

Abstract
Melanocortins (MSH’s) are three structurally related peptides derived from proopiomelanocortin. They regulate several physiologic functions including energy metabolism, appetite, and inflammation. Recent work in rodents has also identified important effects of MSH’s, particularly γ-MSH, on sodium metabolism and blood pressure regulation. Normal rats and mice respond to a high sodium diet with an increase in the plasma concentration of γ-MSH, and remain normotensive, while those with genetic or pharmacologic γ-MSH deficiency become hypertensive on a high sodium diet. This hypertension is corrected by exogenous administration of the peptide. Mice lacking the γ-MSH receptor (the melanocortin 3 receptor, Mc3r) also become hypertensive on a high sodium diet but remain so when administered γ-MSH, and infusions of physiologic levels of the peptide stimulate urinary sodium excretion in normal rats and mice, but not in mice with deletion of Mc3r. The salt-sensitive hypertension in rodents with impaired γ-MSH signaling appears due to stimulation of noradrenergic activity, since plasma noradrenaline is increased and the hypertension is rapidly corrected with infusion of the α-adrenoceptor antagonist phentolamine. In contrast to the antihypertensive property of physiologic levels of γ-MSH, intravenous or intracerebroventircular injections of high levels of the peptide raise blood pressure. This occurs in mice lacking Mc3r, indicating an interaction with some other central receptor. Finally, the salt-sensitive hypertension in rodents with disruption of γ-MSH signaling is accompanied by insulin resistance, an observation which offers a new window into the study of the association of salt-sensitive hypertension with insulin resistance and type II diabetes.
Keywords: Blood pressure, Salt-sensitive hypertension, γ-melanocyte stimulating hormone, Noradrenergic activity, Insulin resistance
Introduction
Melanocortins (melanocyte stimulating hormones, MSH’s) are peptides derived from proopiomelanocortin by proteolytic processing. There are three MSH peptides which share a common core heptapeptide sequence but differ in overall size and in C-terminal amidation. α-and β-MSH were initially identified by their actions on dispersion of melanin in skin of reptiles and amphibians. When the complementary DNA for proopiomelanocortin became available, Nakanishi and colleagues deduced a peptide sequence in the N-terminal region which shared some homology with α-and β-MSH and which was flanked with dibasic amino acid cleavage sites. They named this sequence γ-MSH with the expectation that it would be a secreted peptide with melanin dispersing properties (Nakanishi S., 1979). Although subsequent work indicated little effect of γ-MSH on pigmentation, the peptide has been shown to possess a number of other important qualities. This review will summarize current information on the cardiovascular effects of the melanocortins, with particular emphasis on γ-MSH.
1. Properties of γ-MSH
2.1 Natriuresis
Initial interest in γ-MSH as a peptide with important renal and cardiovascular actions stemmed from the observations of Gruber and colleagues (reviewed in (Gruber and Callahan, 1989)). They were able to demonstrate that γ-MSH was natriuretic when injected in intravenous boluses into hydrated, anesthetized rats in low doses (<64 pmoles) (Lymangrover et al., 1985)but was hypertensive when given in higher amounts (Callahan et al., 1984). The physiologic significance of thenatriuretic property of the peptide became clear in 1987 when it was shown that a γ-MSH-like peptide mediated the reflex natriuresis which occurs after acute unilateral nephrectomy (Lin et al., 1987; Ni et al., 1998). These studies indicated that γ-MSH played a role in this specific situation of natriuresis, and opened up the possibility that it could have a broader role as a component in the maintenance of overall sodium metabolism by participating in the reflex regulation of sodium excretion.
The survival value from an evolutionary standpoint of the postnephrectomy natriuresis would on the face of it seem tobe small, so it became of interest to see if γ-MSH could have a role in longer term adjustments accompanying changes in dietary sodiumintake. Measurement of immunoreactive γ-MSH concentration in plasma of rats fed a high sodium diet (8% NaCl) vs a low sodium diet (0.07%) for ≥ 1 week showed that peptide levels were twice as high in rats fed the high sodium diet compared to those on the low sodium diet(Mayan et al., 1996). No change occurred in plasma adrenocorticotrophic hormone(ACTH) concentration, the major circulating peptide derived from processing of proopiomelanocortin in the anterior lobe of the pituitary. This increase in plasma γ-MSH concentration during ingestion of the high sodium diet was accompanied by an increase in γ-MSH content of the pituitary neurointermediate lobe but not of the anterior lobe. Whole pituitary proopiomelanocortin mRNA abundance increased progressively with duration of the high sodium diet, and in situ hybridization showed that this increase was almost exclusively confined to the neurointermediate lobe(Mayan et al., 1996). These observations, coupled with the natriuretic properties of the peptide (Chen et al., 1997b; Lin et al., 1987; Lymangrover et al., 1985; Ni et al., 1998), indicated that the γ-MSH system could be part of the coordinated response to circumstances of dietary sodium excess, thereby greatly strengthening the argument that it played an important physiological role.
2.2 Renal Receptors for γ-MSH
MSH peptides interact with a family of five receptors, melanocortin MC1 receptor through melanocortin MC5receptor. These are G-protein-coupled receptors with seven membrane-spanning units (Humphreys, 2004; Schioth, 2001; Wikberg et al., 2000). The melanocortin MC1receptor is expressed on skin melanocytes and mediates pigment dispersion by α-MSH, whereas the melanocortin MC2receptor is the ACTH receptor expressed in adrenal cortex and responsible for stimulation of glucocorticoid synthesis and secretion. The melanocortin MC3 and MC4receptors are expressed in brain and other tissues, and information on their function has been gleaned from knockout mouse models lacking one of the receptors. The Mc3rknockout mouse has an alteration in energy metabolism with an increase in body fat content and decrease in muscle mass with no overall change in weight (Butler et al., 2000; Chen et al., 2000), and a role for γ-MSH acting through the melanocortin MC3receptor has been suggested in experimental arthritis (Getting et al., 2006). The Mc4rknockout mouse develops marked obesity and overeating (Huszar et al., 1997), and has been a useful approach to the molecular understanding of appetite control and the determinants of obesity (Ellacott and Cone, 2006). The melanocortin MC5receptor is expressed in a number of tissues including exocrine glands, and deletion of this gene in mice results in a picture of exocrine dysfunction (Chen et al., 1997a). Of these five receptors, γ-MSH has affinity at physiologic concentrations only for the melanocortin MC3receptor, and it has been viewed as the probable endogenous ligand for this receptor. No expression of melanocortin MC1 or MC2 receptor mRNA was found in renal cortex or medulla, but mRNA for the three other receptors could be detected in both regions of the kidney, with roughly similar signal abundance. However, signal intensity of the melanocortin MC3, but not MC4or MC 5receptor, increased dramatically in rats fed the high sodium diet; this increase was confined to the medulla. These changes in melanocortin MC3receptor mRNA abundance induced by the HSD were paralleled by an increase in melanocortin MC3 receptor protein in inner medullary collecting duct cells isolated from rats ingesting the high sodium diet (Ni et al., 2006a), and in whole kidney homogenates from Dahl salt-resistant, but not salt-sensitive, rats (Chandramohan et al., 2009). This increase in melanocortin MC3recesptor expression caused by the high sodium diet had functional significance, since γ-MSH-dependent cAMP production was much greater by collecting duct cells isolated from kidneys of rats ingesting the high sodium diet than from those eating the low salt diet, and intrarenal infusion of γ-MSH to rats fed the high sodium diet led to brisk increases in sodium and cAMP excretion that were not observed when the peptide was infused into kidneys of rats fed the low sodium diet. These results suggest that the γ-MSH system could be an important element in the renal response to high dietary sodium intake: both the plasma concentration of the peptide, reflecting increased synthesis and secretion from the pituitary, and the abundance of its receptor in the kidney, are increased during the ingestion of the high sodium diet.
