

Abstract
Alcohol addiction (alcoholism) is one of the most prevalent substance abuse disorders worldwide. Addiction is thought to arise, in part, from a maladaptive learning process in which enduring memories of drug experiences are formed. However, alcohol (ethanol) generally interferes with synaptic plasticity mechanisms in the CNS and thus impairs various types of learning and memory. Therefore, it is unclear how powerful memories associated with alcohol experience are formed during the development of alcoholism. Here, using brain slice electrophysiology in mice, we show that repeated in vivo ethanol exposure (2 g/kg, i.p., three times daily for 7 d) causes increased susceptibility to the induction of long-term potentiation (LTP) of NMDA receptor (NMDAR)-mediated transmission in mesolimbic dopamine neurons, a form of synaptic plasticity that may drive the learning of stimuli associated with rewards, including drugs of abuse. Enhancement of NMDAR plasticity results from an increase in the potency of inositol 1,4,5-trisphosphate (IP3) in producing facilitation of action potential-evoked Ca2+ signals, which is critical for LTP induction. This increase in IP3 effect, which lasts for a week but not a month after ethanol withdrawal, occurs through a protein kinase A (PKA)-dependent mechanism. Corticotropin-releasing factor, a stress-related neuropeptide implicated in alcoholism and other addictions, further amplifies the PKA-mediated increase in IP3 effect in ethanol-treated mice. Finally, we found that ethanol-treated mice display enhanced place conditioning induced by the psychostimulant cocaine. These data suggest that repeated ethanol experience may promote the formation of drug-associated memories by enhancing synaptic plasticity of NMDARs in dopamine neurons.
Introduction
Despite the large impact of alcohol abuse on society, the neural mechanisms underlying the development of alcoholism, i.e., alcohol addiction, are not well understood. Development of addiction involves a maladaptive form of learning and memory in which drug-related experiences are remembered powerfully, resulting in persistent and uncontrollable drug seeking behavior (Hyman et al., 2006). However, it is well known that alcohol (ethanol) intoxication impairs various types of learning and memory in both humans and animals. Decreases in learning capacity have also been reported in long-term alcoholics and in animals withdrawn from repeated ethanol exposure (Ryback, 1971; Ryabinin, 1998; Stephens et al., 2005). In line with these observations, both acute and chronic exposures to ethanol have been shown to suppress activity-dependent synaptic plasticity, the major neural substrate for learning and memory, in various brain areas (Stephens et al., 2005; Belmeguenai et al., 2008; Xie et al., 2009).
The mesolimbic dopaminergic system that originates in the ventral tegmental area (VTA) is critically involved in the learning of information related to rewards, including drugs of abuse (Schultz, 1998; Hyman et al., 2006). Both natural rewards and drug rewards, such as ethanol, cause release of dopamine in the nucleus accumbens and other limbic structures, which is thought to drive learning by enhancing synaptic plasticity. However, ethanol intoxication may suppress reward-based conditioning (Busse et al., 2004; Cunningham and Gremel, 2006), presumably by hampering synaptic plasticity in dopamine projection areas (Xie et al., 2009). Accumulating evidence indicates that plasticity of glutamatergic transmission onto dopamine neurons within the VTA may also play important roles in the development of drug addiction (Hyman et al., 2006; Kauer and Malenka, 2007). Consistent with this idea, in vivo exposure to ethanol has been shown to produce global enhancement of AMPA receptor (AMPAR)-mediated transmission in VTA dopamine neurons (Saal et al., 2003; Stuber et al., 2008). However, it is not clear whether ethanol experience can promote activity-dependent plasticity of glutamatergic synapses in the VTA.
Plasticity of NMDA receptor (NMDAR)-mediated transmission may be of particular interest, as NMDAR activation in the VTA is necessary for dopamine neuron burst firing and phasic dopamine release in projection areas that occurs in response to rewards or reward-predicting stimuli (Sombers et al., 2009; Zweifel et al., 2009). We have recently reported long-term potentiation (LTP) of NMDAR EPSCs that is induced by sustained glutamatergic input activity paired with postsynaptic burst firing (Harnett et al., 2009). LTP induction requires amplification of action potential (AP)-evoked Ca2+ signals by preceding activation of metabotropic glutamate receptors (mGluRs). This amplification is dependent on Ca2+ release from intracellular stores, where inositol 1,4,5-trisphosphate (IP3) generated by mGluR activation increases Ca2+-induced Ca2+ release triggered by AP-induced Ca2+ influx (Cui et al., 2007).
Long-term ethanol treatment has been shown to produce an enhancement of IP3-mediated Ca2+ signaling in different cell types (Nomura et al., 1996; Saso et al., 1997; Netzeband et al., 2002). In the present study, we examined whether repeated in vivo ethanol exposure promotes mGluR/IP3-dependent plasticity of NMDAR EPSCs in VTA dopamine neurons.
Materials and Methods
Animals.
Male C57BL/6J mice (3–8 weeks old) were obtained from Jackson Laboratory and were housed under a 12 h light/dark cycle (lights on at 7:00 A.M.). Food and water were available ad libitum. All animal procedures were approved by the University of Texas Institutional Animal Care and Use Committee.
In vivo ethanol treatment.
Mice (3–4 weeks old) received intraperitoneal injections of ethanol (2 g/kg, 20% v/v) or an equivalent volume of saline, 3 times per day (3–3.5 h apart) for 7 d (6 g/kg ethanol per day). Mice were returned to the home cage immediately after each injection. It has been shown that intraperitoneal injection of 2 g/kg ethanol produces blood ethanol levels of ∼1.8 mg/ml in C57BL/6J mice (Crabbe et al., 2003), which are comparable to the levels achieved during ethanol drinking (∼1.6 mg/ml) (Elmer et al., 1987; Rhodes et al., 2005). It should also be noted that male C57BL/6J mice consume ∼10–14 g/kg ethanol per day during continuous-access two-bottle choice drinking (Blednov et al., 2005).
Electrophysiology.
Mice were killed by cervical dislocation under halothane or isoflurane anesthesia, and horizontal midbrain slices (190–210 μm) containing the VTA were cut in an ice-cold solution containing (in mm): 205 sucrose, 2.5 KCl, 1.25 NaH2PO4, 7.5 MgCl2, 0.5 CaCl2, 10 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2 (∼305 mOsm/kg) and incubated > 1 h at 35°C in a solution containing (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2 (pH 7.4, ∼295 mOsm/kg). Recordings were made at 34−35°C in the same solution perfused at ∼2.5 ml/min. The pipette solution contained (in mm) 115 K-methylsulfate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2-phosphocreatine (pH 7.25, ∼280 mOsm/kg).
