz. Mass spectra were obtained on either a Applied Biosystems Voyager-DE-STR MALDI-TOF or a high resolution ESI Applied BioSystems Q-Star Elite supported by Grant Number S10RR024605 from the National Center For Research Resources. The spectra is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health. Chemical infrared spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrophotometer fit with an ATR accessory. Surface infrared spectra were obtained on a Bio-Rad FTS 175C Dynamic Alignment FT-IR Spectrometer. Ellipsometry was performed using a Gaertner LSE ellipsometer equipped witha 633 nm HeNe laser fixed at a 70° incidence angle. UV-vis spectra were recorded on a Thermospectronic Biomate 5 spectrophotometer using MeOH as the solvent. An FTA 135 Version 2.0 was used for contact angle measurments. An Entela UVGL-25 4 Watt UV lamp was operated at 365 nm for the photo-deprotection.
Synthesis
Synthesis of 2
2 was synthesized according to literature procedure.32 NaOH (0.49 mL, 50%) was added to tritethylene glycol (8.00 mL, 60.0 mmol) in a 2-necked round bottom flask equipped with a water-jacketed condensor. The mixture was heated to 100 °C for 30 min before adding 11-bromo-1-undecene (2.66 mL, 12.3 mmol) dropwise. The reaction was allowed to proceed for 12 h before being diluted with 50 mL of water, and the resulting aqueous layer was washed with hexanes (3 × 50 mL). The organic layers were combined and dried over MgSO4 and the solvent removed under reduced pressure. 2 was isolated following flash column chromatography (FCC) (Hexanes: ethyl acetate, Rf = 0.2) as a clear oil (2.48 g, 8.21 mmol, 67% yield). NMR δH(500 MHz; CDCl3) 5.82–5.72 (m, 1H, CHCH2), 4.99–4.86 (m, 2H, CHCH2), 3.73–3.52 (m, 12H), 3.41 (t, J = 6.6 Hz, 2H), 2.80 (bs, 1H, OH), 2.02–1.95 (m, 2H, CH2CH2O), 1.59–1.48 (m, 2H, CH2CH2CH2), 1.38–1.19 (m, 12H, CH2CH2CH2) ppm; δC(125 MHz; CDCl3) 139.2, 114.2, 72.6, 71.6, 70.7, 70.7, 70.4, 70.1, 33.8, 29.6, 29.6, 29.5, 29.5, 29.2, 29.0, 26.1 ppm; IR: νmax/cm-1 3456, 2923, 2854, 1640, 1458, 1350, 1295, 1248, 1106, 1069, 993, 908, 722, 675;UV-vis: λmax = 265 nm (ε = 18 cm-1M-1); (m/z (Electrospray) Found: MNa+ (sodium adduct), m/z 325.2346; Calc. for C17H34O4Na; 325.2355.