2. Consequences of Interruption of γ-MSH Signaling
3.1 Genetic Approaches
In view of the evidence just summarized that the γ-MSH system is responsive to dietary sodium intake, it became of interest to examine the consequences of disruption of this system. As mentioned earlier, γ-MSH is derived from pituitary proopiomelanocortin. Processing of proopiomelanocortin into its component peptides is driven by two prohormone convertases, proconvertase 1 (PC1) and proconvertase 2 (PC2) (Fig. 1). PC1 is expressed predominately in anterior lobe corticotrophs and to only a low extent in intermediate lobe (Day et al., 1992; Zhou et al., 1993); it cleaves proopiomelanocortin into the larger peptides ACTH, β-lipotropin, and the large N-terminal fragment labeled pro -γ-MSH. PC2 is more prominent in melanotrophs of the neurointermediate lobe, but is also expressed at low levels in anterior lobe corticotrophs; its action gives rise to the smaller peptides β-endorphin and α-, β-, and γ-MSH (Zhou et al., 1993). Regulation of POMC synthesis and processing in the neurointermediate lobe is mediated primarily by the neurotransmitter dopamine, acting through the dopamine D2 receptor. In this lobe, this receptor is negatively coupled to adenylate cyclase; activation of the receptor inhibits proopiomelanocrtin gene expression and secretion of derived peptides, whereas antagonism of the receptor upregulates its expression (Autelitano et al., 1989; Smith and Funder, 1988). Abundance of PC1 and PC2 messenger RNA and protein move in parallel with proopiomelanocortin messenger RNA in response to dopaminergic stimulation or antagonism (Day et al., 1992; Oyarce et al., 1996). These processing enzymes act in a coordinate manner to mediate specific endoproteolytic cleavages of proopiomelanocortin at dibasic amino acid pairs in a strict temporal order, with PC1 acting initially and PC2 later to generate the smaller peptides secreted from the neurointermedite lobe(Reudelhuber, 2003; Zhou et al., 1993; Zhou et al., 1999) (Fig. 1). As mentioned above, the high sodium diet increased proopiomelanocortin messenger RNA abundance primarily in the neurointermediate lobe (Mayan et al., 1996). We subsequently confirmed this finding and also showed that the high sodium diet also increased the messenger RNA and protein abundance of PC2, again indicating coordinate regulation of the proopiomelanocortin-γ-MSH pathway by the high sodium diet (Chandramohan et al., 2001).
Figure 1.
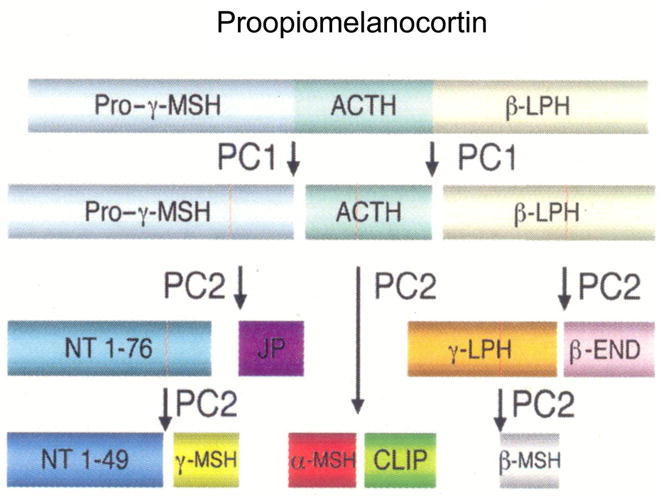
Schematic view of sequential processing of proopiomelanocortin. ACTH, adrenocorticotrophic hormone; LPH, Lipotropin; PC1, Proconvertase 1; PC2, Proconvertase 2; NT, N-terminal peptide; JP, joining peptide; END, endorphin; CLIP, corticotrophin-like intermediate peptide; MSH, melanocyte stimulating hormone. From (Reudelhuber, 2003).
3.1.1 Proconvertase 2-Deficient Mice
Mice with targeted deletion of the PC2 gene were first reported in 1997 by Steiner’s group (Furuta et al., 1997); these mice exhibit a modest impairment in growth and have lower blood glucose concentration than their wild-type littermates, but are otherwise phenotypically normal. They demonstrate defective processing of a number of peptide hormones in addition to proopiomelanocortin (Allen et al., 2001; Berman et al., 2000; Furuta et al., 2001). PC2−/− mice were compared to wild-type mice while ingesting either the low sodium or the high sodium diet for one week. In wild type mice fed the high sodium diet, plasma peptide concentration was more than double the value observed in mice fed the low sodium diet, while mean arterial pressure was not significantly different (Ni et al., 2003) (Fig. 2); these results parallel those described above in normal rats. However, in PC2 knockout mice a different picture emerged. Plasma γ-MSH concentration on the low sodium diet was < 10% of the value seen in wild type mice, and was not any higher in knockout mice on the high sodium diet. Arterial pressure in these mice on the low sodium diet was no different compared to that in wild type mice, but was dramatically increased in knockout mice ingesting the high sodium diet (158 mm Hg) (Fig. 2). These results were confirmed in conscious mice (Ni et al., 2003). Thus, absence of functional PC2 led to γ-MSH deficiency and was accompanied by a marked degree of salt-sensitive hypertension (Ni et al., 2003). To test the role of the γ-MSH deficiency in the hypertension, we infused the peptide intravenously at a low rate and found that it rapidly lowered arterial pressure to normal values, whereas a similar dose of α-MSH was without effect. This blood pressure-lowering effect of γ-MSH resulted from a central site of action, since an even lower dose of the peptide which was without effect when administered intravenously promptly reduced arterial pressure in hypertensive knockout mice when given into the cerebroventricular system (Ni et al., 2003). The hypertension was accompanied by suppression of plasma renin activity and plasma aldosterone concentration (Ni et al., 2003). These results indicate an important central action of γ-MSH to participate in the regulation of blood pressure during ingestion of a high sodium diet. That altered processing of proopiomelanocortin in the face of a high sodium diet could be a more general feature of hypertension are observations that the high sodium diet fails to increase pituitary proopiomelanocortin messenger RNA abundance in Dahl S rats compared to Dahl R (Hao and Rabkin, 1996;Mayan et al., 1993).
Figure 2.

Blood pressure (MAP, top) and immunoreactive (IR) plasma γ-MSH concentration (bottom) in proconvertase 2 (PC2) wild type (+/+) and knockout (−/−) mice (left) and melanocortin 3 receptor (Mc3r) wild type and knockout mice during ingestion of a low sodium (LSD) vs a high sodium (HSD) diet.. From (Ni et al., 2003).
3.1.2 Melanocortin MC3receptor ( Mc3r)-Deficient Mice
We sought to determine if γ-MSH resistance also resulted in a hypertensive phenotype by measuring arterial pressure on the low vs high sodium diet in mice with targeted disruption of the melanocortin MC3or MC 4 receptor. Mc3r−/− mice exhibit a unique metabolic syndrome characterized by an increase in adipose tissue mass without obesity and with reduced energy expenditure (Butler et al., 2000; Chen et al., 2000), while Mc4r−/− mice are phenotypically obese with increased adipose tissue, hyperphagia, and insulin resistance (Huszar et al., 1997). Mc3r−/−mice had a plasma γ-MSH concentration on the low sodium diet that was more than double the value observed in wild type controls, and the value increased even further in knockout mice ingesting the high sodium diet (Fig.2 ). This suggested that these knockouts have a hormone-resistant state. Arterial pressure reflected the results in PC2−/−mice, being somewhat higher in knockout mice on the low sodium diet than in wild type mice, but markedly so in Mc3r−/−mice which had been ingesting the high sodium diet for one week (Ni et al., 2003) (Fig. 2). In contrast to PC2−/−mice, infusion of γ-MSH had no effect on arterial pressure in these Mc3r−/− mice, indicating that the restoration of MAP to normal by γ-MSH administration to hypertensive PC2−/− mice required integrity of this receptor. Mc4r−/−mice were normotensive on both the low and the high sodium diets(Ni et al., 2003). These results demonstrated that resistance to γ-MSH caused by absence of its receptor Mc3r reproduced the phenotype of hypertensive γ-MSH-deficient PC2−/− mice when ingesting the high sodium diet, but the hypertension could not be corrected by administration of exogenous peptide.