Cells were visualized using an upright microscope (Olympus) with infrared/differential interference contrast optics. Recordings were made in the lateral VTA located between 50 and 150 μm from the medial border of the medial terminal nucleus of the accessory optic tract. Putative dopamine neurons were identified by their spontaneous firing (1–5 Hz) with broad APs (>1.2 ms) recorded in cell-attached configuration and large Ih currents (> 200 pA elicited by 1.5 s hyperpolarizing steps from −62 mV to −112 mV) in whole-cell configuration. Whole-cell voltage-clamp recordings were made at a holding potential of −62 mV, corrected for a liquid junction potential of −7 mV. Pipettes with tip resistance of 1.5–2.0 MΩ were used. Series and input resistances were monitored throughout experiments and recordings were discarded if the series resistance increased beyond 20 MΩ or the input resistance dropped below 200 MΩ. A Multiclamp 700A amplifier (Molecular Devices) and AxoGraph X (AxoGraph Scientific) were used to record and collect data, which were filtered at 1–5 kHz and digitized at 2–10 kHz.
Unclamped APs were evoked with 2 ms depolarizing pulses from −62 mV to −7 mV. The time integral of the outward tail current, termed IK(Ca), was calculated after removing a 20 ms window following the depolarizing pulse (expressed in picocoulombs). We have shown previously that IK(Ca) thus measured is completely eliminated by TTX and also by apamin, a selective blocker of small-conductance Ca2+-sensitive K+ (SK) channels, and hence can be used as a readout of AP-induced Ca2+ transients (Cui et al., 2007).
Flash photolysis.
Caged IP3 (25 or 200 μm; Invitrogen) was loaded into the cytosol through the whole-cell pipette. A 1 ms UV pulse was applied using a xenon arc lamp (Cairn Research) to rapidly release IP3. The UV pulse was focused through a 60× objective onto an ∼350-μm-diameter area centered at recorded neurons. The amount of photolysis is known to be proportional to the UV pulse intensity (McCray et al., 1980), which is proportional to the capacitance of the capacitor feeding current to the flash lamp. UV pulse intensity is therefore reported in units of capacitance (50–4050 μF).
LTP experiments.
Synaptic stimuli were applied every 20 s using bipolar tungsten electrodes (∼100 μm tip separation) placed 50–150 μm rostral to the recorded neuron. To isolate NMDAR EPSCs, recordings were performed in the presence of DNQX (10 μm), picrotoxin (100 μm), and eticlopride (100 nm) to block AMPA, GABAA, and D2 dopamine receptors. For LTP induction, sustained synaptic stimulation (70 stimuli at 50 Hz) was paired with a burst of 5 APs at 20 Hz, with the burst onset delayed 1 s from the onset of the synaptic stimulation train. This synaptic stimulation-burst pairing was repeated 10 times every 20 s. Magnitude of LTP was determined by comparing the average EPSC amplitude over the 10 min baseline period with that during another 10 min window ∼30–40 min after LTP induction.
Conditioned place preference.
A conditioned place preference (CPP) box consisting of two distinct compartments, separated by a small middle chamber, was used for conditioning (Med Associates). One compartment had a mesh floor with white walls, while the other had a grid floor with black walls. One day after the 7 d saline/ethanol treatment in the home cage, mice were subjected to a pretest in a conditioning room, in which they were allowed to freely explore the entire CPP box for 20 min. The percentage of time spent in each compartment was determined after excluding the time spent in the middle chamber. Any mice that spent >60% of time in either compartment during the pretest were not used for conditioning. For cocaine CPP, mice were subjected to 2 d conditioning starting the next day, in which one of the two compartments was randomly assigned as the cocaine-paired side in each mouse. Here, mice were given an injection of cocaine (5 mg/kg, i.p.) and confined to one compartment for 30 min in one trial and given a saline injection and confined to the other compartment for 30 min in another trial. The two trials were separated by at least 4 h. The order of cocaine/saline trials was reversed on the second day of conditioning. A 20 min posttest was performed the following day. Mice that spent >8 min in the middle chamber during pretest or posttest were excluded from analysis. CPP score was determined by subtracting the preference for the cocaine-paired side in the pretest from that in the posttest. For ethanol CPP, mice were subjected to 4 d conditioning in which they were given either ethanol (2 g/kg, i.p.) or saline and confined to one compartment for 5 min.
Drugs.
3,4-Dihydroxyphenylglycol (DHPG), CRF, DNQX, eticlopride, and K41498 were obtained from Tocris Bioscience. Caged IP3 was purchased from Invitrogen. All other chemicals were from Sigma-RBI.
Data analysis.
Data are expressed as mean ± SEM. Statistical significance was determined by Student's t test or ANOVA followed by Bonferroni post hoc test. The difference was considered significant at p < 0.05.
Results
In vivo ethanol experience leads to enhanced mGluR-mediated facilitation of AP-evoked Ca2+ signals and increased IP3 sensitivity
mGluR/IP3-induced facilitation of AP-evoked Ca2+ signals is required for the induction of NMDAR LTP in dopamine neurons (Harnett et al., 2009). Thus, we first tested whether in vivo ethanol exposure alters the effect of the mGluR agonist DHPG on AP-evoked Ca2+ signals, which were assessed by measuring SK currents (IK(Ca)) following unclamped APs evoked by brief depolarizing pulses (see Materials and Methods). Recordings were made in midbrain slices prepared from naive C57BL/6J mice and mice that received injections of saline or ethanol (2 g/kg, i.p.) 3 times daily for 7 d (4–5 weeks old at the time of slice preparation). Bath perfusion of DHPG (3 μm) produced small facilitation of IK(Ca) in VTA dopamine neurons from naive or saline-treated mice (Fig. 1A,B). This DHPG effect was significantly increased 1 d after withdrawal from 7 d ethanol treatment. There was no change in the basal IK(Ca) amplitude (Fig. 1C). DHPG effect on IK(Ca) was still elevated 7 d after cessation of ethanol exposure, however it returned to a level comparable to that of the age-matched naive control (8–9 weeks old) after 4–5 weeks of withdrawal (Fig. 1D). Subsequent data for saline- and ethanol-treated mice in the present study were obtained 1 d after 7 d saline/ethanol exposure. DHPG-induced inward currents, which are independent of IP3 or Ca2+ (Guatteo et al., 1999; Cui et al., 2007) and likely involve activation of transient receptor potential-like channels (Tozzi et al., 2003), were not altered by ethanol treatment (Fig. 1E), suggesting that the increase in DHPG effect on IK(Ca) results from changes in IP3 signaling downstream of mGluRs.
