

Abstract
Although clinically distinct diseases, tauopathies and synucleinopathies share common genesis and mechanisms, leading to overlapping degenerative changes within neurons. In human postmortem striatum of Parkinson’s disease [PD] and PD with dementia, we have recently described elevated levels of tauopathy, indexed as increased hyperphosphorylated Tau [p-Tau]. Here we assessed tauopathy in striatum of a transgenic animal model of PD, overexpressing human α-synuclein under the PDGF promoter. At 11 months of age, large and progressive increases in p-Tau in transgenic mice, hyperphosphorylated at sites reminiscent of Alzheimer’s disease, were noted, along with elevated levels of α-synuclein and p-GSK-3β, a major kinase involved in hyperphosphorylation of Tau. Differential Triton X-100 extraction of striata showed the presence of aggregated α-Syn in the Tg mice, along with p-Tau and p-GSK-3β, which was also confirmed through immunohistochemistry. After p-Tau formation, both Tau and MAP1 dissociated from the cytoskeleton, consistent with diminished ability of these cytoskeleton-binding proteins to bind microtubules. Increases in free tubulin and actin were also noted, indicative of cytoskeleton remodeling and destabilization. In vivo magnetic resonance imaging of the transgenic animals showed a reduction in brain volume of transgenic mice indicating substantial atrophy. From immunohistochemical studies, α-synuclein, p-Tau and p-GSK-3β were found to be overexpressed and co-localized in large inclusion bodies, reminiscent of Lewy bodies. The elevated state of tauopathy seen in these PDGF-α-synuclein mice provides further confirmation that Parkinson’s may be a tauopathic disease.
Keywords: Alzheimer’s disease, neurodegeneration, synucleinopathies, tauopathies
Sporadic Parkinson’s disease [PD] is a progressive neurodegenerative disease of unknown etiology [Pollanen et al, 1993], resulting in loss of motor function and degeneration of dopaminergic neurons [Pollanen et al, 1993; Jakes et al, 1994; Forno, 1996; Spillantini et al, 1998; Corti et al 2005]. Alpha-synuclein [α-Syn], a presynaptic protein, is causal in the genesis of PD [Forno, 1996; Spillantini et al, 1998], and gene duplication and triplication of α-Syn are found in sporadic and early onset forms of PD [Singleton et al, 2003], while mutations [A30P, A53T and E46K] in the gene are linked to autosomal dominant familial forms of the disease [Polymeropoulos et al, 1997; Kruger et al, 1998]. Although a soluble protein, under pathological conditions, α-Syn becomes insoluble, self-aggregates and accumulates into intra-neuronal inclusion bodies [Spillantini et al, 1998; Masliah et al, 2000; El-Agnaf et al, 1998; Gosavi et al, 2002]. Similar to α-Syn, Tau is also a highly soluble protein that becomes insoluble by pathological hyperphosphorylation, leading to tauopathies, including Alzheimer’s disease (AD), [Dickson et al, 2002; Esper et al, 2007; Gasparini et al, 2007; Wadia & Lang, 2007; Gong & Iqbal, 2008; Ling et al, 2010; Ludolph et al, 2009; Williams & Lees, 2009].
Whereas both primary synucleinopathies and tauopathies have distinct clinical features, significant overlap exists between these neurodegenerative diseases, particularly manifest in the variable co-morbidity of dementia and Parkinsonism [Murray et al, 2005]. Moreover, p-Tau is often found in Lewy bodies in PD, while α-Syn pathology is seen in Alzheimer’s disease [Crews et al, 2009]. There is increasing evidence that suggests a direct interaction between these proteins [Giasson et al, 2003].
Using the MPTP mouse neurotoxin model of PD, we recently demonstrated that increases in α-Syn can initiate and sustain Tau hyperphosphorylation in vivo and in vitro [Duka et al, 2006; Duka & Sidhu, 2006; Kozikowski et al, 2006; Duka et al, 2009]. The hyperphosphorylation of Tau was absolutely dependent on the presence of α-Syn, as indexed by lack of p-Tau formation in MPTP-treated α-Syn−/− mice or in cells lacking α-Syn. Our more recent studies indicates that α-Syn induces p-Tau formation through specific activation and recruitment of p-GSK-3β, a kinase known to hyperphosphorylate Tau at distinct sites in AD, which itself becomes activated through autophosphorylation at Tyr216 [Duka et al, 2009]. Importantly, we also found tauopathy in postmortem brains of PD and PDD patients, where we found high levels of p-Tau, α-Syn and p-GSK-3β in striatum, but not in the inferior frontal gyrus, suggesting a different anatomical distribution of Tau pathology in PD patients as compared to AD patients [Wills et al, 2010].
In the current study, we have investigated the state of tauopathy in a transgenic mouse model of PD that over-expresses the human α-Syn transgene under the control of the platelet derived growth factor [PDGF] promoter [Rockenstein et al, 2002]. Our results indicate a spontaneous age-dependent development of tauopathy in these mice, and provide further support for the concept that PD is a form of tauopathy.
Methods
Materials
The antibodies used in this study are: anti-Tau MAB361 from Millipore [Temecula, CA]; anti-Tau Neurofibrillary Tangles Marker AHB0042 and anti-tau (pS262), Biosource Invitrogen [Carlsbad, CA]; anti-α-Syn CAT# 610787, anti-GSK-3β CAT# 612313 and anti-pGSK-3B [purified mouse anti-GSK-3B (pY216) CAT # 612313], from BD Transduction Labs [San Jose, CA]; anti-β-actin SC-1616 from Santa Cruz Biotechnology, Inc. [ Santa Cruz, CA]; The CP-13, PHF-1 and MC1 antibodies [recognizing Tau-Ser202, Tau-Ser396/404 and conformational-sensitive antibody, respectively] were gifts from Dr. Peter Davies [New York]; anti-α-Tubulin T6074 from Sigma Aldrich [St. Louis, MO]; GAPDH antibodies (14C10) were from Cell Signaling Technology (Danvers, Massachusetts); mouse anti-Tyrosine Hydroxylase Alexa Fluor 488 Conjugated Monoclonal MAB5280X from Chemicon International [Billerica, MA]; rabbit polyclonal to MAP1 ab25954 from Abcam Inc. [Cambridge, MA].