3.2 Pharmacologic Approach
We used a pharmacologic approach to interfere with neurointermediate lobe processing of proopiomelanocortin without perturbing other systems dependent on PC2. As discussed earlier, the major pathway regulating neurointermediate lobe function involves dopaminergic suppression. We treated male rats with the dopamine agonist bromocriptine (5 mg/kg intraperitoneally by daily injection) for one week while they were ingesting either the low or the high sodium diet, and compared the results to those in vehicle-treated rats. Vehicle-treated rats on the high sodium diet showed an elevation in plasma γ-MSH concentration and neurointermediate lobe γ-MSH content compared to rats ingesting the low sodium diet(Mayan et al., 2003) as seen earlier (Mayan et al., 1996); arterial pressure did not differ in the two groups. Bromocriptine treatment produced opposite results. Neither plasma γ-MSH concentration nor neurointermediate lobe γ-MSH content was elevated in bromocriptine-treated rats on the high compared with low sodium diet values and, interestingly, arterial pressure was significantly higher in the high sodium diet animals (132±3 vs 100±3 mm Hg in low sodium diet rats, p <.001) (Mayan et al., 2003). Thus, dopaminergic stimulation with bromocriptine produced relative γ-MSH deficiency during ingestion of the high sodium diet, and this was accompanied by the development of salt-sensitive hypertension. As was true with γ-MSH-deficient PC2−/−mice, infusion of the peptide at a physiologically relevant rate restored arterial pressure to control values very quickly (Mayan et al., 2003). Also like the PC2−/− mice, the hypertensive rats o n the high sodium diet had the expected suppression of plasma renin activity; plasma atrial natriuretic peptide concentration was also appropriately elevated on the high sodium diet (Mayan et al., 2003). These results indicate that pharmacologic treatment known to interfere with proopiomelanocortin processing results in γ-MSH deficiency, and that this deficiency is associated with the occurrence of salt-sensitive hypertension. This finding strengthens the contention that hypertension in PC2−/−mice is a reflection of impaired proopiomelanocortin processing into γ-MSH, since pharmacologic induction of deficiency of this peptide results in the development of salt-sensitive hypertension.
3.3 Mechanism of the Salt-Sensitive Hypertension
The mechanism(s) by which interruption of γ-MSH signaling causes hypertension is not yet clear. Plasma renin activity and plasma aldosterone concentration were equivalently suppressed, and plasma atria natriuretic peptide stimulated, during ingestion of the high sodium diet in PC2−/−mice and in bromocriptine -treated rats (Mayan et al., 2003; Ni et al., 2003), arguing that these important determinants of blood pressure were not involved. Intravenously infused γ-MSH increased sodium excretion in hypertensive PC2−/− mice as arterial pressure was falling to normal levels, but the rapidity of the blood pressure-lowering effect of the peptide makes a reduction in plasma volume from the natriuresis seem unlikely. Moreover, cerebroventricular administration of the peptide led to even more rapid correction of arterial pressure in doses which had no effect given intravenously, so that natriuresis could not be the mechanism by which blood pressure was lowered. However, the natriuretic property of the peptide could certainly interact with its blood pressure-lowering action to participate in the overall long-term regulation of the circulation and body fluid volumes. The central site of action of the peptide given to γ-MSH-deficient mice suggests that it may possibly reduce central sympathetic outflow; data presented below support this possibility.
The time course of the development of this salt-sensitive hypertension was studied in rats implanted intraabdominally with radiotransmitters connected to a catheter inserted into the distal aorta. After recovery from surgery, rats were again anesthetized and a microosmotic pump placed subcutaneously in the back. In half the rats (n=6), this pump contained NDP-γ-MSH ([Norleu3,D-Phe6]-γ-MSH) dissolved in normal saline delivered to the interstitial space at an estimated rate of 12 pmol/h; this compound is a stable analog of γ-MSH (Hruby et al., 2007) and possesses equivalent natriuretic properties (Chen et al., 1997b; Ni et al., 1998). The other six rats received pumps with normal saline alone. All rats were then fed the high sodium diet and received daily intraperitoneal injections of bromocriptine, 5 mg/kg. Blood pressure and heart rate were recorded by telemetry for one hour each morning for three weeks; after 14 days the bromocriptine injections were discontinued. Rats receiving normal saline by osmotic pump developed hypertension by Day 4; this reached a peak arterial pressure of ~130 –135 mm Hg by Day 7, where it remained stable until the cessation of the bromocriptine injections after Day 14 (Fig. 3) (Ni and Humphreys, 2007). At this time arterial pressure started to fall, reaching initial values by Day 18. In contrast to these vehicle-treated rats, the animals receiving NDP-γ-MSH remained normotensive throughout the whole 21-day study period. No changes in heart rate were observed in either group.
Figure 3.

Mean arterial pressure and heart rate in conscious, unrestrained rats ingesting a high sodium diet (HSD) while receiving daily injections of bromocriptine intraperitoneally, 0.5 mg/kg. The rats were intstrumented with radiotelemetry transmitters for the recording; they were implanted with osmotic miniumps to deliver normal saline vehicle (filled circles) or the stable γ-MSH analog NDP-γ-MSH. Bromocriptine injections were discontinued after 14 days. From (Ni and Humphreys, 2007).
These results demonstrate several things about the salt-sensitive hypertension in this model of γ-MSH deficiency. First, the hypertension resulting from the interaction of the high sodium diet with bromocriptine administration takes several days to develop, and a similar period of time to resolve on discontinuation of the drug administration (Fig. 3). Since rats adjust their rate of sodium excretion to match intake with a short half time of ~2–3 hrs (Holtzman et al., 1988)vs ~24 h in humans (Sagnella et al., 1990; Strauss et al., 1958), this observation suggests that renal sodium retention and attendant plasma volume expansion may not be the sole underlying pathogenetic mechanism of the hypertension as predicted by the model developed by Guyton and colleagues (Guyton, 1991). Second, the results indicate that continuous administration of the γ-MSH analog NDP-γ-MSH prevented the development of hypertension in bromocriptine-treated rats fed the high sodium diet. This observation parallels one we reported in PC2−/−mice (Ni et al., 2003), and provides further evidence of the causal role of γ-MSH deficiency in the salt-sensitive hypertension. Third, the constancy of heart rate in vehicle-treated rats despite the higher MAP raises the possibility of altered baroreflex responses in these animals with γ-MSH deficiency. Finally, the similarity of blood pressure in these conscious, unrestrained rats to the value measured in anesthetized animals (Mayan et al., 2003) indicates that the stressors of anesthesia and surgical preparation do not contribute appreciably to the hypertension.