Figure 1.

mGluR-dependent facilitation of AP-evoked Ca2+ signals is augmented after withdrawal from repeated ethanol exposure. A, Example traces of IK(Ca) illustrating the effects of DHPG (3 μm) on IK(Ca) in cells from saline- and ethanol-treated mice (1 d withdrawal). B, Summary bar graph demonstrating that in vivo ethanol exposure augmented DHPG-induced facilitation of IK(Ca) (naive group, 7 cells from 4 mice; saline group, 10 cells from 5 mice; ethanol group, 14 cells from 8 mice; F(2,28) = 6.25, p < 0.01, one-way ANOVA). *p < 0.05 versus naive and saline groups. C, The size of basal IK(Ca) was not altered after ethanol treatment. D, Summary graph depicting DHPG effect on IK(Ca) after different periods of ethanol withdrawal [1 d withdrawal group, 14 cells from 8 mice; 7 d withdrawal group, 7 cells from 5 mice; 4–5 week withdrawal group, 10 cells from 5 mice; naive (>8 weeks old) group, 9 cells from 5 mice]. E, DHPG-induced inward currents were not affected by ethanol treatment. Right, Example traces of DHPG-induced currents in cells from saline- and ethanol-treated mice. DHPG was perfused at the time indicated. The data in C and E were obtained from the same cells shown in B.
To more directly examine the alterations in IP3 signaling, we next performed flash photolysis of caged IP3 (200 μm) and measured the resulting SK-mediated outward current (IIP3) (Morikawa et al., 2000). The concentration of IP3 released was varied by applying different UV flash intensities, which were determined by the capacitance (up to 4050 μF) of the capacitor in the photolysis system (see Materials and Methods). IP3 concentration–response curves thus constructed exhibited a leftward shift after in vivo ethanol exposure (Fig. 2A,B). The average EC50 value [i.e., the UV intensity (expressed in microfarads) producing half-maximal current] was significantly lower in ethanol-treated mice compared with controls (Fig. 2C), while there was no difference in the maximal IIP3 amplitude (Fig. 2D). Therefore, repeated ethanol exposure increases IP3 sensitivity in VTA dopamine neurons.
Figure 2.

In vivo ethanol exposure increases IP3 sensitivity. A, Traces of IIP3 evoked with different UV pulse intensities (150, 450, 1350, and 4050 μF) in VTA neurons from saline- and ethanol-treated mice. These cells were loaded with caged IP3 (200 μm). UV flashes were applied at the time indicated by the arrow. B, Averaged concentration (UV flash intensity)–response (IIP3) curves from control and ethanol-treated mice. The IIP3 amplitude was normalized to the maximal value (estimated from fit to a logistic equation) in each cell. Data from naive and saline-treated mice were pooled as a control group (control group, 13 cells from 7 naive mice and 6 cells from 4 saline-treated mice; ethanol group, 16 cells from 10 mice; group, F(1,99) = 12.7, p < 0.01; flash intensity, F(3,99) = 643.1, p < 0.0001; group × flash intensity, F(3,99) = 7.45, p < 0.001, mixed two-way ANOVA). Dashed lines are fit to a logistic equation. ***p < 0.001 versus control. C, Summary bar graph showing that EC50 values were reduced in ethanol-treated mice (t33 = 3.88, p < 0.001, unpaired t test). D, The maximal IIP3 amplitude was not altered by in vivo ethanol treatment.
Repeated ethanol exposure and acute CRF application both enhance IP3 signaling via protein kinase A
It has been shown that in vivo ethanol exposure causes upregulation of the adenylyl cyclase-cAMP-PKA (protein kinase A) pathway in the mesolimbic dopamine system (Ortiz et al., 1995; Melis et al., 2002). PKA-mediated phosphorylation of IP3 receptors (IP3Rs) increases their IP3 sensitivity (Wagner et al., 2008). Thus, we tested the effect of forskolin, a potent activator of adenylyl cyclase, on IIP3 in naive mice. Bath perfusion of forskolin (3 μm) caused a significant increase in IIP3 evoked with an EC50 flash intensity, while there was minimal enhancement of IIP3 evoked with a maximal flash intensity (Fig. 3A,B). Therefore, activation of the cAMP-PKA pathway increases IP3R sensitivity in a manner analogous to repeated ethanol exposure. We further tested forskolin (3–10 μm) on AP-evoked IK(Ca) and found that it had no effect (Fig. 3C,D), similarly to the lack of effect of in vivo ethanol treatment on IK(Ca) (Fig. 1C).
Figure 3.

Stimulation of the cAMP-PKA pathway increases IP3 sensitivity. A, Representative traces of IIP3 in control (gray) and forskolin (3 μm; black). Traces on the left were elicited with an EC50 flash intensity, while those on the right represent maximal IIP3 evoked in the same cell. The cell was loaded with caged IP3 (200 μm). B, Effects of forskolin on IIP3 evoked with EC50 or maximal flash intensities are plotted in 6 cells (t5 = 9.07, p < 0.001, paired t test). C, Example traces of IK(Ca) in control (gray) and forskolin (10 μm; black). D, Summary time graph demonstrating that forskolin (3–10 μm) failed to affect IK(Ca) (n = 5).
Repeated ethanol exposure increases CRF levels in the brain (Sarnyai et al., 2001; Heilig and Koob, 2007). Furthermore, it has been shown that activation of CRF2 receptors potentiates mGluR-induced intracellular Ca2+ release via a PKA-dependent mechanism in dopamine neurons (Riegel and Williams, 2008). Therefore, we next examined the effects of CRF on IP3 signaling in saline- and ethanol-treated mice. In these experiments, dopamine neurons were filled with a low concentration of caged IP3 (25 μm) and subjected to a subthreshold UV flash (100 μF) that produced no measurable outward current by itself. This subthreshold flash caused significant facilitation of IK(Ca) when applied 50 ms before an AP (Fig. 4A). The flash intensity was held constant to compare the magnitude of IK(Ca) facilitation caused by the same concentration of IP3 in different cells. In saline-treated animals, bath perfusion of CRF (300 nm) significantly increased the magnitude of IP3-induced facilitation of IK(Ca) (Fig. 4A,C). Consistent with the results from DHPG experiments (Fig. 1B), IP3-induced facilitation of IK(Ca) was significantly larger in ethanol-treated mice compared with saline-treated controls (Fig. 4A–C). Furthermore, CRF was able to cause a robust additional increase in IK(Ca) that was already greatly facilitated by IP3 in ethanol-treated mice. Treatment with the PKA inhibitor H89 (10 μm, >1 h preincubation plus intracellular application through whole-cell pipette) reversed the increase in IK(Ca) facilitation following in vivo ethanol exposure and also completely abolished the effect of CRF. Notably, CRF application or H89 treatment had no effect on basal IK(Ca) not facilitated by IP3 (Fig. 4D; also note the overlap of gray and orange traces in Fig. 4A,B). We further confirmed that the augmentation of IP3 effect by CRF was suppressed by prior application of the selective CRF2 receptor antagonist K41498 (100–300 nm) in ethanol-treated mice (Fig. 4E). K41498 itself had no effect on IP3-induced facilitation of IK(Ca), showing the absence of functional CRF tone in brain slices. Together, these data demonstrate that previous in vivo ethanol exposure and acute CRF application converge on PKA to amplify the facilitatory effect of IP3 on AP-evoked Ca2+ signals.