Animals
All studies with animals were conducted under strict guidelines of the National Institutes of Research and were approved by Georgetown University Animal Care and Use Committee. Hemizygous mice overexpressing α-Syn driven by the platelet-derived growth factor [PDGF] promoter were imported (from E. Masliah, University of California San Diego, CA). For all experiments, hemizygous PDGF-α-Syn mice were bred with wildtype (WT) mice (C57BL/6 × DBA/2 F1; B6D2F1/J) obtained from Jackson Labs to produce both WT and PDGF- α-Syn littermates, and a breeding colony was established as described previously [25].
Postmortem tissue
Postmortem tissue was provided by the Sun Health Research Institute Brain donation program (Sun City, AZ) and included samples from PD cases that, antemortem, showed no evidence of dementia (and neuropathologically confirmed to be absent of AD pathology or cortical Lewy Bodies). Clinical evaluation and neuropathological diagnosis of these cases have been published in greater detail elsewhere [Joyce et al, 2002]. The average postmortem interval is ~3 hours. Data in this study were as follows, PD patients: 6 male and 3 female, ages 74–90, with mean age of 80.3 years; control group, 7 males and 5 females, ages 63–89, with mean age of 80.7 years. Since no gender differences were observed, data were pooled together.
Isolation of cytoskeletal-free and cytoskeleton-associated fractions
Tissues were extracted and separated into cytosksleton-free and cytoskeleton-associated fractions as described previously (Duka et al, 2006). Briefly, tissues were homogenized in buffer containing 80 mM PIPES (pH 6.8), 1 mM MgCl2, 2 mM EGTA, 0.1 mM EDTA, 0.1% Triton X-100 and 30% glycerol. Lysates were incubated at 37°C for 10 minutes prior to centrifugation at room temperature at 14,000 × g for 20 min. The supernatant contained cytoskeleton-free fractions. The pellet was re-suspended in 2% SDS, 5 mM EDTA, 5 mM EGTA, 10 % glycerol, 0.25 M Tris-HCl (pH 6.8), incubated at room temperature on an inverter for 60 min, and sonicated 3 × 30 secs at room temperature with a Branson Sonifier 250, representing the cytoskeleton-associated fraction.
Preparation of Triton X-100 soluble and insoluble fractions
The aggregation state of α-Syn was analyzed based on its differential solubility in 1% Triton X-100, as described elsewhere (Zhou & Freed, 2003). Briefly, tissues were extracted in buffer containing 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1% Triton X-100, protease inhibitor cocktail tablets (Complete Mini, EDTA-free; Roche Diagnostics GmbH, Germany) and phosphatase inhibitors (Halt Protease Inhibitor Cocktail; Pierce). Lysates were incubated for 30 min on ice, followed by centrifugation at 15,000 × g for 60 min at 4°C. The pellet and supernatant were collected as the Triton X-100-insoluble and soluble fractions, respectively. The Triton X-100-insoluble pellets were redissolved in the previously described lysis buffer containing 2% SDS.
Co-Immunoprecipitation analyses
Human tissues from normal controls and PD brains were suspended in lysis buffer [10%, w/v; 50 mM Tris-HCl, pH 7.5, 5mM KCl, protease inhibitor cocktail tablets (Complete Mini, EDTA-free; Roche Diagnostics GmbH, Germany) and phosphatase inhibitors (Halt Protease Inhibitor Cocktail; Pierce) and 1 mM phenylmethylsulfonyl fluoride] and lysed by homogenization and hypotonic shock on ice for 10 min. After homogenization of the lysates, proteins were solubilized in a Dounce homogenizer [1 mg/ml protein] in a modified RIPA buffer [in mM: Tris, 50, pH 7.5; NaCl, 150; 0.4% Triton X-100, 0.5% deoxycholate sodium salt, 0.5% Nonidet P-40, dithiothreitol, 1; EDTA, 1; EGTA, 1; PMSF, 1 mM) and incubated on ice for 30 min. The solubilized lysates were centrifuged [18,500 × g, 20 min at 4°C] and solubilized lysates [500–700 μL assay] were precleared for 30 min with 50 100 μL of protein A/G+ agarose beads (sc-2003; Santa Cruz Biotechnology, CA, USA) with gentle rocking. Lysates were centrifuged at 18,500 × g for 20 min at 4 °C, and the supernatant removed carefully. Supernatants were incubated with 1:100 dilution of α-Syn antibody and incubated overnight with gentle end-to-end rocking at 4 °C. Protein A/G+ agarose beads [100 μl] were added and tubes were shaken gently for an additional 90 min. Following centrifugation [18,500 × g for 20 min at 4 °C], beads were washed 4 times in RIPA buffer and suspended directly in Laemmli buffer and proteins analyzed by Western blots.
Immunoblot analysis of mouse and human tissues
Western blot analyses of mouse tissue homogenates were performed as previously described [Duka et al, 2006; Duka et al 2009]. Briefly, tissue samples were loaded on 10–20% Tris-HCl Criterion gels (Bio-Rad) and transferred to PVDF blots. Blots were blocked with 20 mM Tris-buffered saline, pH 7.6 containing 0.1% Tween 20 (TBST) and 5% (wt/vol) non-fat dry milk for 1 hour at room temperature. Blots were subsequently incubated overnight at 4°C with mouse tau-5 (1:500) antibody and anti-tau Neurofibrillary Tangles Marker (1:500), to detect total human tau. Western blots were developed with a wide range of specific human Tau antibodies that recognize the protein at different phosphorylation sites, including: CP13 (pS202) (1:500), PHF-1 (pS396/404) (1:1000) and pS262 (1:500). Total Glycogen Synthase Kinase-3β (GSK-3β) was probed for with mouse GSK-3β antibody (1:500) and phospho-GSK-3β was probed for using mouse phospho-specific (pY216) antibody (1:500). Alpha-Tubulin was probed for using mouse monoclonal anti-alpha tubulin antibody (1:4000), and tyrosine hydroxylase was probed for using TH (F-11) mouse monoclonal IgG2a antibody (1:1000). To probe for α-Syn, samples were run on 10–20% Tris HCl Criterion gels (Bio-Rad) and immunoblotted with mouse α-synuclein (1:500) antibody. All proteins were normalized to total Tau (1:500), β-actin (1:500) or GAPDH (1:500). After incubation for 2 hours at room temperature with HRP-conjugated secondary antibodies (1:3000; Santa Cruz), proteins were revealed by enhanced chemiluminescence (Perkin Elmer). Images were scanned by Scanner EPSON Perfection V700 Photo and then optically quantified using ImageJ.