4. Central Actions of Exogenous Melanocortin Peptides on Blood Pressure
4.1 γ-MSH
As mentioned earlier, initial work drew attention to the property of γ-MSH to elevate blood pressure and heart rate when injected intravenously(reviewed in (Gruber and Callahan, 1989)). This was due to central stimulation of sympathetic outflow, since the effect was blocked by pithing and by adrenergic blocking drugs (Gruber and Callahan, 1989). This action was in contrast to α-MSH, which exerted no effect on blood pressure or heart rate when injected intravenously. The basis for this difference between the two peptides lies in their structure at the C terminus: the C-terminus of α-MSH consists of Lys-Pro-Val, while in γ1-MSH it is Arg-Phe-NH2 and in γ2-MSH it is Arg-Phe-Gly. Structure-function studies quickly identified that this Arg-Phe sequence in γ-MSH was critical for the hypertensinogenic action of the peptide: synthetic peptides lacking the sequence did not raise blood pressure, whereas truncated peptides which retained the C-terminal Arg-Phe sequence possessed potent blood pressure elevating properties (Nijsen et al., 2000; Van Bergen et al., 1995; Van Bergen et al., 1997; Van Bergen et al., 1996). The truncated peptides which elevated blood pressure in vivo had little or no activity at the melanocortin MC3or MC 4 receptors in vitro (Wikberg et al., 2000), and inhibitors of these receptors, agouti protein, SHU9119 or SHU9005, did not interfere with the effect of γ-MSH to raise blood pressure in intact animals (Kunos G, 1997; Li et al., 1996). These observations all suggested that this action of γ-MSH was mediated not by melanocortin MC3 or MC4receptors but rather by an entirely different type of receptor expressed in the CNS. Attention was directed to a Phe-Met-Arg-Phe-NH2 (FMRFamide) gated sodium channel. FMRFamide is a cardioexcitatory peptide initially isolated from ganglia of the clam Macrocallista nimbosa(Price and Greenberg, 1977). In 1995, the cloning of an amiloride sensitive, FMRFamide gated sodium channel from snail nervous tissue was reported (Lingueglia et al., 1995) and was found to be the receptor responsible for FMRFamide’s cardioexcitatory properties. Studies in the Xenopus expression system showed that the channel caused a large inward sodium current when exposed to FMRFamide; this current was blocked by amiloride (Lingueglia et al., 2006). This channel has been implicated in the development of salt-sensitive hypertension in rats (Huang and Leenen, 2002; Nishimura et al., 2000). The conclusions from these pharmacologic studies have recently been confirmed in Mc3r−/−and Mc4r−/−mice: blood pressure and heart rate increased equivalently following γ-MSH injection in to both wild type and knockout mice (Ni et al., 2006b), supporting the conclusion that these receptors are not involved in the sympathetic stimulation resulting from intravenous γ2-MSHinjection. However, injection of benzamil, an amiloride analog, directly into the lateral cerebral ventricle of wild type mice completely blocked the increase in blood pressure and heart rate following γ-MSH injection (Ni et al., 2006b). This strongly suggests that the peptide interacts with the FMRFamide gated sodium channel to cause sympathoexcitation. Microinjection of γ-MSH into the nucleus of the solitary tract actually lowers blood pressure through reduction in SNS activity (De Wildt et al., 1994; Li et al., 1996); since the NTS is a key relay site in the baroreflex arc, this raises the possibility that γ-MSH participates in baroreflex function.
These observations help to explain the earlier observations of Gruber and colleagues that low doses of γ-MSH were natriuretic whereas higher doses raised blood pressure through sympathetic stimulation (Gruber and Callahan, 1989; Lymangrover et al., 1985): low concentrations of the peptide interact with renal melanocortin MC3 receptors to cause natriuresis without activating the FMRFamide sodium channel in the CNS, while higher concentrations activate this channel and cause increased sympathetic outflow which overrides any natriuretic effect via the renal melanocortin MC3receptor. They also help to resolve the apparent paradox between the protective effect of γ-MSH to prevent salt-sensitive hypertension during ingestion of a high sodium diet and its hypertensinogenic action via the FMRFamide gated sodium channel: physiologic concentrations of the peptide during ingestion of a high sodium diet interact with melanocortin MC3 receptors in the CNS, perhaps in the nucleus of the solitary tract, to act as a brake on sympathetic activation normally accompanying the high sodium diet, and in addition lead to natriuresis, while supraphysiologic concentrations are required for interaction with the sodium channel and the development of hypertension from increased sympathetic nervous systemactivity.
4.2 α-MSH
In contrast to γ-MSH, α-MSH has no effect on blood pressure when injected intravenously, but causes hypertension when administered directly into the cerebroventricular system (Hill and Dunbar, 2002; Matsumura et al., 2002)due to sympathetic excitation. These results were confirmed in Mc4r wild type and knockout mice: an intravenous dose of this peptide had no effect on blood pressure, but caused ~20 mm Hg increase in blood pressure when injected into a lateral cerebral ventricle. This response was not observed in Mc4r−/− mice, in which no change in blood pressure occurred and confirming that sympathetic activation by centrally administered α-MSH was mediated by the Mc4r (Ni et al., 2006b).
5. γ-MSH and Glucose Metabolism
5.1 Interruption of γ-MSH Signaling
5.1.1 Genetic Models
A close association between salt-sensitive hypertension and insulin resistance has been recognized for more than twenty years, although the basis for this association is not clear (Ferrannini et al., 1987; Fujita, 2007; Reaven et al., 1996). The Mc3r−/− mouse exhibits mild insulin resistance which may require an increase in dietary fat to be fully expressed (Butler, 2006; Chen et al., 2000). Since interruption of γ-MSH signaling leads to hypertension during ingestion of a high sodium diet, it became logical to ask if rodent models of impaired γ-MSH signaling and salt-sensitive hypertension are accompanied by impaired glucose metabolism. Fasting blood glucose and insulin concentrations did not differ between wild type and knockout mice on the normal sodium diet, and were not affected by the high sodium diet in wild type mice. However, both PC2−/−and Mc3r−/− mice fed the high sodium diet developed fasting hyperglycemia and hyperinsulinemia in association with hypertension (Fig. 4) (Ni and Humphreys, 2008). Glucose tolerance tests in the Mc3r knockouts showed a significantly greater area under the curve when ingesting the high compared to a normal sodium diet, and an insulin tolerance test demonstrated a blunted decrease in plasma glucose concentration in Mc3r−/− fed the high sodium diet compared to the other groups. No major impairment in insulin secretion was identified (Ni and Humphreys, 2008). These results indicate that the high sodium diet leads to the development of impaired glucose metabolism, with the characteristics of insulin resistance, as well as hypertension in these mice with impaired γ-MSH signaling. PC2−/− mice exhibit delayed processing of proinsulin to insulin, with an increase in the ratio of proinsulin to insulin in both pancreas and serum (Furuta et al., 1998), and it is possible that the hyperinsulinemia observed during ingestion of the high sodium diet could reflect an increase in proinsulin rather than insulin. However, the normal plasma concentration of insulin in PC2−/−while ingesting the normal sodium diet, and the nearly identical development of hyperglycemia and hyperinsulinemia during ingestion of the high sodium diet in Mc3r−/− mice with a normal complement of PC2, argue strongly against this possibility.
Figure 4.

Mean arterial pressure (MAP, mm Hg), heart rate (HR), blood glucose, and plasma insulin concentrations in proconvertase 2 (PC2) wild type (+/+) and knockout (−/−) and Mc3r wild type and knockout mice during ingestion of a normal sodium (NSD) or high sodium diet (HSD). Both knockout strains of mice developed hyperglycemia and hyperinsulinemia as well as hypertension while ingesting the high sodium diet. From (Ni and Humphreys, 2008).
5.1.2 Pharmacologic Model
To determine if this abnormal glucose metabolism was a function of altered γ-MSH signaling, or reflected some other consequence of the genetically modified mouse models, the bromocriptine-treated rat model was studied during ingestion of the high sodium diet. Bromocriptine given systemically interacts with dopaminergic receptors throughout the body and has an acute renal vasodilatory and hypotensive action in normal rats (Stier et al., 1982). It also lowers blood pressure in the spontaneously hypertensive rat (SHR) (Kanayama et al., 1987; Nagahama et al., 1984; Oguro et al., 1992; Racz et al., 1986; van den Buuse and Lambrechts, 1989), DOCA-salt hypertensive rats (Nagahama et al., 1985), and normal and hypertensive humans (Franchi et al., 2001; Kok et al., 2006; Mannelli et al., 1984; Sowers, 1981). The possible basis for this hypotensive action is a reduction in sympathetic activity, as plasma and urine norepinephrine levels are reduced in most studies in rats (Kanayama et al., 1987; Nagahama et al., 1984; 1985; Oguro et al., 1992; Racz et al., 1986)and humans (Carey et al., 1983; Mannelli et al., 1984; Sowers, 1981). Thus, the development of salt-sensitive hypertension after one week’s treatment with bromocriptineis perhaps surprising given these reports of its hypotensive action, and is an indicator of the importance of the accompanying γ-MSH deficiency coupled with the high sodium diet in blood pressure regulation. Male rats fed the high sodium diet and treated with bromocriptine had fasting hyperglycemia (blood glucose 116±4 vs 102±2 mg/dL, p <0.01) and hyperinsulinemiaas well as hype rtension (133±3 vs 103±3 mm Hg, p <0.001) (Ni et al., 2009; Van Dijk et al., 2006), paralleling the results in the PC2−/−and Mc3r−/− mouse models. This result too is surprising, since bromocriptine has been shown to ameliorate glucose metabolism and increase insulin sensitivity in obese humans (Kok et al., 2006; Pijl et al., 2000)and the obese Syrian hamster (Luo et al., 1999). Indeed, the drug is the subject of a therapeutic trial in patients with type 2 diabetes mellitus (Gaziano et al., 2010). Bromocriptine treatment in the rat appears to interact with the high sodium diet in a manner not present with either treatment alone and indeed overcomes the hypotensive and insulin -sensitizing actions of bromocriptine seen during ingestion of more normal amounts of dietary sodium.