Figure 4.

CRF amplifies the increase in IP3 effect on AP-evoked Ca2+ signals produced by in vivo ethanol exposure. A, Representative traces of IK(Ca), evoked by itself (gray and orange traces) or paired with preceding photolysis of caged IP3 (25 μm; black and red traces), in a saline-treated mouse. A low-intensity UV flash (100 μF) was applied at the arrow (50 ms before the AP). The magnitude of IP3-induced facilitation of IK(Ca) is determined by comparing gray and black traces in control solution and by comparing orange and red traces in CRF (300 nm). Note that bath perfusion of CRF, which failed to affect IK(Ca) itself, was capable of augmenting IP3-induced facilitation. B, Representative traces of IK(Ca), evoked as in A, from an ethanol-treated mouse. IP3 produced robust facilitation of IK(Ca) (black trace), which was further augmented by CRF (red trace). Treatment with H89 (10 μm) largely suppressed IP3-induced IK(Ca) facilitation and abolished the CRF effect. C, Summary bar graph plotting the magnitude of IP3-induced facilitation of IK(Ca) under the conditions illustrated in A and B (saline group, 8 cells from 4 mice; ethanol group, 6 cells from 4 mice; ethanol group recorded in H89: 6 cells from 5 mice; group: F(2,17) = 11.4, p < 0.001; CRF: F(1,17) = 21.1, p < 0.001; group × CRF: F(2,17) = 4.38, p < 0.05, mixed two-way ANOVA). *p < 0.05, **p < 0.01, ***p < 0.001. D, Summary bar graph depicting the size of IK(Ca) (without IP3). These data were from the same cells shown in C. E, The CRF2 receptor antagonist K41498 blocked the augmentation of IP3-induced facilitation of IK(Ca) by CRF (5 cells from 3 ethanol-treated mice).
In vivo ethanol experience promotes NMDAR plasticity
We next examined whether repeated ethanol exposure affects the induction of NMDAR LTP, which requires mGluR/IP3-induced Ca2+ signal facilitation and is gated by PKA activity (Harnett et al., 2009). Pharmacologically isolated NMDAR EPSCs were recorded at −62 mV in physiological Mg2+ (1.2 mm) solution (see Materials and Methods). Previous studies using C57BL/6J mice have found no global changes in NMDAR function in VTA dopamine neurons after repeated in vivo injections of ethanol (Brodie, 2002; Hopf et al., 2007). Therefore, we normalized synaptic stimulation intensity using the NMDAR EPSC amplitude (∼20–30 pA; saline group: 26 ± 2 pA, 8 cells from 7 mice; ethanol group: 27 ± 3 pA, 5 cells from 5 mice). Following 10 min baseline recording, LTP was induced by repetitively pairing (10 times every 20 s) sustained synaptic stimulation (70 stimuli at 50 Hz) with a burst of 5 APs at 20 Hz in the postsynaptic neuron. While this pairing protocol resulted in relatively small LTP of NMDAR EPSCs in saline-treated controls, ethanol-treated animals exhibited significantly larger magnitude of LTP (Fig. 5A–C). Before running the LTP induction protocol, facilitation of IK(Ca) by synaptic stimulation was assessed in each neuron by evoking an AP at 60 ms after the end of a 1 s stimulation train. We found that facilitation of IK(Ca) by synaptic stimulation was significantly increased in neurons from ethanol-treated animals (Fig. 5D). Furthermore, the magnitude of NMDAR LTP was positively correlated with that of IK(Ca) facilitation across neurons from both groups of mice (r = 0.61, p < 0.05) (Fig. 5E). These data demonstrate that repeated ethanol exposure enhances the induction of NMDAR plasticity, most likely via an increase in mGluR-mediated facilitation of AP-evoked Ca2+ signals.
Figure 5.
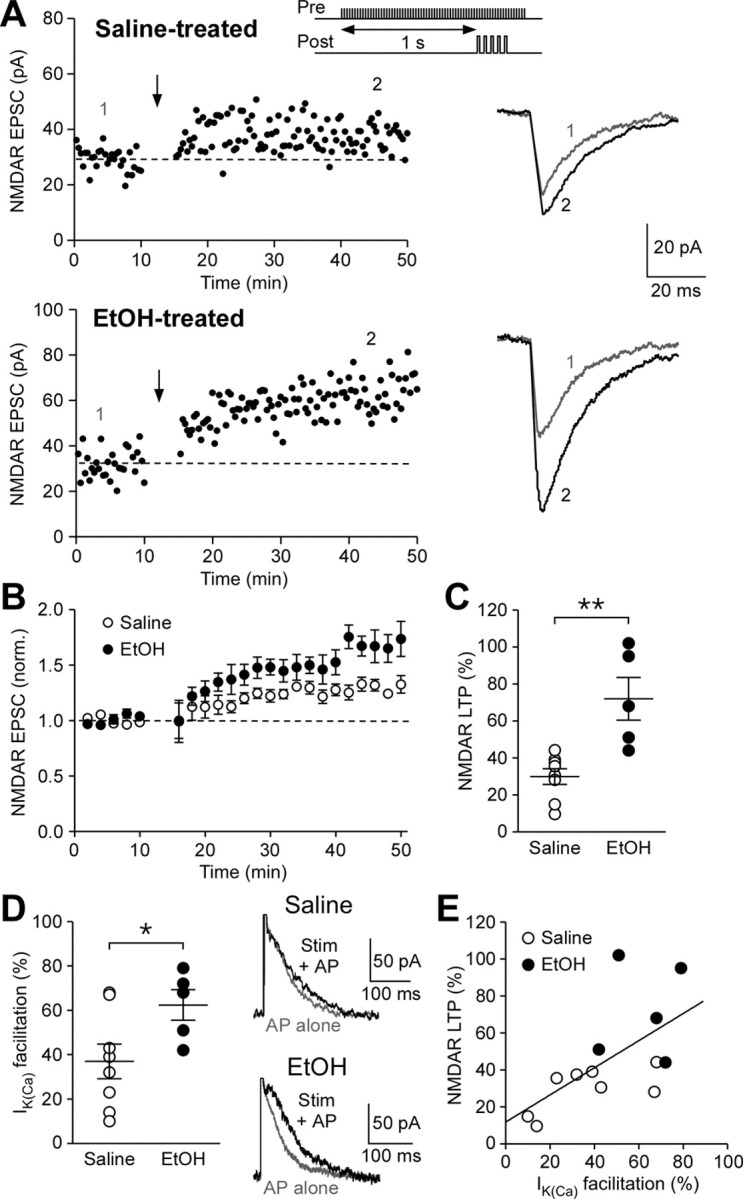
NMDAR-mediated transmission becomes more susceptible to LTP induction after repeated ethanol exposure. A, Example experiments to induce NMDAR LTP in saline- and ethanol-treated mice. Time graphs of NMDAR EPSC amplitude are shown on the left. The LTP induction protocol, which consisted of synaptic stimulation-burst pairing (top inset), was delivered at the time indicated by the arrow. Traces of NMDAR EPSCs at times indicated by numbers in the time graphs are shown on the right. B, Summary time graph of NMDAR LTP experiments (saline group, 8 cells from 7 mice; ethanol group, 5 cells from 5 mice). C, Summary graph plotting the magnitude of NMDAR LTP in saline- and ethanol-treated mice (t11 = 4.04, p < 0.01, unpaired t test). D, Summary graph showing that the magnitude of IK(Ca) facilitation produced by preceding synaptic stimulation was larger in ethanol-treated mice (t11 = 2.25, p < 0.05, unpaired t test). Example traces illustrating synaptic facilitation of IK(Ca) are shown on the right. E, The magnitude of NMDAR LTP is plotted versus the magnitude of synaptic facilitation of IK(Ca) in the cells shown in. Solid line is a linear fit to all data points from both saline- and ethanol-treated mice. The data summarized in B–E were all from the same cells.