Synthesis of 3
Alkane-PEG 2 (2.7 g, 8.9 mmol) was dissolved in 90 mL of dry dichloromethane (DCM). To that solution, N-hydroxyphthalimide (1.74 g, 10.6 mmol) was added, followed by triphenylphosphine (2.79 g, 10.6 mmol). After ensuring complete dissolution of the regeants, DIAD (1.9 mL, 9.8 mmol) was added dropwise over 5 min, and the reaction was allowed to stir over 18 h. The solvent was removed in vacuo, and the residue resuspended in hexanes. The triphenylphosphine oxide byproduct crystallized and was isolated by filtration. The solvent was evaporated in vacuo. A white residue (4.0 g, 8.9 mmol, >99% yield) was obtained after purification by FCC (hexanes:ethyl acetate, 3:1, Rf = 0.7). NMR δH(500 MHz; CDCl3) 7.85-7.79 (m, 2H, Ar-H), 7.76-7.71 (m, 2H, Ar-H), 5.85–5.77 (m, 1H CHCH2), 4.97-4.87 (m, 2 H, CH2CH), 3.86-3.80 (m, 2H), 3.66-3.59 (m, 2H), 3.57-3.44 (m, 6H),3.41 (t, J = 7.0 Hz, 2H), 2.80 (bs, 1H, OH)2.02-1.97 (m, 2 H, CH2CH2O) 1.57–1.46 (m, 2H, CH2CH2O), 1.38–1.16 (m, 12H, CH2CH2CH2) ppm; δC(125 MHz; CDCl3) 163.6, 139.3, 134.5, 129.1, 123.6, 114.2, 76.9, 71.6, 70.9, 70.7, 70.6, 70.1, 69.4, 33.9, 29.7, 29.7, 29.6, 29.2, 29.0, 28.7, 25.9, 21.5 ppm;IR: νmax/cm-12924, 2854, 1790, 1731, 1639, 1611, 1524, 1467, 1374, 1325, 1292, 1257, 1186, 1109, 1083, 1033, 996, 978, 953, 908, 877, 787, 699; UV-vis: λmax = 310 nm (ε = 455 cm-1M-1); m/z (Electrospray) Found: MNa+ (sodium adduct), m/z 470.2491; Calc. for C25H37NO6Na; 470.2519
Synthesis of 4
Hydrazine hydrate (0.9 mL, 20 mmol) was added to product 3 (1.64 g, 3.66 mmol) dissolved in 20 mL of DCM. The solution was stirred vigorously for 8 h. Monitoring the reaction by TLC indicated that starting material remained and additional hydrazine (0.9 mL, 20 mmol) was added. The solution stirred for an additional 24 h. The reaction mixture was concentrated and the product purified by FCC (hexanes:ethyl acetate, 2:1, Rf = 0.2) to give a clear oil (0.96 mg, 3.0 mmol, 83% yield). NMR δH(500 MHz; CDCl3)5.85–5.77 (m, 1H, CHCH2), 5.01–4.91 (m, 2H, CHCH2), 3.85–3.84 (m, 2H, CH2ON), 3.69–3.63 (m, 8H, OCH2CH2O), 3.59–3.57 (m, 2H, OCH2CH2O), 3.44 (t, J = 6.8 Hz, 2H, CH2CH2O), 2.05–2.01 (m, 2H, CH2CH2O), 1.60–1.54 (m, 2H, CH2CH2CH2), 1.39–1.27 (m, 12H, CH2CH2CH2) ppm; δC(125 MHz; CDCl3) 139.2, 114.1, 74.8, 71.6, 70.6, 70.6, 70.1, 69.6, 33.8, 29.6, 29.5, 29.5, 29.4, 29.1, 28.9, 26.1 ppm; IR: νmax/cm-1 2924, 2854, 1640, 1591, 1458, 1349, 1458, 1349, 1296, 1245, 1200, 1106, 1041, 993, 908, 846, 723;UV-vis: λmax = 252 nm (ε = 28 cm-1M-1); m/z (Electrospray) Found: M+1, m/z 318.2585; Calc. for C17H35NO4; 318.2644.
Synthesis of 8
8 was synthesized according to a literature procedure.31 Triton B (3.3 mL, 8 mmol) was added to nitroethyl benzene (1.08 mL, 8 mmol). Paraformaldehyde (245 mg, 8.1 mmol) was added, and the reaction was heated to 60 °C for 6 h. The reaction was concentrated in vacuo and neutralized with 5% aqueous HCl followed by extraction with ethyl acetate (3 × 10mL). The material was dried over MgSO4 and the solvent was removed under reduced pressure. 8 was isolated following FCC (CH2Cl2, Rf = 0.2). NMR δH(500 MHz, CDCl3)7.75 (dd, J=8.3, 1.2 Hz, 1H, Ar-H), 7.60-7.55 (m, 1H, Ar-H), 7.50 (dd, J=8.1, 1.5 Hz, 1H, Ar-H), 7.365 (m, 1H, Ar-H), 3.84-3.76 (m, 2H, CH2), 3.56-3.48 (m, 1H, CH), 1.70 (br s, 1H,OH), 1.33 (d, J=6.8 Hz, 3H, CH3) ppm; δC(125 MHz; CDCl3) 138.2, 132.8, 128.5, 128.3, 127.3, 124.2, 67.9, 36.5, 17.7 ppm; IR: νmax/cm-1 3375, 3073, 2973, 2877, 1719, 1607, 1577, 1519, 1480, 1465, 1454, 1351, 1299, 1244, 1194, 1164, 1085, 1055, 1034, 1011, 976, 954, 875, 851, 782, 746, 709, 680, 662;UV-vis: λmax = 250 nm (ε = 4126 cm-1M-1), 390 nm (ε = 471 cm-1M-1);m/z (MALDI-TOF) Found: MNa+ (sodium adduct), m/z 204.47; Calc. for C27H44N2O8Na; 204.06.