5.2 Mechanism of Abnormal Glucose Metabolism
The relationship between the hypertension and the fasting hyperglycemia was examined in bromocriptine-treated vs vehicle –treated rats fed the high sodium diet. Infusion of γ2-MSH (0.8 pmol/min intravenously) rapidly lowered arterial pressure, as had been shown previously (Mayan et al., 2003) so that after 15 min blood pressure was no different from that seen in vehicle-treated rats. Of interest, fasting blood glucose was also corrected in an equivalent manner, indicating a close association of these two consequences of γ-MSH deficiency(Fig. 5). The same results occurred when blood pressure was lowered with the adrenergic receptor blocking agent phentolamine (3 μg/kg/min): after only 15 min infusion the hypertension and the hyperglycemia of the bromocriptine-high sodium diet rats was no different from vehicle -treated rats. This was not the case when the peripheral vasodilator hydralazine (0.1 μg/kg/min) was used to lower blood pressure. As with the studies infusing γ2-MSH and phentolamine, hydralazine lowered pressure to the same level as vehicle -treated rats but had no significant effect on blood glucose concentration after 15 min; after 60 min blood pressure remained stable, and blood glucose concentration had fallen only trivially (Ni et al., 2009) (Fig. 5). These results lead to several important conclusions. First, the hypertension and hyperglycemia of bromocriptine-high sodium diet rats are both rapidly corrected with administration of γ2-MSH or phentolamine. This suggests that a common adrenergic mechanism may underlie both consequences of the drug/diet treatment. Second, the peripherally acting vasodilator hydralazine was successful in lowering blood pressure but did not have a major effect on blood glucose concentration. This suggests that a current hypothesis stating that insulin resistance reflects impaired blood flow to skeletal muscle (Duplain et al., 2001; Laakso et al., 1990; Steinberg et al., 1994)may not fully explain the observations shown in hydralazine-infused rats. Third, the hyperinsulinemia in bromocriptine-treated rats ingesting the high sodium diet could itself be a stimulus for increased sympathetic outflow via a central mechanism (Muntzel et al., 2007; Muntzel et al., 1994). Consistent with increased sympathetic nerve activity is the observation that plasma noradrenaline concentration was twice as high in bromocriptine-treated rats eating the high sodium diet as in the other groups; infusion of γ2-MSHa t a low rate (0.4 pmol/min) lowered plasma noradrenaline concentration from 184±37 to 98±17 pg/ml as blood pressure fell from 131±3 to 104 ±3 mm Hg (Ni et al., 2009). Peripheral vasoconstriction and sympathetically-and insulin -mediated increase sin tubular sodium reabsorption (Baum, 1987; DeFronzo et al., 1975; DiBona and Kopp, 1997)could both contribute to the hypertension.
Figure 5.

Mean arterial pressure (mm Hg, left) and blood glucose concentration (mg/dL, right) in bromocriptine-(closed circles) or vehicle- (open circles) treated rats fed the high sodium diet. Bromocriptine-treated rats were hypertensive; intravenous infusion of γ2-MSH (top), phentolamine (middle), or hydralazine (bottom) at the infusion rates indicated rapidly corrected the hypertension. However, only γ-MSH and phentolamine corrected the hyperglycemia; hydralazine had a trivial effect on blood glucose concentration despite its effect on blood pressure. *, bromocriptine value significantly greater than vehicle, P < 0.01 or greater; †, value significantly different from corresponding control value, P < 0.01 or greater, repeated-measures ANOVA. From (Ni et al., 2009).
Bromocriptine treatment has endocrine actions in addition to suppression of pituitary proopiomelanocortin processing. Its primary use in clinical medicine has been to suppress release of prolactin, and studies in both rats and humans indicate that it does cause reductions in plasma prolactin concentration (Kok et al., 2006; Mannelli et al., 1984; Nagahama et al., 1984; 1985; Ni et al., 2009; Stier et al., 1982). Prolactin infusion leads to natriuresis in the rat through an inhibition of proximal tubular Na, K-ATPase, an effect blocked by a dopamine D1 receptor antagonist (Ibarra et al., 2005). Reduced prolactin levels after bromocriptine treatment could favor sodium retention and lead to hypertension. Consistent with dopaminergic regulation of proximal tubule Na, K-ATPase is the observation that bromocriptine itself stimulates activity of this enzyme in vitro(Hussain et al., 1997), which normally would be expected to increase sodium reabsorption. These actions of bromocriptine do not seem to be major contributors to the salt-sensitive hypertension, since the blood pressure is rapidly restored to normal during infusion of γ2-MSH. Bromocriptine treatment has also been shown to increase vasopressin messenger RNA in the pituitary neural lobe(Carter et al., 1993). This peptide hormone can stimulate natriuresis in some conditions (Humphreys et al., 1970)and is a participant in the regulation of blood pressure (Johnston, 1985). Although each of these pathways in bromocriptine-treated rats could influence blood pressure during the ingestion of a high sodium diet, the currently available data argue for a major role of increased sympathetic nerve activity in the abnormal glucose metabolism observed in rodents with impaired γ-MSH signaling during ingestion of a high sodium diet. By the same token, increased sympathetic drive must be a major determinant of the hypertension in these rodents. A high sodium diet activates neurons in brain regions associated with cardiovascular control, including the nucleus of the solitary tract and the caudal and rostral ventrolateral medulla (Adams et al., 2007; Bealer and Metcalf, 2005; Isogai et al., 2005). Such increased sympathetic outflow stimulated by a high sodium diet therefore provides a link between the salt-sensitive hypertension and the abnormal glucose metabolism in rodents with impaired γ-MSH signaling.
6. Overview of the γ-MSH System
The findings reported above indicate that the γ-MSH system plays an important role in both sodium and glucose metabolism. This is shown schematically in Fig. 6. In the normal circumstance, a high sodium diet stimulates an increase in both circulating and presumably central γ-MSH levels. This increase in γ-MSH serves to counteract the hypertensinogenic potential of the high sodium diet centrally and may participate in the decrease in renal tubular sodium reabsorption to bring sodium excretion into balance with the elevated sodium intake. This prevents a major increase in plasma volume and helps to maintain a normal blood pressure. However, when synthesis and secretion of γ-MSH are impaired ( PC2−/−mice, bromocriptine treatment in rats) or signaling interrupted (Mc3r−/−mice), the high sodium diet now manifests its hypertensinogenic potential, presumably through activation of sympathetic nervous activity to result in hypertension. This situation is also accompanied by the development of abnormal glucose metabolism with the characteristics of insulin resistance. This scheme integrates the numerous observations summarized above, and provides a platform on which to base further experiments to characterize these interactions more fully. As yet unknown is the function of this system in humans in response to manipulations of dietary salt intake and whether disruption of this system could represent one cause of salt-sensitive hypertension. These questions are clearly important to address in future studies.