Immunohistochemistry
IHC analysis of mouse brain coronal sections was performed as previously described [Duka et al, 2006], with slight modifications. Briefly, mouse brains from 22 month old control wild-type and age-matched PDGF-α-Syn were perfused with 4% PFA, and prepared in a sequential sucrose gradient, from 10% to a final 30% sucrose soak. 5 μm sections were washed, permeabilized, and stained in the following manner. Each slice was washed 3 times in 1 mg/ml NaBr2, 1 X PBS pH 7.4, for 5 min at room temperature. Following the NaBr2 auto-fluorescencequenching treatment, each slice was washed 6 X, for 10 min in 1 X PBS pH 7.4, 1% Triton X-100 followed by blocking for 1hr at room temperature in 1 X PBS pH 7.4, 1% Triton X-100, 10% FCS. Antibodies were conjugated primarily to the appropriate fluorophore as indicated using the following reagents and manufacturer’s protocol: Lighting-Link FITC #707-0030, Lighting-Link Rhodamine #710-0030, Lighting-Link Texas Red #714-0030 (Novus Biologicals Littleton, CO). Incubation with primary antibody occurred at 4°C, overnight in the dark, in blocking buffer using the following concentrations for either single or dual staining with the following fluorophore conjugated antibodies: anti-α-Syn, Texas Red 1:750; Anti-p-GSK-3β (pY216), Texas Red; Anti-p-GSK-3β (pY216), FITC 1:500; PHF-1, FITC 1:500; MC1, Rhodamine 1:500. Following staining, each slice was washed 3 X in 1 X PBS pH 7.4, 1% Triton X-100 at room temperature, incubated for 30 min in blocking buffer, and washed a final 3X in 1X PBS pH 7.4, 1% Triton X-100. Stained slices were mounted to Fischer Scientific Superfrost standard microscope slides using Fluoromount-DAPI. Fluorescence images were captured using a laser scanning confocal microscope (Olympus FV300). Paired images between WT and Tg tissue for all figures were collected at the same laser power, gain, and offset settings. Postcollection processing was performed using ImageJ and applied uniformly to all paired images.
Magnetic resonance imaging
MRI was performed on a 7.0 Tesla Bruker horizontal spectrometer/imager with a 20 cm bore equipped with 100 gauss/cm microimaging gradients and run by Paravision 4.0 software in the Preclinical Imaging Research Laboratory of the Lombardi Cancer Center, Georgetown University. Mice were anesthetized using 1.5% isoflurane and 30% nitrous oxide, positioned in a custom-made stereotaxic animal holder with temperature and respiration control and imaged in a 35 mm birdcage radiofrequency volume coil. The imaging protocol used was a T1-weighted Turbo RARE (rapid acquisition with rapid enhancement) three-dimensional imaging sequence. Imaging parameters were matrix: 256x256x256, TR: 500 ms, TE: 7.3 ms, number of averages: 1, number of echoes: 1, Rare factor: 8, FOV: 5.3 × 3.0 × 3.0 cm. Volume measurements were performed as described previously [Fricke et al, 2006] with the use of Paravision 4.0 software by defining threshold values that segmented brain mass from structures such as the ventricles and determining the pixels and area corresponding to the brain on each slice.
Statistical Analysis
Results were expressed as mean ± S.E.M. and statistically analyzed by the student’s t test between two groups.Statistical significance was accepted at the [P < 0.05] level.
Results
Age-dependent changes in overexpression of α-Syn, p-Tau and p-GSK-3β in PDGF-α-Syn transgenic mice
The expression of α-Syn as a function of age was examined by Western blots in transgenic mice over-expressing the human α-Syn transgene under the control of the PDGF promoter [Rockenstein et al, 2002]. In 4 month old transgenic animals, no increases were seen in levels of α-Syn, hyperphosphorylated Tau, or Tyr216-phosphorylated GSK-3β, when compared to 4 month old litter-mate non-transgenic controls [Data not shown]. In the 11 month old transgenic animals, however, there was increased expression of α-Syn compared to age-matched non-transgenic litter-mate controls [Fig. 1A]. These data indicate an age-dependent increase in α-Syn levels in the overexpressing transgenic mice.
Figure 1. Western blots of protein levels in striata of PDGF-α-Syn overexpressing transgenic mice.
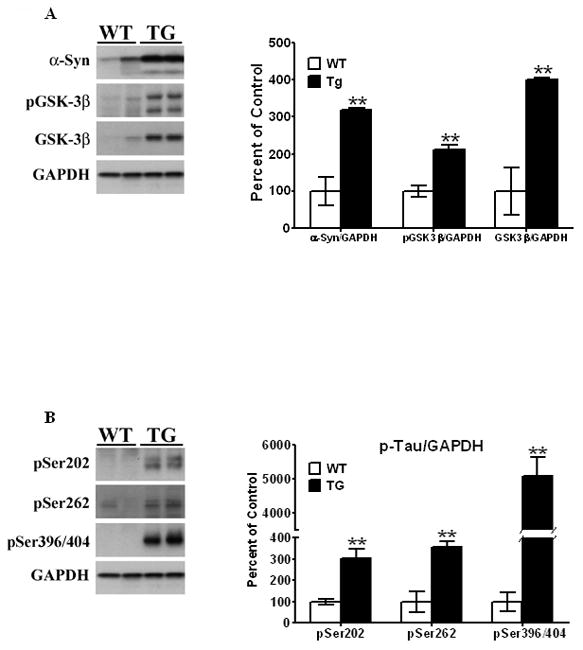
Striata from PDGF-α-Syn transgenic mice and litter-mate non-transgenic mice [WT] were solubilized in RIPA buffer and analyzed by Western blots for α-Syn [A], p-GSK- 3β [A], and p-Tau [B] levels. α-Syn, p-GSK-3β and p-Tau levels were all expressed relative to GAPDH. All values are expressed as percent change relative to changes observed in WT control animals. Results are from 3–4 animals per group; [*, P < 0.05] and [**, P < 0.01] compared to age-matched WT animals. All blots are representative of samples.