Synthesis of 9
Disuccinimidyl carbonate (390 mg, 1.5 mmol) was dissolved in 5 mL of dry DMF. 8 (180 mg, 0.99 mmol) was added followed by triethylamine (0.77 mL, 5.5 mmol). The reaction was stirred for 18 h and concentrated in vacuo. A dark red oil 9 (220 mg, 0.68 mmol, 69% yield) was isolated following FCC (hexanes:ethyl acetate, 1:1, Rf = 0.4). NMR δH(500 MHz; CDCl3)7.82 (dd, J = 8.0, 1.3 Hz, 1H, Ar), 7.63–7.60 (m, 1H, Ar), 7.50 (dd, J = 7.9, 1.2 Hz, 1H, Ar), 7.43–7.40 (m, 1H, Ar), 4.56–4.48 (m, 2H, CH2), 3.82–3.78 (m, 1H, CH), 2.85 (s, 4H, Su), 1.43 (d, J = 7.0 Hz, 3H, CH3) ppm; δC(125 MHz; CDCl3) 168.7, 151.5, 150.0, 135.9, 133.1, 128.6, 128.4, 128.0, 124.6, 74.5, 33.4, 25.5, 17.5 ppm; IR: νmax/cm-1 2936, 1812, 1788, 1736, 1668, 1609, 1577, 1523, 1458, 1430, 1386, 1355, 1257, 1199, 1088, 1048, 1022, 991, 942, 853, 813, 786, 750, 712, 658;UV-vis: λmax = 251 nm (ε = 3855 cm-1M-1), 372 nm (ε = 458 cm-1M-1);m/z (MALDI-TOF) Found: MK+ (potassium adduct), m/z 361.12; Calc. for C27H44N2O8K; 361.04.
Synthesis of 5
The hydroxylamine 4 (230 mg, 0.73 mmol) was dissolved in 10 mL of DCM. The activated ester 9 (250 mg, 0.77 mmol) was added followed by triethylamine (0.41 mL, 2.9 mmol). The reaction was allowed to stir over 18 h in the dark. The reaction was concentrated and purified by FCC (hexanes:ethyl acetate, 2:1, Rf = 0.4) with a gradient solvent system from 2:1 to 1:1 hexanes:ethyl acetate to afford a pale yellow oil 5 (370 mg, 71 mmol, 97% yield). NMR δH(500 MHz; CDCl3) 7.96 (bs, 1H, NH), 7.74 (dd, J = 8.1, 1.0 Hz, 1H, Ar), 7.58–7.55 (m, 1H, Ar), 7.48–7.46 (m, 1H, Ar), 7.38–7.35 (m, 1H, Ar), 5.81–5.80 (m, 1H, CHCH2), 5.00–4.96 (m, 1H, CHCH2), 4.93–4.91 (m, 1H, CHCH2), 4.33–4.26 (m, 2H, CH2CHCH3), 3.95–3.94 (m, 2H, CH2ON), 3.71–3.61 (m, 9H, OCH2CH2O, CHCH3), 3.56–3.54 (m, 2H, OCH2CH2O), 3.42 (t, J = 6.8 Hz, 2H, CH2CH2O), 2.05–2.01 (m, 2H, CH2CH2O), 1.57–1.54 (m, 2H, CH2CH2CH2), 1.36–1.27 (m, 15H, CH2CH2CH2) ppm; δC(125 MHz; CDCl3) 156.9, 150.5, 139.2, 137.0, 132.6, 