Figure 6.

Schematic view of the role of γ-MSH during ingestion of a high sodium diet. See Section 6 for details.
Acknowledgments
Work reported in this article was supported in part by grant HL68871 from the National Institutes of Health, USA.
Footnotes
Submitted in conjunction with the 6th International Melanocortin Meeting, Utrecht, The Netherlands, July 8-11, 2010
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Madden CJ, Sved AF, Stocker SD. Increased dietary salt enhances sympathoexcitatory and sympathoinhibitory responses from the rostral ventrolateral medulla. Hypertension. 2007;50:354–359. doi: 10.1161/HYPERTENSIONAHA.107.091843. [DOI] [PubMed] [Google Scholar]
- Allen RG, Peng B, Pellegrino MJ, Miller ED, Grandy DK, Lundblad JR, Washburn CL, Pintar JE. Altered processing of pro-orphanin FQ/nociceptin and pro-opiomelanocortin-derived peptides in the brains of mice expressing defective prohormone convertase 2. J Neurosci. 2001;21:5864–5870. doi: 10.1523/JNEUROSCI.21-16-05864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autelitano DJ, Lundblad JR, Blum M, Roberts JL. Hormonal regulation of POMC gene expression. Annu Rev Physiol. 1989;51:715–726. doi: 10.1146/annurev.ph.51.030189.003435. [DOI] [PubMed] [Google Scholar]
- Baum M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J Clin Invest. 1987;79:1104–1109. doi: 10.1172/JCI112925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bealer SL, Metcalf CS. Increased dietary sodium enhances activation of neurons in the medullary cardiovascular pathway during acute sodium loading in the rat. Auton Neurosci. 2005;117:33–40. doi: 10.1016/j.autneu.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Berman Y, Mzhavia N, Polonskaia A, Furuta M, Steiner DF, Pintar JE, Devi LA. Defective prodynorphin processing in mice lacking prohormone convertase PC2. J Neurochem. 2000;75:1763–1770. doi: 10.1046/j.1471-4159.2000.0751763.x. [DOI] [PubMed] [Google Scholar]
- Butler AA. The melanocortin system and energy balance. Peptides. 2006;27:281–290. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- Callahan MF, Kirby RF, Lymangrover JR, Johnson AK, Gruber KA. Cardiovascular mechanisms of gamma (gamma) 2-MSH. Clin Exp Hypertens A. 1984;6:1727–1730. doi: 10.3109/10641968409046067. [DOI] [PubMed] [Google Scholar]
- Carey RM, Van Loon GR, Baines AD, Kaiser DL. Suppression of basal and stimulated noradrenergic activities by the dopamine agonist bromocriptine in man. J Clin Endocrinol Metab. 1983;56:595–602. doi: 10.1210/jcem-56-3-595. [DOI] [PubMed] [Google Scholar]
- Carter DA, Pardy K, Murphy D. Regulation of vasopressin gene expression: changes in the level, but not the size, of vasopressin mRNA following endocrine manipulations. Cell Mol Neurobiol. 1993;13:87–95. doi: 10.1007/BF00712991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramohan G, Durham N, Sinha S, Norris K, Vaziri ND. Role of gamma melanocyte-stimulating hormone-renal melanocortin 3 receptor system in blood pressure regulation in salt-resistant and salt-sensitive rats. Metabolism. 2009;58:1424–1429. doi: 10.1016/j.metabol.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Chandramohan G, Ni XP, Kalinyak JE, Humphreys MH. Dietary sodium modulates mRNA abundance of enzymes involved in pituitary processing of proopiomelanocortin. Pituitary. 2001;4:231–237. doi: 10.1023/a:1020746414046. [DOI] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell. 1997a;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Chen XW, Ying WZ, Valentin JP, Ling KT, Lin SY, Wiedemann E, Humphreys MH. Mechanism of the natriuretic action of gamma-melanocyte-stimulating hormone. Am J Physiol. 1997b;272:R1946–1953. doi: 10.1152/ajpregu.1997.272.6.R1946. [DOI] [PubMed] [Google Scholar]
- Day R, Schafer MK, Watson SJ, Chretien M, Seidah NG. Distribution and regulation of the prohormone convertases PC1 and PC2 in the rat pituitary. MolEndocrinol. 1992;6:485 –497. doi: 10.1210/mend.6.3.1316544. [DOI] [PubMed] [Google Scholar]
- De Wildt DJ, Van der Ven JC, Van Bergen P, De Lang H, Versteeg DH. A hypotensive and bradycardic action of gamma 2-melanocyte-stimulating hormone (gamma 2-MSH) microinjected into the nucleus tractus solitarii of the rat. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:50–56. doi: 10.1007/BF00178205. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–855. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. 1997;77:75–197. doi: 10.1152/physrev.1997.77.1.75. [DOI] [PubMed] [Google Scholar]
- Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- Ellacott KL, Cone RD. The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond B Biol Sci. 2006;361:1265–1274. doi: 10.1098/rstb.2006.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, Buzzigoli G, Bonadonna R, Giorico MA, Oleggini M, Graziadei L, Pedrinelli R, Brandi L, Bevilacqua S. Insulin resistance in essential hypertension. N Engl J Med. 1987;317:350–357. doi: 10.1056/NEJM198708063170605. [DOI] [PubMed] [Google Scholar]
- Franchi F, Lazzeri C, Barletta G, Ianni L, Mannelli M. Centrally mediated effects of bromocriptine on cardiac sympathovagal balance. Hypertension. 2001;38:123–129. doi: 10.1161/01.hyp.38.1.123. [DOI] [PubMed] [Google Scholar]
- Fujita T. Insulin resistance and salt-sensitive hypertension in metabolic syndrome. Nephrol Dial Transplant. 2007;22:102–107. doi: 10.1093/ndt/gfm409. [DOI] [PubMed] [Google Scholar]
- Furuta M, Carroll R, Martin S, Swift HH, Ravazzola M, Orci L, Steiner DF. Incomplete processing of proinsulin to insulin accompanied by elevation of Des-31,32 proinsulin intermediates in islets of mice lacking active PC2. J Biol Chem. 1998;273:3431–3437. doi: 10.1074/jbc.273.6.3431. [DOI] [PubMed] [Google Scholar]
- Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci U S A. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta M, Zhou A, Webb G, Carroll R, Ravazzola M, Orci L, Steiner DF. Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J Biol Chem. 2001;276:27197–27202. doi: 10.1074/jbc.M103362200. [DOI] [PubMed] [Google Scholar]
- Gaziano JM, Cincotta AH, O’Connor CM, Ezrokhi M, Rutty D, Ma ZJ, Scranton RE. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33:1503–1508. doi: 10.2337/dc09-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getting SJ, Lam CW, Chen AS, Grieco P, Perretti M. Melanocortin 3 receptors control crystal-induced inflammation. Faseb J. 2006;20:2234–2241. doi: 10.1096/fj.06-6339com. [DOI] [PubMed] [Google Scholar]
- Gruber KA, Callahan MF. ACTH-(4–10) through gamma-MSH: evidence for a new class of central autonomic nervous system-regulating peptides. Am J Physiol. 1989;257:R681–694. doi: 10.1152/ajpregu.1989.257.4.R681. [DOI] [PubMed] [Google Scholar]
- Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- Hao J, Rabkin SW. Differences in pituitary expression of proopiomelanocortin in Dahl salt-resistant and salt-sensitive rats on a high salt diet. Can J Physiol Pharmacol. 1996;74:657–662. [PubMed] [Google Scholar]