Previous ethanol experience enhances subsequent cocaine-induced place conditioning
Enhanced NMDAR plasticity in VTA dopamine neurons may facilitate the learning of environmental stimuli associated with rewards, including drugs of abuse (Ahn et al., 2010). Thus, we tested the effect of repeated ethanol exposure on subsequent reward learning using cocaine-induced CPP, a form of behavioral conditioning that is dependent on NMDARs in the VTA (Harris et al., 2004; Zweifel et al., 2008). Mice were first treated with either saline or ethanol (2 g/kg, i.p.) for 7 d, as described above. Then, after initial preference for the two compartments of the CPP box was determined on the pretest day, mice were subjected to 2 d conditioning in which cocaine (5 mg/kg, i.p.) was paired with one compartment while saline was paired with the other compartment on each day. During the CPP posttest, ethanol-treated mice exhibited a significantly greater increase in preference for the cocaine-paired compartment (Fig. 6A,B). These data demonstrate that repeated ethanol exposure enhances subsequent learning of cocaine-associated environmental stimuli.
Figure 6.

Previous ethanol exposure promotes cocaine-induced CPP. A, Changes in the preference for the cocaine-paired side after 2 d conditioning are shown for saline- and ethanol-treated mice (saline group, t6 = 4.46, p < 0.01; ethanol group, t8 = 5.39, p < 0.001, paired t test). B, Summary bar graph showing that cocaine CPP is enhanced in ethanol-treated mice (t14 = 2.35, p < 0.05, unpaired t test). C, Changes in the preference for the ethanol-paired side after 4 d conditioning are shown for saline- and ethanol-treated mice (saline group, t6 = 0.16, p = 0.88; ethanol group, t7 = 1.10, p = 0.31, paired t test). D, Summary bar graph of ethanol CPP experiments (t13 = 0.78, p = 0.45, unpaired t test).
We further asked whether repeated ethanol exposure could affect subsequent ethanol CPP. To test this, mice underwent 4 d CPP conditioning with ethanol (2 g/kg, i.p.) after 7 d saline/ethanol treatment. Neither group developed significant preference for the ethanol-paired compartment (Fig. 6C,D), in agreement with a previous report showing that C57BL/6J mice do not readily develop ethanol CPP (Cunningham et al., 1992).
Discussion
It is well known that acute ethanol exposure inhibits NMDAR function, thus causing suppression of many forms of synaptic plasticity that are dependent on NMDAR activation in the CNS (Lovinger et al., 1989; Ron, 2004). In contrast, long-term ethanol exposure has been shown to produce upregulation of NMDAR function/expression in the hippocampus (Nelson et al., 2005), amygdala (Roberto et al., 2006), and striatum (Wang et al., 2010), although it is not clear whether this leads to an enhancement of NMDAR-dependent synaptic plasticity (Stephens et al., 2005; Xia et al., 2006; Sabeti and Gruol, 2008). In the present study, we found that VTA dopamine neurons exhibit increased susceptibility to the induction of mGluR/IP3-dependent LTP of NMDAR EPSCs after repeated ethanol exposure in vivo. Our finding provides an important insight into the neural plasticity mediating the learning component of alcoholism.
Repeated ethanol exposure enhances IP3R sensitivity
The increase in IP3-induced Ca2+ signaling observed in ethanol-treated mice is most likely due to increased PKA phosphorylation of IP3Rs, as it can be mimicked by activation of the cAMP/PKA pathway by forskolin and reversed by the PKA inhibitor H89. Changes in gene expression and protein levels of multiple components of the cAMP/PKA pathway have been found in tissue samples from human alcoholic brain (Yamamoto et al., 2001; Mayfield et al., 2002). Furthermore, upregulation of the cAMP/PKA pathway, presumably resulting from chronic activation of Gi-coupled receptors, such as D2 dopamine receptors or μ-opioid receptors, appears to be a common neuroadaptation observed in the mesolimbic system and other brain areas following exposure to different drugs of abuse (Hyman et al., 2006). Indeed, ethanol is known to elevate dopamine levels within the VTA, activating D2 autoreceptors on dopamine neurons (Yan et al., 1996; Kohl et al., 1998). Ethanol-induced dopamine release in the VTA has also been shown to activate presynaptic D1 receptors on glutamatergic terminals, which may further increase somatodendritic dopamine release via glutamatergic excitation of dopamine neurons (Deng et al., 2009; Xiao et al., 2009). Furthermore, we have recently reported a similar PKA-dependent increase in IP3R sensitivity in dopamine neurons following in vivo exposure to amphetamine (Ahn et al., 2010). Thus, repetitive stimulation of D2 autoreceptors as a consequence of drug-induced dopamine release within the VTA may be a potential mechanism mediating the enhancement of IP3R function.