128.1, 127.4, 124.1, 114.1, 75.4, 71.5, 70.6, 70.5, 70.4, 70.0, 69.2, 69.1, 33.8, 33.3, 29.5, 29.5, 29.4, 29.4, 29.1, 28.9, 26.0, 17.6 ppm; IR: νmax/cm-1 3252, 2925, 2855, 1740, 1639, 1609, 1578, 1524, 1460, 1352, 1298, 1242, 1103, 1033, 995, 909, 852, 784, 749, 710, 662;UV-vis: λmax = 252 nm (ε = 3338 cm-1M-1), 330 nm (ε = 503 cm-1M-1); m/z (Electrospray) Found: MNa+ (sodium adduct), m/z 547.2846; Calc. for C27H44N2O8Na; 547.2995.
Synthesis of 6
Thioacetic acid (0.26 mL, 4 mmol) was added to a solution of 5 (370 mg, 0.71 mmol) in 2.6 mL of methanol. AIBN (5 mg, 0.03 mmol) was added, and the reaction mixture was heated to 70 °C for 9 h. The solution was concentrated and purified by FCC (hexanes:ethyl acetate, 2:1, Rf = 0.2) to give impure product 6 as a pale yellow oil. This was used directly in the next step.
Synthesis of 7
A solution of 6 (320 mg, 0.53 mmol) in 20 mL of 0.1 M HCl in EtOH was prepared and refluxed for 3 h. The solution was cooled and stirred an additional 3 h. The reaction mixture was concentrated and purified by FCC (hexanes: ethyl acetate, 2:1, Rf = 0.4) to give 7 (110 mg, 0.20 mmol, 28% yield over two steps). NMR δH(500 MHz; CDCl3)7.95 (bs, 1H, NH), 7.74 (dd, J = 8.1, 1.2 Hz, 1H, Ar), 7.58–7.55 (m, 1H, Ar), 7.48–7.46 (m, 1H, Ar), 7.38–7.35 (m, 1H, Ar), 4.31–4.20 (m, 2H, CH2CHCH3), 3.95–3.86 (m, 2H, CH2ON), 3.78–3.50 (m, 11H, OCH2CH2O, CH2CHCH3), 3.42 (t, J = 6.8 Hz, 2H, CH2CH2O), 2.52–2.50 (m, 2H, CH2SH), 1.67–1.51 (m, 4H, CH2CH2O, CH2CH2SH), 1.42–1.15 (m, 17H, CH2CH2CH2) ppm; δC(125 MHz; CDCl3) 156.9, 150.5, 137.0, 132.6, 128.1, 127.4, 124.1, 84.7, 76.1, 75.4, 71.5, 70.5, 70.5, 70.4, 70.0, 69.2, 69.1, 34.0, 33.8, 33.5, 33.3, 29.5, 29.5, 29.5, 29.4, 29.0, 28.3, 28.2, 26.0, 24.6, 24.5, 17.6 ppm;IR: νmax/cm-1 3252 (NH), 2925 and 2854 (CH), 1734 (C=O), 1526 (CNO2) 1464 (NH),1243 (N-CO-O), 1114 vs (C-O); UV-vis: λmax = 252 nm (ε = 5581 cm-1M-1);m/z (Electrospray) Found: MNa+ (sodium adduct), m/z 581.2873; Calc. for C27H46N2O8SNa; 581.2873.