- Hill C, Dunbar JC. The effects of acute and chronic alpha melanocyte stimulating hormone (alphaMSH) on cardiovascular dynamics in conscious rats. Peptides. 2002;23:1625–1630. doi: 10.1016/s0196-9781(02)00103-1. [DOI] [PubMed] [Google Scholar]
- Holtzman EJ, Braley LM, Williams GH, Hollenberg NK. Kinetics of sodium homeostasis in rats: rapid excretion and equilibration rates. Am J Physiol. 1988;254:R1001–1006. doi: 10.1152/ajpregu.1988.254.6.R1001. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Cai M, Cain JP, Mayorov AV, Dedek MM, Trivedi D. Design, synthesis and biological evaluation of ligands selective for the melanocortin-3 receptor. Curr Top Med Chem. 2007;7:1107–1119. doi: 10.2174/156802607780906645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BS, Leenen FH. Brain amiloride-sensitive Phe-Met-Arg-Phe-NH(2)--gated Na(+) channels and Na(+)-induced sympathoexcitation and hypertension. Hypertension. 2002;39:557–561. doi: 10.1161/hy02t2.103004. [DOI] [PubMed] [Google Scholar]
- Humphreys MH. Gamma-MSH, sodium metabolism, and salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2004;286:R417–430. doi: 10.1152/ajpregu.00365.2003. [DOI] [PubMed] [Google Scholar]
- Humphreys MH, Friedler RM, Earley LE. Natrriuresis produced by vasopressin or hemorrhage during water diuresis in the dog. Am J Physiol. 1970;219:658–665. doi: 10.1152/ajplegacy.1970.219.3.658. [DOI] [PubMed] [Google Scholar]
- Hussain T, Abdul-Wahab R, Lokhandwala MF. Bromocriptine stimulates Na+, K(+)-ATPase in renal proximal tubules via the cAMP pathway. Eur J Pharmacol. 1997;321:259–263. doi: 10.1016/s0014-2999(97)00039-3. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Ibarra F, Crambert S, Eklof AC, Lundquist A, Hansell P, Holtback U. Prolactin, a natriuretic hormone, interacting with the renal dopamine system. Kidney Int. 2005;68:1700–1707. doi: 10.1111/j.1523-1755.2005.00586.x. [DOI] [PubMed] [Google Scholar]
- Isogai O, Tsukamoto K, Masubuchi Y, Tomioka S, Suzuki T, Kawato H, Yajima Y, Kasamaki Y, Ito S, Kanmatsuse K. High salt diet enhances cardiovascular responses from the nucleus tractus solitarius and ventrolateral medulla of Sprague-Dawley rats. Clin Exp Hypertens. 2005;27:33–44. doi: 10.1081/ceh-200044252. [DOI] [PubMed] [Google Scholar]
- Johnston CI. Vasopressin in circulatory control and hypertension. J Hypertens. 1985;3:557–569. doi: 10.1097/00004872-198512000-00001. [DOI] [PubMed] [Google Scholar]
- Kanayama Y, Kohno M, Takaori K, Itoh S, Yasunari K, Takeda T. Involvement of sympathetic nervous system inhibition in the hypotensive effect of bromocriptine in spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1987;14:141–144. doi: 10.1111/j.1440-1681.1987.tb00969.x. [DOI] [PubMed] [Google Scholar]
- Kok P, Roelfsema F, Frolich M, van Pelt J, Stokkel MP, Meinders AE, Pijl H. Activation of dopamine D2 receptors simultaneously ameliorates various metabolic features of obese women. Am J Physiol Endocrinol Metab. 2006;291:E1038–1043. doi: 10.1152/ajpendo.00567.2005. [DOI] [PubMed] [Google Scholar]
- Kunos G, LSJ, Varga K, Archer P, Kesterson RA, Cone RD, Hruby VJ, Sharma SD. Novel neural pathways of cardiovascular control α– by and γ-MSH. Fundam Clin Pharmacol. 1997;11(Suppl 1):44s–483. [Google Scholar]
- Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest. 1990;85:1844–1852. doi: 10.1172/JCI114644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Varga K, Archer P, Hruby VJ, Sharma SD, Kesterson RA, Cone RD, Kunos G. Melanocortin antagonists define two distinct pathways of cardiovascular control by alpha- and gamma-melanocyte-stimulating hormones. J Neurosci. 1996;16:5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Chaves C, Wiedemann E, Humphreys MH. A gamma-melanocyte stimulating hormone-like peptide causes reflex natriuresis after acute unilateral nephrectomy. Hypertension. 1987;10:619–627. doi: 10.1161/01.hyp.10.6.619. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Champigny G, Lazdunski M, Barbry P. Cloning of the amiloride-sensitive FMRFamide peptide-gated sodium channel. Nature. 1995;378:730–733. doi: 10.1038/378730a0. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Deval E, Lazdunski M. FMRFamide-gated sodium channel and ASIC channels: a new class of ionotropic receptors for FMRFamide and related peptides. Peptides. 2006;27:1138–1152. doi: 10.1016/j.peptides.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Luo S, Liang Y, Cincotta AH. Intracerebroventricular administration of bromocriptine ameliorates the insulin-resistant/glucose-intolerant state in hamsters. Neuroendocrinology. 1999;69:160–166. doi: 10.1159/000054415. [DOI] [PubMed] [Google Scholar]
- Lymangrover JR, Buckalew VM, Harris J, Klein MC, Gruber KA. Gamma-2MSH is natriuretic in the rat. Endocrinology. 1985;116:1227–1229. doi: 10.1210/endo-116-3-1227. [DOI] [PubMed] [Google Scholar]
- Mannelli M, Delitala G, De Feo ML, Maggi M, Cuomo S, Piazzini M, Guazzelli R, Serio M. Effects of different dopaminergic antagonists on bromocriptine-induced inhibition of norepinephrine release. J Clin Endocrinol Metab. 1984;59:74–78. doi: 10.1210/jcem-59-1-74. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Tsuchihashi T, Abe I, Iida M. Central alpha-melanocyte-stimulating hormone acts at melanocortin-4 receptor to activate sympathetic nervous system in conscious rabbits. Brain Res. 2002;948:145–148. doi: 10.1016/s0006-8993(02)03045-7. [DOI] [PubMed] [Google Scholar]
- Mayan H, Ling KT, Kalinyak JE, Sharma A, Wiedemann E, Humphreys MH. Modulation by dietary salt intake of pituitary proopiomelanocortin (POMC) messenger RNA (mRNA) abundance in normotensive and hypertensive rats. Hypertension. 1993;22:417. [Google Scholar]
- Mayan H, Ling KT, Lee EY, Wiedemann E, Kalinyak JE, Humphreys MH. Dietary sodium intake modulates pituitary proopiomelanocortin mRNA abundance. Hypertension. 1996;28:244–249. doi: 10.1161/01.hyp.28.2.244. [DOI] [PubMed] [Google Scholar]
- Mayan H, Ni XP, Almog S, Humphreys MH. Suppression of gamma-melanocyte-stimulating hormone secretion is accompanied by salt-sensitive hypertension in the rat. Hypertension. 2003;42:962–967. doi: 10.1161/01.HYP.0000097601.83235.F8. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Crespo R, Joseph T, Onwumere O. Dietary salt loading exacerbates the increase in sympathetic nerve activity caused by intravenous insulin infusion in rats. Metabolism. 2007;56:373–379. doi: 10.1016/j.metabol.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol. 1994;267:R1350–1355. doi: 10.1152/ajpregu.1994.267.5.R1350. [DOI] [PubMed] [Google Scholar]
- Nagahama S, Chen YF, Oparil S. Mechanism of the depressor effect of bromocriptine in the spontaneously hypertensive rat. J Pharmacol Exp Ther. 1984;228:370–375. [PubMed] [Google Scholar]
- Nagahama S, Chen YF, Oparil S. Enhanced depressor effect of bromocriptine in the DOCA/NaCl hypertensive rat. Am J Physiol. 1985;249:H64–70. doi: 10.1152/ajpheart.1985.249.1.H64. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, IA, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- Ni XP, Bhargava A, Pearce D, Humphreys MH. Modulation by dietary sodium intake of melanocortin 3 receptor mRNA and protein abundance in the rat kidney. Am J Physiol Regul Integr Comp Physiol. 2006a;290:R560–567. doi: 10.1152/ajpregu.00279.2005. [DOI] [PubMed] [Google Scholar]