We have previously shown an increase in p-Tau, hyperphosphorylated at Ser396/404 and Ser262 in striata of the MPTP-treated mouse model of PD [Duka et al, 2006; Duka et al 2009]. In this study we wished to ascertain whether transgenic mouse models of PD could spontaneously accumulate p-Tau, even in the absence of any toxic insult. Therefore, we examined p-Tau levels in 11 month old PDGF-α-Syn transgenic mice.
Robust and significant increases were seen for p-Tau at all sites of hyperphosphorylation measured in striata of the 11 month old PDGF-α-Syn Tg mice, compared to age-matched control wild-type animals [Fig. 1B]. Thus, there was a 205% increase in levels of p-Tau hyperphosphorylated at Ser202 [t5 = 4.024, P = 0.010], pSer262 levels were significantly elevated by 255% [t5 = 4.66, P = 0.0055], and pSer396/404 Tau, detected by PHF-1 antibodies, was increased 50-fold [t5 = 7.76, P = 0.0006] relative to wild type controls [Fig. 1B]. It should be noted that in the mammalian nervous system, Tau protein exists as different splice variants ranging in size from ~55–65 kDa, and the intensity of these bands on gels can vary from animal to animal, and on the extraction protocols used and on the antibodies. Thus, we often observe a doublet or a smear of bands using p-Tau antibodies, and we therefore calculate p-Tau as the multiple bands that appear at 55–65 kDa.
We next examined levels of p-GSK-3β, phosphorylated at Tyr216, an enzyme we have previously shown to be recruited and activated by α-Syn in the hyperphosphorylation of Tau in the MPTP/MPP+ models of parkinsonism [Duka et al, 2006; Duka et al 2009]. In the 11 month old transgenic mouse, p-GSK-3β levels were elevated by 112% [t5 = 4.79, P = 0.0087] compared to age-matched wild-type control animals [Fig. 1A]. The anti-p-GSK-3β antibody recognizes two bands: an upper band corresponding to the alpha subunit and a lower band corresponding to the beta subunit of GSK-3β. Therefore, the appearance of the upper band, p-GSK-3α can be variable. In our calculations, only the lower band is used to determine p-GSK-3β levels. Total GSK-3β levels were also elevated compared to control animals [~300%, t5 = 6.29, P = 0.0033], possibly a compensatory increase due to the increased levels of Tyr216 phosphorylation.
These data are indicative of an age-dependent increase in expression of α-Syn in striata of the PDGF-α-Syn overexpressing mice, which is accompanied by increased hyperphosphorylation of Tau along with increased levels of activated p-GSK-3β, suggesting a spontaneous development of tauopathy in the Tg mice and a tight linkage between increased levels of α-Syn and development of tauopathy.
Increases in α-Syn, p-Tau and p-GSK-3β in striata of postmortem PD brains
To assess how the α-Syn pathology seen in the α-Syn overexpressing mice compared to changes seen in humans having Parkinson’s disease, α-Syn was measured in postmortem striata of 12 control subjects [Fig. 2A] and 9 patients clinically diagnosed with PD, as described in Methods. Tissues from these subjects were part of a larger study using 22 controls and 17 PD samples [Wills et al, 2010], except that in former study PIPES/SDS fractions were combined and analyzed, while in the current study fewer samples from RIPA-solubilized fractions were used. In all PD samples tested, higher [165%, t10 = 7.37, P = 0.0007] levels of α-Syn were present compared to age-matched controls.
Figure 2. Neurochemical changes of proteins in striata of postmortem human striata from PD patients and age-matched controls.
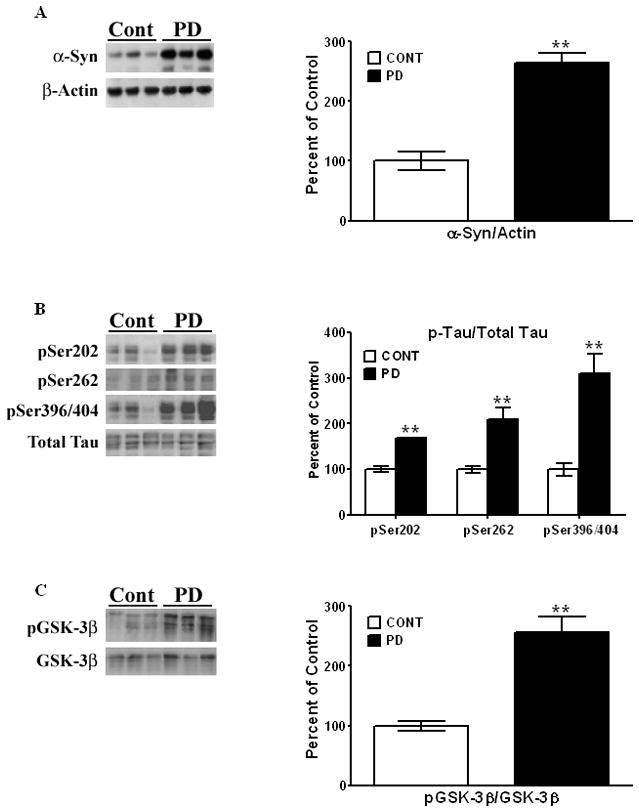
Human striata were solubilized in RIPA buffer. [A]. Levels of α-Syn were estimated by Western blots and expressed relative to β-actin. p-Tau [B] and p-GSK-3β levels [C] were measured as described in the legend to Figure 1. All values are expressed as percent change relative to the control, non-diseased group. Results are from 6 controls and 6 PD patients. [*, P < 0.01] compared to striata from the control group. All blots are representative of samples.
We also examined p-Tau levels in striata from control and PD patients. Large significant increases of 210% [t10 = 4.63, P = 0.0009] in the levels of pSer396/404, detected by the PHF-1 antibody, were observed in PD striata compared to striata from control subjects [Fig. 2B]. An increase of ~70% [t10 = 3.78, P = 0.0036] was seen for pSer202, in PD striata compared to controls, while an increase of ~110% [t10 = 3.97, P = 0.0026] was observed for pSer262 [Fig. 2B].
We next examined p-GSK-3β levels and found an increase of 156% in PD brains compared to age-matched controls [Fig. 2C], which was highly significant [t10 = 5.91, P = 0.0001]. The increase in p-GSK-3β, along with increased levels of p-Tau at the different serine residues, is consistent with tauopathy in striata of PD patients.