Self assembled monolayer (SAM) formation and reactivity
SAM 7
A gold wafer was immersed in hot piranha solution ( H2O2:H2SO4) for 5 min CAUTION:Piranha solution reacts violently with organic materials, rinsed with Milli-Q water and ethanol, and dried under a stream of argon. The wafer was then exposed to a 5 mM ethanolic solution of 7 at 23°C for 36 h. The surface was then rinsed with 5mL of ethanol and dried under argon before analysis via contact angle (76±2°) and IR spectroscopy to confirm SAM 7. IR: νmax/cm-1 2915 and 2845 (CH), 1720 (C=O), 1530 and 1350 (NO2), 1490 (NH), 1260 (N-CO-O), 1130 (C-O). Ellipsometry measurements gave a surface thickness of 2.4±0.3 nm.
Photo-deprotection of SAM 7
SAM 7 was exposed to a 365 nm hand held UV lamp for 3 hat a distance of 1.5 cm under ambient conditions before washing with 5mL of ethanol and drying under a stream of argon. Deprotection of SAM 7 was confirmed by contact angle (62±7°) and IR: νmax/cm-1 2920 and 2850 (CH), 1490 (NH2),1130 (C-O).
Ethyl levulinate conjugation to DP-SAM 7
DP-SAM 7 was exposed to a 3 mM ethanolic solution of ethyl levulinate at 60 °C for 3 h. The resulting surface was washed with 5 mL of ethanol and dried under a stream of argon. Conjugation to the surface was confirmed by contact angle (64±4°) and IR: νmax/cm-1 2920 and 2850 (CH), 1715 (C=O), 1640 (C=N), 1130 (C-O).
Supplementary Material
Acknowledgments
This research was funded by the National Science Foundation Career Award CHE–0645793. RJM acknowledges a fellowship from the NSF IGERT: Materials Creation Training Program (MCTP)–DGE-0654431 and the California Nanosystems Institute. ZPT thanks the NIH sponsered Biotechnology Training in Biomedical Sciences and Engineering Program and the Jonsson Comprehensive Cancer Center (JCCC) for fellowships. HDM thanks the Alfred P. Sloan Foundation for additional funding. The authors would like to thank Steven Jonas for his aid with the ellipsometry measurements.
Footnotes
Electronic supplementary information (ESI) available: 1H NMR spectra of compounds 2-5, 7-9. See DOI: 10.1039/b000000x/
References
- 1.Bhadriraju K, Chen CS. Drug Discovery Today. 2002;7:612–620. doi: 10.1016/s1359-6446(02)02273-0. [DOI] [PubMed] [Google Scholar]
- 2.Fields S. Science. 2001;291:1221–1224. doi: 10.1126/science.291.5507.1221. [DOI] [PubMed] [Google Scholar]
- 3.MacBeath G, Schreiber SL. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 4.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 5.Mrksich M, Sigal GB, Whitesides GM. Langmuir. 1995;11:4383–4385. [Google Scholar]
- 6.Wilson DS, Nock S. Angew Chem Int Ed. 2003;42:494–500. doi: 10.1002/anie.200390150. [DOI] [PubMed] [Google Scholar]
- 7.Christman KL, Enriquez-Rios VD, Maynard HD. Soft Matter. 2006;2:928–939. doi: 10.1039/b611000b. [DOI] [PubMed] [Google Scholar]
- 8.Walter G, Bussow K, Lueking A, Glokler J. Trends Mol Med. 2002;8:250–253. doi: 10.1016/s1471-4914(02)02352-3. [DOI] [PubMed] [Google Scholar]
- 9.Frutos AG, Brockman JM, Corn RM. Langmuir. 2000;16:2192–2197. [Google Scholar]
- 10.Pitt WG, Young BR, Cooper SL. Colloids and Surfaces. 1987;27:345–355. [Google Scholar]