- Ni XP, Butler AA, Cone RD, Humphreys MH. Central receptors mediating the cardiovascular actions of melanocyte stimulating hormones. J Hypertens. 2006b;24:2239–2246. doi: 10.1097/01.hjh.0000249702.49854.fa. [DOI] [PubMed] [Google Scholar]
- Ni XP, Humphreys MH. Prevention of salt-induced hypertension by an analog of gamma-melanocyte-stimulating hormone in the rat. Am J Hypertens. 2007;20:862–865. doi: 10.1016/j.amjhyper.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Ni XP, Humphreys MH. Abnormal glucose metabolism in hypertensive mice with genetically interrupted gamma-melanocyte stimulating hormone signaling fed a high-sodium diet. Am J Hypertens. 2008;21:1284–1287. doi: 10.1038/ajh.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni XP, Kesterson RA, Sharma SD, Hruby VJ, Cone RD, Wiedemann E, Humphreys MH. Prevention of reflex natriuresis after acute unilateral nephrectomy by melanocortin receptor antagonists. Am J Physiol. 1998;274:R931–938. doi: 10.1152/ajpregu.1998.274.4.R931. [DOI] [PubMed] [Google Scholar]
- Ni XP, Pearce D, Butler AA, Cone RD, Humphreys MH. Genetic disruption of gamma-melanocyte-stimulating hormone signaling leads to salt-sensitive hypertension in the mouse. J Clin Invest. 2003;111:1251–1258. doi: 10.1172/JCI16993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni XP, van Dijk C, Pearce D, Humphreys MH. Evidence for a noradrenergic mechanism causing hypertension and abnormal glucose metabolism in rats with relative deficiency of gamma-melanocyte-stimulating hormone. Exp Physiol. 2009;94:867–876. doi: 10.1113/expphysiol.2009.046748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijsen MJ, de Ruiter GJ, Kasbergen CM, Hoogerhout P, de Wildt DJ. Relevance of the C -terminal Arg-Phe sequence in gamma(2)-melanocyte-stimulating hormone (gamma(2)-MSH) for inducing cardiovascular effects in conscious rats. Br J Pharmacol. 2000;131:1468–1474. doi: 10.1038/sj.bjp.0703709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Ohtsuka K, Takahashi H, Yoshimura M. Role of FMRFamide-activated brain sodium channel in salt-sensitive hypertension. Hypertension. 2000;35:443–450. doi: 10.1161/01.hyp.35.1.443. [DOI] [PubMed] [Google Scholar]
- Oguro M, Takeda K, Itoh H, Takesako T, Tanaka M, Takenaka K, Hirata M, Nakata T, Tanabe S, Hayashi J, et al. Role of sympathetic nerve inhibition in the vasodepressor effect of bromocriptine in normotensive and hypertensive rats. Jpn Circ J. 1992;56:943–949. doi: 10.1253/jcj.56.943. [DOI] [PubMed] [Google Scholar]
- Oyarce AM, Hand TA, Mains RE, Eipper BA. Dopaminergic regulation of secretory granule-associated proteins in rat intermediate pituitary. J Neurochem. 1996;67:229–241. doi: 10.1046/j.1471-4159.1996.67010229.x. [DOI] [PubMed] [Google Scholar]
- Pijl H, Ohashi S, Matsuda M, Miyazaki Y, Mahankali A, Kumar V, Pipek R, Iozzo P, Lancaster JL, Cincotta AH, DeFronzo RA. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care. 2000;23:1154–1161. doi: 10.2337/diacare.23.8.1154. [DOI] [PubMed] [Google Scholar]
- Price DA, Greenberg MJ. Structure of a molluscan cardioexcitatory neuropeptide. Science. 1977;197:670–671. doi: 10.1126/science.877582. [DOI] [PubMed] [Google Scholar]
- Racz K, Kuchel O, Buu NT. Bromocriptine decreases blood pressure of spontaneously hypertensive rats without affecting the adrenomedullary synthesis of catecholamines. J Cardiovasc Pharmacol. 1986;8:676–680. [PubMed] [Google Scholar]
- Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities--the role of insulin resistance and the sympathoadrenal system. N Engl J Med. 1996;334:374–381. doi: 10.1056/NEJM199602083340607. [DOI] [PubMed] [Google Scholar]
- Reudelhuber TL. Salt-sensitive hypertension: if only it were as simple as rocket science. J Clin Invest. 2003;111:1115–1116. doi: 10.1172/JCI18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnella GA, Markandu ND, Singer DR, MacGregor GA. Kinetics of renal sodium excretion during changes in dietary sodium intake in man--an exponential process? Clin Exp Hypertens A. 1990;12:171–178. doi: 10.3109/10641969009074726. [DOI] [PubMed] [Google Scholar]
- Schioth HB. The physiological role of melanocortin receptors. Vitam Horm. 2001;63:195–232. doi: 10.1016/s0083-6729(01)63007-3. [DOI] [PubMed] [Google Scholar]
- Smith AI, Funder JW. Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- Sowers JR. Dopaminergic control of circadian norepinephrine levels in patients with essential hypertension. J Clin Endocrinol Metab. 1981;53:1133–1137. doi: 10.1210/jcem-53-6-1133. [DOI] [PubMed] [Google Scholar]
- Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stier CT, Jr, Cowden EA, Allison ME. Effects of bromocriptine on single nephron and whole-kidney function in rats. J Pharmacol Exp Ther. 1982;220:366–370. [PubMed] [Google Scholar]
- Strauss M, Lamdin E, Smith W, Bleifer D. Surfeit and deficit of sodium. A kinetic concept of sodium excretion. Arch Intern Med. 1958;102:527–536. doi: 10.1001/archinte.1958.00260210013003. [DOI] [PubMed] [Google Scholar]
- Van Bergen P, Janssen PM, Hoogerhout P, De Wildt DJ, Versteeg DH. Cardiovascular effects of gamma-MSH/ACTH-like peptides: structure-activity relationship. Eur J Pharmacol. 1995;294:795–803. doi: 10.1016/0014-2999(95)00657-5. [DOI] [PubMed] [Google Scholar]
- Van Bergen P, Kleijne JA, De Wildt DJ, Versteeg DH. Different cardiovascular profiles of three melanocortins in conscious rats; evidence for antagonism between gamma 2-MSH and ACTH-(1–24) Br J Pharmacol. 1997;120:1561–1567. doi: 10.1038/sj.bjp.0701065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bergen P, Van Der Vaart JG, Kasbergen CM, Versteeg DH, De Wildt DJ. Structure-activity analysis for the effects of gamma-MSH/ACTH-like peptides on cerebral hemodynamics in rats. Eur J Pharmacol. 1996;318:357–368. doi: 10.1016/s0014-2999(96)00806-0. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, Lambrechts AC. Bromocriptine-induced decrease in blood pressure in conscious spontaneously hypertensive rats: evidence for a peripheral site of action. J Pharm Pharmacol. 1989;41:644–646. doi: 10.1111/j.2042-7158.1989.tb06549.x. [DOI] [PubMed] [Google Scholar]
- Van Dijk C, Ni XP, Humphreys MH. Correction of blood pressure in hyupertensive rats with γ–melanocyte stimulating hormone (γ-MSH) deficiency rapidly corrects altered glucose homeostasis (abstract) J Am Soc Nephrol. 2006;17:212A. [Google Scholar]
- Wikberg JE, Muceniece R, Mandrika I, Prusis P, Lindblom J, Post C, Skottner A. New aspects on the melanocortins and their receptors. Pharmacol Res. 2000;42:393–420. doi: 10.1006/phrs.2000.0725. [DOI] [PubMed] [Google Scholar]
- Zhou A, Bloomquist BT, Mains RE. The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. J Biol Chem. 1993;268:1763–1769. [PubMed] [Google Scholar]
- Zhou A, Webb G, Zhu X, Steiner DF. Proteolytic processing in the secretory pathway. J Biol Chem. 1999;274:20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]