The basal IK(Ca) was not altered by repeated ethanol exposure or by modulation of the cAMP/PKA pathway with forskolin or H89, suggesting that PKA does not significantly affect voltage-gated Ca2+ channels responsible for AP-induced Ca2+ influx. PKA-mediated phosphorylation is known to modulate multiple types of voltage-gated Ca2+ channels (Hell et al., 1995; Dolphin, 1996), including L-type Ca2+ channels that are highly expressed in dopamine neurons and play an important role in AP firing (Rajadhyaksha et al., 2004; Chan et al., 2007). However, PKA sensitivity of the pore-forming α1 subunit of L-type channels appears to be determined by alternative splicing and the presence of certain auxiliary subunits (Hell et al., 1993; Liang and Tavalin, 2007). To our knowledge, the precise splice variant and subunit composition of L-type channels in dopamine neurons are unknown.
Interactions between the CRF system and ethanol-induced neuroadaptations
An extensive body of work has implicated stress and the CRF system in the acquisition, expression, and reinstatement of drug-seeking behaviors (Sarnyai et al., 2001; Heilig and Koob, 2007). For example, stress-induced enhancement of the acquisition of cocaine CPP can be reversed by systemic administration of a CRF receptor antagonist (Kreibich et al., 2009). Furthermore, acute stress has been shown to more effectively increase opiate self-administration when the stressor and the self-administration session are closely paired in time and not when the stressor is applied several hours after the end of the session, suggesting that stress can strengthen learned associations that contribute to drug taking behavior (Shaham, 1993). Recent evidence also indicates direct interactions between CRF and dopamine systems within the VTA. Indeed, stress-induced CRF release in the VTA can trigger reinstatement of cocaine-seeking behavior (Wise and Morales, 2010), which could be mediated by direct effects of CRF on intrinsic excitability or glutamatergic excitation of dopamine neurons (Ungless et al., 2003; Wanat et al., 2008). Our finding that CRF potentiates IP3-induced facilitation of IK(Ca) is in agreement with a previous study demonstrating that CRF increases mGluR-mediated intracellular Ca2+ release in dopamine neurons (Riegel and Williams, 2008). These CRF effects on Ca2+ signaling occur via the cAMP/PKA pathway, which also mediates the effects of repeated in vivo exposure to ethanol and amphetamine, as described above. Despite this shared mechanism of action, the enhancement of IP3 sensitivity by ethanol exposure does not occlude the facilitation of AP-evoked Ca2+ signals by CRF. On the contrary, a robust facilitation of IP3-mediated Ca2+ release can be achieved when acute CRF is combined with prior ethanol exposure. PKA regulation of IP3Rs may, therefore, represent an important site of convergence where the stress system interacts with drug-induced neuroadaptations, thereby promoting drug-induced learning and behaviors. In this regard, it should be noted that increased CRF levels are observed during ethanol withdrawal in multiple brain areas of rodents (Sarnyai et al., 2001; Heilig and Koob, 2007) as well as in the CSF of alcohol-dependent humans (Adinoff et al., 1996). Thus, although CRF tone was not detected in VTA slices in the present study, VTA CRF levels may be elevated in vivo during ethanol withdrawal.
Previous studies have found that CRF can activate the phospholipase C (PLC) pathway in dopamine neurons (Ungless et al., 2003; Wanat et al., 2008). Since PLC activation leads to the generation of IP3, CRF could be expected to facilitate IK(Ca) itself through this pathway, as agonists of mGluRs and other PLC-coupled receptors do (Cui et al., 2007). However, CRF failed to facilitate IK(Ca) unless APs were combined with preceding IP3 flash photolysis in the present study. This result suggests that coupling of CRF receptors to the PLC pathway may not be strong enough to produce a sufficient amount of IP3 to cause IK(Ca) facilitation by itself.
Repeated ethanol exposure enhances NMDAR plasticity and learning of drug-associated stimuli
Extensive studies have shown that in vivo exposure to drugs of abuse, including ethanol, causes global potentiation of AMPAR, but not NMDAR, function in VTA dopamine neurons (Brodie, 2002; Saal et al., 2003; Hopf et al., 2007; Kauer and Malenka, 2007; Stuber et al., 2008). This generalized increase in the sensitivity of dopamine neurons to AMPAR-mediated excitation is thought to result in increased dopamine release in the nucleus accumbens and other dopamine neuron projection areas, thereby enhancing activity-dependent synaptic plasticity in those areas (Wolf et al., 2004). In contrast, the present study demonstrates that ethanol exposure induces neuroadaptation within the VTA that facilitates activity-dependent plasticity of NMDAR-mediated glutamatergic transmission onto dopamine neurons, as has been shown previously with amphetamine exposure (Ahn et al., 2010). NMDARs are thought to play a critical role in triggering phasic dopamine neuron responses to reward-predicting cues (Sombers et al., 2009; Zweifel et al., 2009). Interestingly, in vivo exposure to ethanol and other drugs of abuse has been shown to impair LTP induction at GABAergic synapses in VTA dopamine neurons (Guan and Ye, 2010; Niehaus et al., 2010; but also see Melis et al., 2002). Since GABAergic inhibition can effectively suppress NMDAR-induced burst firing (Lobb et al., 2010), these two forms of metaplasticity, i.e., enhancement of NMDAR LTP and impairment of GABA LTP, may work in concert to promote the development of conditioned dopamine neuron responses to environmental stimuli associated with drug experience.
NMDAR activation in the VTA is required for the learning of drug-associated cues assessed with a CPP paradigm (Harris et al., 2004; Zweifel et al., 2008). In accordance with this idea, acute ethanol exposure, which inhibits NMDARs (Lovinger et al., 1989), has been shown to interfere with CPP when administered immediately before conditioning sessions (Cunningham and Gremel, 2006). In contrast, our CPP experiments show that the same repeated ethanol treatment that enhances NMDAR LTP also enhances subsequent learning of cocaine-associated cues. Previous studies performing long-term ethanol preexposure did not find an enhancement of cocaine or ethanol CPP (Le Pen et al., 1998; Busse et al., 2005). However, these studies both used significantly different ethanol treatment protocols than the one used here, involving much fewer ethanol injections or drinking of ethanol-containing solutions. These discrepancies suggest that fewer doses or more gradual intake of ethanol may not be sufficient to induce the neuroadaptations underlying the enhanced CPP seen in the present study. This type of “cross-sensitization” of learning shown in our study has been demonstrated with other drugs of abuse using CPP or operant conditioning paradigms (Lett, 1989; Horger et al., 1992), which may account for the concurrent use of different types of drugs frequently seen in alcoholics and drug addicts (Pennings et al., 2002; Dani and Harris, 2005). Sensitization of IP3Rs in the VTA may thus be a common drug-induced neuroadaptation among alcohol and other drugs of abuse that acts to drive the formation of powerful memories of stimuli encountered during the initial days to weeks of drug experience and withdrawal.