- 11.Soderquist ME, Walton AG. J Colloid Interface Sci. 1980;75:386–397. [Google Scholar]
- 12.Onclin S, Mulder A, Huskens J, Ravoo BJ, Reinhoudt DN. Langmuir. 2004;20:5460–5466. doi: 10.1021/la049561k. [DOI] [PubMed] [Google Scholar]
- 13.Smith JC, Lee KB, Wang Q, Finn MG, Johnson JE, Mrksich M, Mirkin CA. Nano Lett. 2003;3:883–886. [Google Scholar]
- 14.Dawson PE, Muir TW, Clarklewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 15.Muir TW. Annu Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 16.Kleinert M, Winkler T, Terfort A, Lindhorst TK. Org Biomol Chem. 2008;6:2118–2132. doi: 10.1039/b801595c. [DOI] [PubMed] [Google Scholar]
- 17.Yousaf MN, Mrksich M. J Am Chem Soc. 1999;121:4286–4287. [Google Scholar]
- 18.Rozkiewicz DI, Gierlich J, Burley GA, Gutsmiedl K, Carell T, Ravoo BJ, Reinhoudt DN. Chembiochem. 2007;8:1997–2002. doi: 10.1002/cbic.200700402. [DOI] [PubMed] [Google Scholar]
- 19.Yeo WS, Yousaf MN, Mrksich M. J Am Chem Soc. 2003;125:14994–14995. doi: 10.1021/ja038265b. [DOI] [PubMed] [Google Scholar]
- 20.Hodneland CD, Lee YS, Min DH, Mrksich M. Proc Natl Acad Sci U S A. 2002;99:5048–5052. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemieux GA, Bertozzi CR. Trends Biotechnol. 1998;16:506–513. doi: 10.1016/s0167-7799(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 22.Kochendoerfer GG. Curr Opin Chem Biol. 2005;9:555–560. doi: 10.1016/j.cbpa.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem Int Ed Engl. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 24.Boncheva M, Scheibler L, Lincoln P, Vogel H, Akerman B. Langmuir. 1999;15:4317–4320. [Google Scholar]
- 25.Christman KL, Broyer RM, Tolstyka ZP, Maynard HD. J Mater Chem. 2007;17:2021–2027. [Google Scholar]
- 26.Christman KL, Schopf E, Broyer RM, Li RC, Chen Y, Maynard HD. J Am Chem Soc. 2009;131:521–527. doi: 10.1021/ja804767j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan EWL, Yu LP. Langmuir. 2002;18:311–313. [Google Scholar]
- 28.Chan EWL, Park S, Yousaf MN. Angew Chem Int Ed. 2008;47:6267–6271. doi: 10.1002/anie.200800166. [DOI] [PubMed] [Google Scholar]
- 29.Park S, Yousaf MN. Langmuir. 2008;24:6201–6207. doi: 10.1021/la8005663. [DOI] [PubMed] [Google Scholar]
- 30.Hasan A, Stengele KP, Giegrich H, Cornwell P, Isham KR, Sachleben RA, Pfleiderer W, Foote RS. Tetrahedron. 1997;53:4247–4264. [Google Scholar]
- 31.Bhushan KR, DeLisi C, Laursen RA. Tetrahedron Lett. 2003;44:8585–8588. [Google Scholar]
- 32.Chirakul P, Perez-Luna VH, Owen H, Lopez GP, Hampton PD. Langmuir. 2002;18:4324–4330. [Google Scholar]
- 33.Schlick TL, Ding ZB, Kovacs EW, Francis MB. J Am Chem Soc. 2005;127:3718–3723. doi: 10.1021/ja046239n. [DOI] [PubMed] [Google Scholar]
- 34.Palegrosdemange C, Simon ES, Prime KL, Whitesides GM. J Am Chem Soc. 1991;113:12–20. [Google Scholar]
- 35.Alang Ahmad SA, Wong LS, ul-Haq E, Hobbs JK, Leggett GJ, Micklefield J. J Am Chem Soc. 2009;131:1513–1522. doi: 10.1021/ja807612y. [DOI] [PubMed] [Google Scholar]
- 36.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772–4779. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.