Triton X-100 solubilization of striatal proteins
Previous studies have shown a differential solubility of α-Syn in 1% Triton X-100, and aggregates of α-Syn could be isolated upon centrifugation [Zhou & Freed, 2003]. To test whether aggregates of α-Syn were formed in striata of the α-Syn Tg mice, striatal tissues from 11 month old animals were extracted in Triton X-100, and this protein was examined in both soluble and insoluble fractions [Fig. 3A]. Compared to wild type non-transgenic animals, levels of α-Syn were significantly increased [7-fold, t5 = 12.99, P < 0.0001] in Triton X-100-insoluble fractions extracted from the α-Syn Tg mice, while no changes were observed in α-Syn present in soluble fractions, as compared to wild-type non-Tg animals.
Figure 3. Triton X-100 extraction of striatal lysates from PDGF-α-Syn overexpressing transgenic mice.
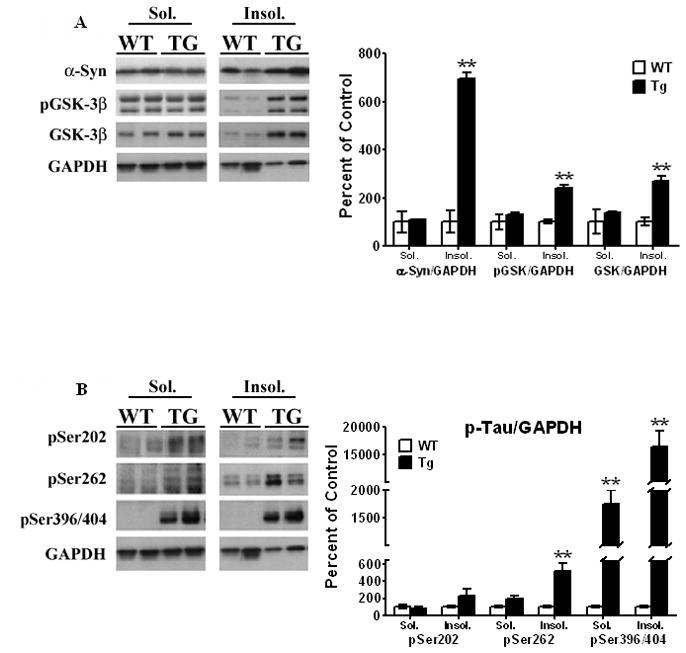
Striata from 11 month old transgenic mice and age-matched litter mates were extracted in Triton X-100 and soluble and insoluble fractions were isolated as described under Methods. [A] α-Syn, p-GSK-3β and [B] p-Tau levels were all expressed relative to GAPDH. All values are expressed as percent change relative to changes observed in WT control animals. Results are from 3–4 animals per group; [*, P < 0.05] and [**, P < 0.01] compared to age-matched WT animals. All blots are representative of samples.
When similar studies were conducted with p-GSK-3β [Fig. 3A], there was a significant increase [138%, t5 = 8.03, P = 0.0005] in the levels of p-GSK-3β in the Triton X-100-insoluble fraction, with no differences in the soluble fraction. The presence of p-GSK-3β in the insoluble fraction was unexpected considering p-GSK-3β is a soluble protein, and its presence in the insoluble fraction suggests that it has either become aggregated or that a strong co-interaction of the kinase with α-Syn exists, causing it to co-segregate with α-Syn in the insoluble fraction. In earlier studies, using a MPTP-treated mouse model which develops tauopathy, we have shown that activated p-GSK-3β co-immunoprecipitates with α-Syn and with pSer396/404 Tau [Duka et al, 2009], suggesting that the presence of p-GSK-3β in the insoluble fraction may be due to increased protein:protein interaction with α-Syn, causing it to co-aggregate with α-Syn. There were also increased levels of total, non-phosphorylated GSK-3β [168%, t5 = 5.011, P = 0.0041] in the Triton X-100 insoluble fractions [Fig. 3A]. These findings suggest that total GSK-3β is capable of forming complexes with α-Syn even prior to its autophosphorylation, and indeed, formation of such complexes between α-Syn and GSK-3β may be an integral step by which α-Syn causes the activation of GSK-3β, converting it to the activated p-GSK-3β form.
When we examined p-Tau levels in the Triton X-100 soluble and insoluble fractions [Fig. 3B], we found large but statistically insignificant increases of pSer202 Tau in the insoluble fractions [124%, t5 = 1.24, P = 0.27]. For Ser262 Tau, no changes in the soluble fraction were noted, while there was 5-fold increase in insoluble fractions [t5 = 4.04, P = 0.0099]. For pSer396/404 Tau, we found elevated levels of this form of p-Tau to be present in both the soluble and insoluble fractions [Fig. 3B]. Thus, in the soluble fraction, pSer396/404 Tau levels were significantly [t5 = 5.68, P = 0.0023] increased by 16-fold, whereas in the insoluble fraction it was further increased by ~150-fold compared to wild type animals [t5 = 4.69, P = 0.0054]. Our previous studies have also shown co-interactions of α-Syn with pSer396/404 [Duka et al 2006; Duka et al, 2009], and the presence of p-Tau in the insoluble extracts is highly suggestive of interactions between these two proteins, resulting in its co-aggregation with α-Syn in the Triton X-100-insoluble fractions.
Immunohistochemical co-localization and distribution
The aggregation and co-localization of α-Syn with pGSK-3β and p-Tau was confirmed by immunohistochemical [IHC] staining of the striatum of PDGF-α-Syn mice [Fig. 4A - C]. An overall total increase in levels of α-Syn, pGSK-3β, and p-Tau was seen in the striatum of PDGF-α-Syn mice as compared to wild-type control striatum. Dual staining was performed to analyze the co-localization of the following protein combinations: α-Syn:p-Tau, α-Syn:p-GSK-3β and p-Tau:p-GSK-3β. Each protein combination co-localized together in a single large peri-nuclear, aggregate formation in the transgenic striatum, as opposed to the multiple small puncta and more diffuse cytoplasmic distribution seen in the WT striatum. This large aggregate seen in the Tg striatum is reminiscent of pathogenic Lewy bodies in shape, size, and cellular location.