Footnotes
This work was supported by National Institutes of Health Grant AA015521. B.E.B. was supported by a National Research Service Award.
References
- Adinoff B, Anton R, Linnoila M, Guidotti A, Nemeroff CB, Bissette G. CSF concentrations of corticotropin-releasing hormone (CRH) and diazepam-binding inhibitor (DBI) during alcohol withdrawal and abstinence. Neuropsychopharmacology. 1996;15:288–295. doi: 10.1016/0893-133X(95)00212-V. [DOI] [PubMed] [Google Scholar]
- Ahn KC, Bernier BE, Harnett MT, Morikawa H. IP3 receptor sensitization during in vivo amphetamine experience enhances NMDA receptor plasticity in dopamine neurons of the ventral tegmental area. J Neurosci. 2010;30:6689–6699. doi: 10.1523/JNEUROSCI.4453-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmeguenai A, Botta P, Weber JT, Carta M, De Ruiter M, De Zeeuw CI, Valenzuela CF, Hansel C. Alcohol impairs long-term depression at the cerebellar parallel fiber-Purkinje cell synapse. J Neurophysiol. 2008;100:3167–3174. doi: 10.1152/jn.90384.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Metten P, Finn DA, Rhodes JS, Bergeson SE, Harris RA, Crabbe JC. Hybrid C57BL/6J × FVB/NJ mice drink more alcohol than do C57BL/6J mice. Alcohol Clin Exp Res. 2005;29:1949–1958. doi: 10.1097/01.alc.0000187605.91468.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MS. Increased ethanol excitation of dopaminergic neurons of the ventral tegmental area after chronic ethanol treatment. Alcohol Clin Exp Res. 2002;26:1024–1030. doi: 10.1097/01.ALC.0000021336.33310.6B. [DOI] [PubMed] [Google Scholar]
- Busse GD, Lawrence ET, Riley AL. The modulation of cocaine-induced conditioned place preferences by alcohol: effects of cocaine dose. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:149–155. doi: 10.1016/j.pnpbp.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Busse GD, Lawrence ET, Riley AL. The effects of alcohol preexposure on cocaine, alcohol and cocaine/alcohol place conditioning. Pharmacol Biochem Behav. 2005;81:459–465. doi: 10.1016/j.pbb.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Cotnam CJ, Cameron AJ, Schlumbohm JP, Rhodes JS, Metten P, Wahlsten D. Strain differences in three measures of ethanol intoxication in mice: the screen, dowel and grip strength tests. Genes Brain Behav. 2003;2:201–213. doi: 10.1034/j.1601-183x.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- Cui G, Bernier BE, Harnett MT, Morikawa H. Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci. 2007;27:4776–4785. doi: 10.1523/JNEUROSCI.0139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Gremel CM. Proximal ethanol pretreatment interferes with acquisition of ethanol-induced conditioned place preference. Pharmacol Biochem Behav. 2006;85:612–619. doi: 10.1016/j.pbb.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Niehus DR, Malott DH, Prather LK. Genetic differences in the rewarding and activating effects of morphine and ethanol. Psychopharmacology (Berl) 1992;107:385–393. doi: 10.1007/BF02245166. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Deng C, Li KY, Zhou C, Ye JH. Ethanol enhances glutamate transmission by retrograde dopamine signaling in a postsynaptic neuron/synaptic bouton preparation from the ventral tegmental area. Neuropsychopharmacology. 2009;34:1233–1244. doi: 10.1038/npp.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- Elmer GI, Meisch RA, George FR. Differential concentration-response curves for oral ethanol self-administration in C57BL/6J and BALB/cJ mice. Alcohol. 1987;4:63–68. doi: 10.1016/0741-8329(87)90062-0. [DOI] [PubMed] [Google Scholar]
- Guan YZ, Ye JH. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving mu-opioid receptors. Neuropsychopharmacology. 2010;35:1841–1849. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knöpfel T. Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopamine neurons that is independent from calcium mobilization. J Neurophysiol. 1999;82:1974–1981. doi: 10.1152/jn.1999.82.4.1974. [DOI] [PubMed] [Google Scholar]
- Harnett MT, Bernier BE, Ahn KC, Morikawa H. Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron. 2009;62:826–838. doi: 10.1016/j.neuron.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–847. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Wong ST, Warner C, Snutch TP, Catterall WA. Differential phosphorylation of two size forms of the neuronal class C L-type calcium channel alpha 1 subunit. J Biol Chem. 1993;268:19451–19457. [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Martin M, Chen BT, Bowers MS, Mohamedi MM, Bonci A. Withdrawal from intermittent ethanol exposure increases probability of burst firing in VTA neurons in vitro. J Neurophysiol. 2007;98:2297–2310. doi: 10.1152/jn.00824.2007. [DOI] [PubMed] [Google Scholar]
- Horger BA, Giles MK, Schenk S. Preexposure to amphetamine and nicotine predisposes rats to self-administer a low dose of cocaine. Psychopharmacology (Berl) 1992;107:271–276. doi: 10.1007/BF02245147. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kohl RR, Katner JS, Chernet E, McBride WJ. Ethanol and negative feedback regulation of mesolimbic dopamine release in rats. Psychopharmacology (Berl) 1998;139:79–85. doi: 10.1007/s002130050692. [DOI] [PubMed] [Google Scholar]
- Kreibich AS, Briand L, Cleck JN, Ecke L, Rice KC, Blendy JA. Stress-induced potentiation of cocaine reward: a role for CRF R1 and CREB. Neuropsychopharmacology. 2009;34:2609–2617. doi: 10.1038/npp.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pen G, Duterte-Boucher D, Daoust M, Costentin J. Pre-exposure to alcohol does not sensitize to the rewarding effects of cocaine. Neuroreport. 1998;9:2887–2891. doi: 10.1097/00001756-199808240-00038. [DOI] [PubMed] [Google Scholar]
- Lett BT. Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology (Berl) 1989;98:357–362. doi: 10.1007/BF00451687. [DOI] [PubMed] [Google Scholar]
- Liang Y, Tavalin SJ. Auxiliary beta subunits differentially determine pka utilization of distinct regulatory sites on Cav1.3 L type Ca2+ channels. Channels (Austin) 2007;1:102–112. doi: 10.4161/chan.4284. [DOI] [PubMed] [Google Scholar]
- Lobb CJ, Wilson CJ, Paladini CA. A dynamic role for GABA receptors on the firing pattern of midbrain dopaminergic neurons. J Neurophysiol. 2010;104:403–413. doi: 10.1152/jn.00204.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Mayfield RD, Lewohl JM, Dodd PR, Herlihy A, Liu J, Harris RA. Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J Neurochem. 2002;81:802–813. doi: 10.1046/j.1471-4159.2002.00860.x. [DOI] [PubMed] [Google Scholar]
- McCray JA, Herbette L, Kihara T, Trentham DR. A new approach to time-resolved studies of ATP-requiring biological systems; laser flash photolysis of caged ATP. Proc Natl Acad Sci U S A. 1980;77:7237–7241. doi: 10.1073/pnas.77.12.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20:RC103. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL. Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res. 2005;1048:69–79. doi: 10.1016/j.brainres.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Netzeband JG, Schneeloch JR, Trotter C, Caguioa-Aquino JN, Gruol DL. Chronic ethanol treatment and withdrawal alter ACPD-evoked calcium signals in developing Purkinje neurons. Alcohol Clin Exp Res. 2002;26:386–393. [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci. 2010;32:108–117. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Higashi K, Hoshino M, Saso K, Itou M, Hoek JB. Effect of glutathione on inositol 1,4,5-triphosphate-induced Ca2+ release in permeabilized hepatocytes from control and chronic ethanol-fed rats. Alcohol Clin Exp Res. 1996;20:325A–329A. [PubMed] [Google Scholar]
- Ortiz J, Fitzgerald LW, Charlton M, Lane S, Trevisan L, Guitart X, Shoemaker W, Duman RS, Nestler EJ. Biochemical actions of chronic ethanol exposure in the mesolimbic dopamine system. Synapse. 1995;21:289–298. doi: 10.1002/syn.890210403. [DOI] [PubMed] [Google Scholar]
- Pennings EJ, Leccese AP, Wolff FA. Effects of concurrent use of alcohol and cocaine. Addiction. 2002;97:773–783. doi: 10.1046/j.1360-0443.2002.00158.x. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Husson I, Satpute SS, Küppenbender KD, Ren JQ, Guerriero RM, Standaert DG, Kosofsky BE. L-type Ca2+ channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci. 2004;24:7464–7476. doi: 10.1523/JNEUROSCI.0612-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Williams JT. CRF facilitates calcium release from intracellular stores in midbrain dopamine neurons. Neuron. 2008;57:559–570. doi: 10.1016/j.neuron.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- Ron D. Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist. 2004;10:325–336. doi: 10.1177/1073858404263516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryabinin AE. Role of hippocampus in alcohol-induced memory impairment: implications from behavioral and immediate early gene studies. Psychopharmacology (Berl) 1998;139:34–43. doi: 10.1007/s002130050687. [DOI] [PubMed] [Google Scholar]
- Ryback RS. The continuum and specificity of the effects of alcohol on memory. A review. Q J Stud Alcohol. 1971;32:995–1016. [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sabeti J, Gruol DL. Emergence of NMDAR-independent long-term potentiation at hippocampal CA1 synapses following early adolescent exposure to chronic intermittent ethanol: role for sigma-receptors. Hippocampus. 2008;18:148–168. doi: 10.1002/hipo.20379. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev. 2001;53:209–243. [PubMed] [Google Scholar]
- Saso K, Moehren G, Higashi K, Hoek JB. Differential inhibition of epidermal growth factor signaling pathways in rat hepatocytes by long-term ethanol treatment. Gastroenterology. 1997;112:2073–2088. doi: 10.1053/gast.1997.v112.pm9178701. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Shaham Y. Immobilization stress-induced oral opioid self-administration and withdrawal in rats: role of conditioning factors and the effect of stress on “relapse” to opioid drugs. Psychopharmacology (Berl) 1993;111:477–485. doi: 10.1007/BF02253539. [DOI] [PubMed] [Google Scholar]
- Sombers LA, Beyene M, Carelli RM, Wightman RM. Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J Neurosci. 2009;29:1735–1742. doi: 10.1523/JNEUROSCI.5562-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DN, Ripley TL, Borlikova G, Schubert M, Albrecht D, Hogarth L, Duka T. Repeated ethanol exposure and withdrawal impairs human fear conditioning and depresses long-term potentiation in rat amygdala and hippocampus. Biol Psychiatry. 2005;58:392–400. doi: 10.1016/j.biopsych.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hopf FW, Hahn J, Cho SL, Guillory A, Bonci A. Voluntary ethanol intake enhances excitatory synaptic strength in the ventral tegmental area. Alcohol Clin Exp Res. 2008;32:1714–1720. doi: 10.1111/j.1530-0277.2008.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci. 2003;18:2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–3596. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, Morales M. A ventral tegmental CRF-glutamate-dopamine interaction in addiction. Brain Res. 2010;1314:38–43. doi: 10.1016/j.brainres.2009.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Xia JX, Li J, Zhou R, Zhang XH, Ge YB, Ru Yuan X. Alterations of rat corticostriatal synaptic plasticity after chronic ethanol exposure and withdrawal. Alcohol Clin Exp Res. 2006;30:819–824. doi: 10.1111/j.1530-0277.2006.00095.x. [DOI] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC, 3rd, Li KY, Krnjević K, Zhou C, Ye JH. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology. 2009;34:307–318. doi: 10.1038/npp.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie GQ, Wang SJ, Li J, Cui SZ, Zhou R, Chen L, Yuan XR. Ethanol attenuates the HFS-induced, ERK-mediated LTP in a dose-dependent manner in rat striatum. Alcohol Clin Exp Res. 2009;33:121–128. doi: 10.1111/j.1530-0277.2008.00818.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Pohli S, Durany N, Ozawa H, Saito T, Boissl KW, Zöchling R, Riederer P, Böning J, Götz ME. Increased levels of calcium-sensitive adenylyl cyclase subtypes in the limbic system of alcoholics: evidence for a specific role of cAMP signaling in the human addictive brain. Brain Res. 2001;895:233–237. doi: 10.1016/s0006-8993(00)03260-1. [DOI] [PubMed] [Google Scholar]
- Yan QS, Reith ME, Jobe PC, Dailey JW. Focal ethanol elevates extracellular dopamine and serotonin concentrations in the rat ventral tegmental area. Eur J Pharmacol. 1996;301:49–57. doi: 10.1016/0014-2999(96)00018-0. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]