Figure 4. Immunohistochemical co-localization and distribution.
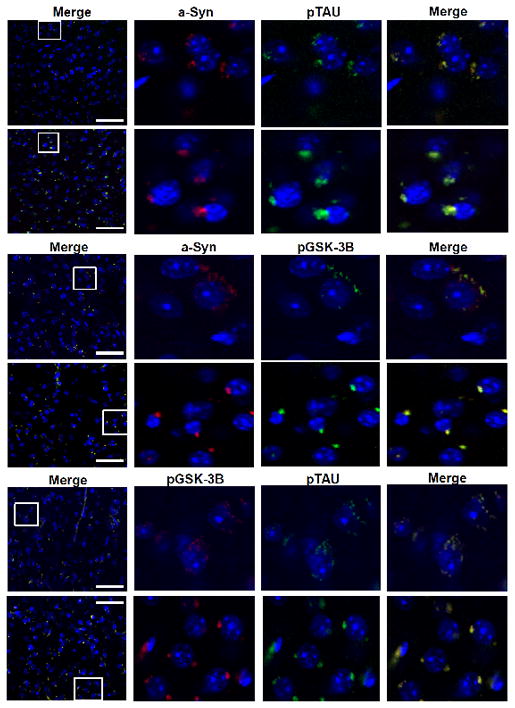
(Upper Panel) Striatum stained with α-Syn (Red) and PHF-1 Tau (Green) with DAPI as merged, higher magnification with single antibody(s) and higher magnification merged. (Middle Panel) Striatum stained with α-Syn (Red) and p-GSK-3β (Green) with DAPI as merged, higher magnification with single antibody(s) and higher magnification merged. (Lower Panel) Striatum stained with p-GSK-3β (Red) and PHF-1 (Green) with DAPI as merged, higher magnification with single antibody(s) and higher magnification merged. White boxes highlighted on left panel indicate areas shown at higher magnification in right panel(s). Scale bar: 50uM.
Assessment of α-Syn, p-Tau and pGSK-3β binding to cytoskeleton proteins
Both α-Syn and Tau are microtubule binding proteins [Abraha et al, 2000; Alonso et al, 2001], and our earlier data showed that α-Syn is also capable of binding to the actin cytoskeleton [Jeannotte & Sidhu, 2008]. In order to ascertain how pathological changes in α-Syn and p-Tau in the 11 month old α-Syn Tg affect the ability of these proteins to bind to cytoskeleton proteins, cytoskeleton-free and cytoskeleton-associated fractions were isolated from the striatum of these mice [Methods], and individual proteins were analyzed by Western blots [Fig. 5].
Figure 5. Association of striatal proteins to cytosketon-free and cytoskeleton-bound fractions.
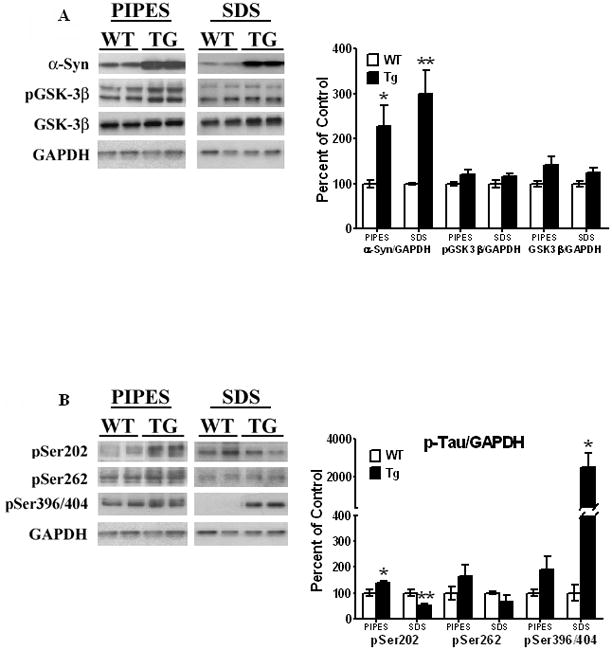
Striata from 11 month old animals and age-matched litter mates were extracted in PIPES buffer and centrifuged, as described under Methods. The supernatant obtained upon PIPES extraction represented the cytoskeleton-free fraction, while the PIPES-insoluble pellet was further solubilized in SDS buffer and represented the cytoskeleton-bound fractions. [A] α-Syn and p-GSK-3β, and [B] p-Tau proteins were analyzed by Western blots, as described in legend to Fig. 3. All values are expressed as percent change relative to changes observed in WT control animals. Results are from 5–6 animals per group; [*, P < 0.05] and [**, P < 0.01] compared to age-matched WT animals. All blots are representative of samples.
In the cytoskeleton-free PIPES-extracted fractions, we found significant increases [of 128%, t9 = 2.43, P = 0.038] in α-Syn levels in the Tg mice when compared to the wild-type controls [Fig. 5A]. Moreover, in cytoskeleton-associated SDS-extracted fractions, large increases [of 200%, t9 = 3.49, P = 0.0068] of α-Syn were seen in the α-Syn Tg mice compared to the wild-type mice. For p-GSK-3β, higher levels [22%] of this protein were seen in cytoskeleton-free fractions, with just a 16% increase in cytoskeletal-associated fractions, in Tg mice compared to wild-type animals, but the changes were not significant [t9 = 1.77, P = 0.11; t9 = 1.45, P = 0.18, respectively]. GSK-3β followed suit, as it was insignificantly increased in both cytoskeletal-free fractions [by 42%, t9 = 2.00, P = 0.077] and cytoskeletal-bound fractions [by 26%, t9 = 2.19, P = 0.056]. As previously mentioned, the increase in GSK-3β may be a compensatory increase to off-set changes in p-GSK-3β levels.
Variable increases in p-Tau were observed in both the cytoskeleton-free and cytoskeleton-associated fractions in α-Syn Tg mice, as compared to wild-type mice [Fig. 5B]. Thus, in the cytoskeleton-free fraction, pSer202 Tau was significantly increased by 38% [t9 = 2.35, P = 0.044], while pSer396/404 and pSer262 were both insignificantly increased [90%, t9 = 1.53, P = 0.16; 65%, t9 = 1.21, P = 0.26, respectively]. In the cytoskeleton-associated fractions, a largely significant increase in pSer396/404 Tau was observed [25-fold] as compared to wild- type animals [t9 = 2.91, P = 0.02]. For pSer202, however, we found a significant [t9 = 4.04, P = 0.0099] decrease [by 48%] in the levels of this protein associated with the cytoskeleton [Fig. 5B]. pSer262 was insignificantly decreased [t9 = 1.14, P = 0.28] by 37%. When Tau becomes hyperphosphorylated, it loses its ability to bind to microtubules [Sengupta et al, 1998; Abraha et al, 2000; Alonso et al, 2001], and the presence of these p-Tau proteins in the cytoskeleton-free fractions indicates that p-Tau has dissociated from the microtubule cytoskeleton. The presence of p-Tau in the cytoskeletal fractions indicates that Tau becomes hyperphosphorylated when it is still bound to the microtubule cytoskeleton and prior to its dissociation from microtubules.
Cytoskeletal remodeling in PDGF-α-Syn
We next examined whether cytoskeletal elements were altered and undergoing remodeling in the striatum of the 11 month old mice [Fig. 6], by analyzing cytoskeletal proteins [actin and tubulin], as well as microtubule-associated proteins [MAP1 and total Tau]. In cytoskeleton-free fractions, we saw a small but significant increase [24%, t5 = 3.04, P = 0.029] in MAP1, while cytoskeleton-associated levels of this protein were unchanged. We observed increases in levels of total Tau in the PDGF-α-Syn transgenic mice in both the cytoskeleton-free and the cytoskeleton-associated fractions [~110, t9 = 2.89, P = 0.018; 725%, t12 = 2.40, P = 0.034, respectively]. The increases in MAP1 and total Tau in the cytoskeleton-free fraction suggests that a substantial level of these microtubule-associated proteins, which stabilize the microtubules, are no longer bound to the microtubule cytoskeleton, and may lead to its instability.
Figure 6. Cytoskeletal remodeling in PDGF-α-Syn mice.
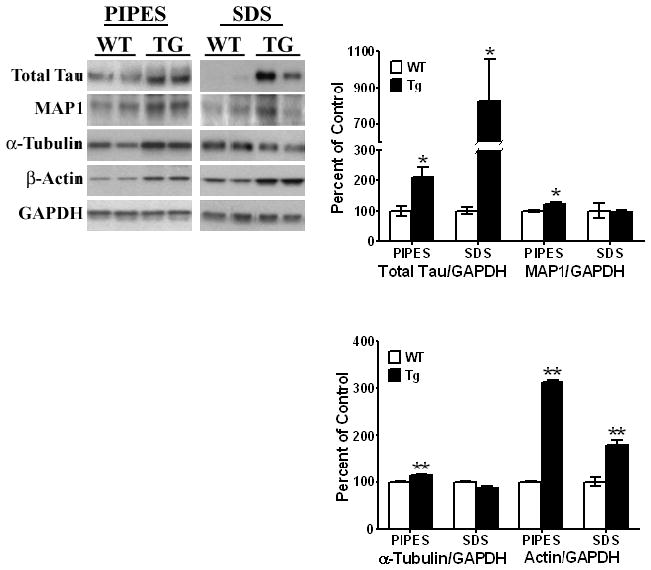
Striata from 11 month old animals and age-matched litter mates were extracted to obtain isolations of cytoskeletal-free and cytoskeleton-associated fractions, as described under Methods. Total Tau, α-tubulin, β-actin and MAP1 proteins were analyzed by Western Blots and expressed relative to GAPDH. All values are shown as a percent change relative to changes observed in WT control animals. Results are from 3–6 animals per group; [*, P < 0.05] and [**, P < 0.01] with Student’s t-tests comparing age-matched WT and Tg animals. All blots are representative of samples.
In cytoskeleton-free fractions of the PDGF-α-Syn Tg mice, we observed a small but significant increase [13%, t5 = 5.29, P = 0.0032] in the levels of free α-tubulin, indicating that α-tubulin was being dissociated from the microtubules [Fig. 6]. In cytoskeleton-associated fractions, no significant changes in α-tubulin were observed. Increases [~200%, t5 = 44.15, P < .0001] in β-actin were seen in cytoskeleton-free fractions isolated from Tg mice. This was accompanied by increases [79%, t5 = 5.38, P = 0.0003] in levels of β-actin associated with the cytoskeleton, which could be a compensatory increase. The presence of elevated levels of free α-tubulin and β-actin in the cytoskeleton-free fractions suggests that both the microtubule and actin cytoskeletons are destabilized in the PDGF-α-Syn overexpressing mice, and that there is substantial cytoskeleton remodeling in the striatum of these mice.
Striatum dopaminergic Neuronal Death and p-Tau Aggregation
To examine the effect of α-Syn expression levels on viability of dopaminergic neurons, we stained for tyrosine hydroxylase [TH, Fig 7A]. Using a conjugate labeled antibody specific for tyrosine hydroxylase [TH], higher levels of positively stained TH fibers were found to be present in wild type striatum versus the Tg mice, suggesting loss of dopaminergic processes into the striatum [Fig. 7B]. Interestingly, this was not accompanied by a decrease in dopaminergic neurons in basal ganglia or Substantia nigra, total dopaminergic neurons in these regions of the transgenic mice were similar to those of wild type mice [Fig. 7B].
Figure 7. Immunostaining of TH neurons.
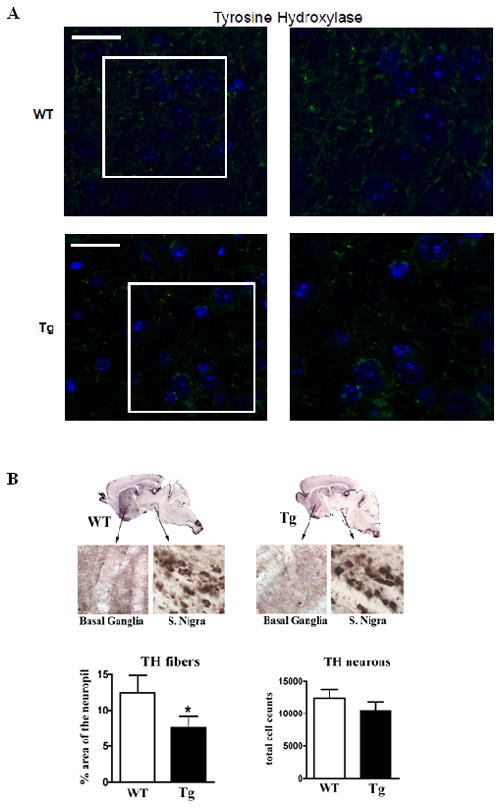
Immunostaining of transgenic [Tg] and wild type [Wt] mice was conducted as described in Methods. [A] Staining for TH (Green), DAPI (Blue) at lowest magnification (left) to highest magnification (right). White boxes highlighted on left panel indicate areas shown at higher magnification in right panel(s). Scale bar: 10uM. [B] TH-positive neurons in coronal sections of transgenic and wild type mice were analyzed. The data are from 6 mice each [12 months of age]. The counts in the SN were done by sterology, and no significant differences were found between transgenic and wild type mice.
Prior to aggregation, p-Tau undergoes a conformational change whereby the N- and C-terminus are brought together into a paperclip-like structure [Jeganathan et al, 2008]; such conformational changes are best detected by the MC1 antibodies developed to simultaneously detect antigenic epitopes in both termini of the aggregate-prone, pathogenic version of p-Tau. Immunostaining for MC1 in striatum clearly showed increased staining in the Tg versus the WT, confirming the presence of the aggregated p-Tau [Fig 8]. Using MC1, we show that the Tg striatum has a greater presence of MC1 positive staining cell bodies than the WT mice.
Figure 8. Immunostaining demonstrating conformational changes in p-Tau in transgenic mice.
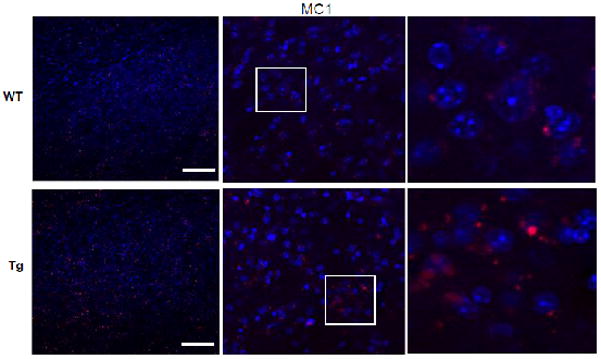
Staining of p-Tau using the conformational-sensitive MC1 antibody (Red) was conducted as described in Methods, with DAPI (Blue) at lowest magnification (left) to highest magnification (right). White boxes highlighted on left panel indicate areas shown at higher magnification in right panel(s). Scale bar: 70uM.
Co-Immunoprecipitation of α-Syn with p-Tau and p-GSK-3β in human tissues
The co-localization of α-Syn with p-Tau [pSer396/404] and p-GSK-3β seen in immunohistochemistry is highly suggestive of an interaction between these three proteins. Indeed, we have previously shown in mice striatum, through co-immunoprecipitation [co-IP] studies, that α-Syn can co-IP p-Tau [Duka et al, 2006] and p-GSK-3β [Duka et al, 2009]. In order to test whether human α-Syn can similarly co-IP p-Tau and p-GSK-3β, we conducted co-IP using lysates from control nondiseased and PD tissues [Fig. 9]. Anti-α-Syn antibodies co-IPed α-Syn protein from both control and PD lysates, with higher levels of α-Syn co-immunoprecipitated in PD than control lysates. p-Tau, hyperphsphorylated at pSer396/404 was also detected in the immunoprecipitates from both control and PD samples, with higher levels seen in PD [Fig. 9]. These data indicate that pSer396/404 Tau and α-Syn form stable complexes via protein:protein interactions. The higher levels of p-Tau co-IPed in the PD samples compared to controls are consistent with higher levels of p-Tau present in PD. Similar results were also obtained with p-GSK-3β, where α-Syn antibodies co-IPed this protein in both control and PD lysates, indicating an interaction between these two proteins [Fig.9]. The higher levels of p-GSK-3β seen in PD samples are also consistent with higher levels of this protein in this fraction compared to that of control.
Figure 9. Co-Immunoprecipitation studies of human samples using α-Syn antibodies.
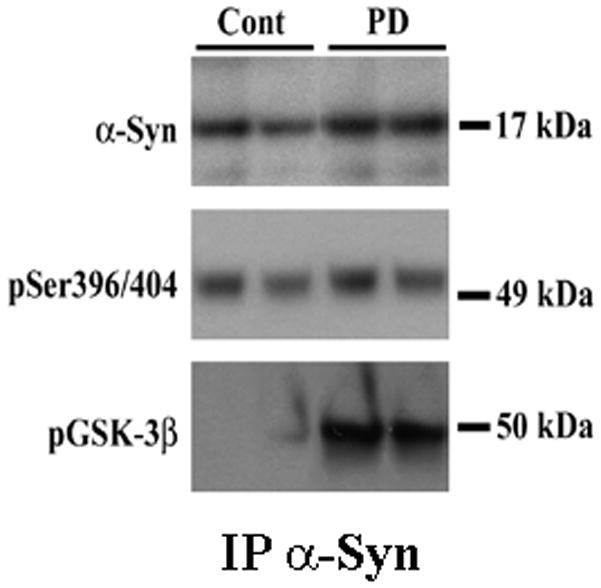
Lysates from control and PD striatum were prepared and subject to co-IP studies using α-Syn antibodies as described under Materials and Methods. Proteins present in the washed immunoprecipitates were analyzed by Western blots. Higher levels of α-Syn, p-Tau [pSer396/404] and p-GSK-3β were found present in the lysates from PD compared to control brains, consistent with increased expression of these proteins in the diseased brains.
In vivo volumetric molecular resonance imaging [MRI] in PDGF-α-Syn overexpressing mice
Volumetric MRI studies in 18–22 month old PDGF-α-Syn overexpressing Tg mice and age-matched WT controls were conducted as described in Methods [Fig. 10]. The results show decreases in whole brain volumetry in the Tg mice, as compared to the WT, suggestive of morphological and cytoarchitectural changes of brain structures consistent with atrophy of the brain in these mice. In WT animals, whole brain volumetry was 0.4526 + 0.002 cm3, which was significantly [t2 = 8.59, P = 0.013] decreased by 9% to 0.4137 + 0.003 cm3 indicative of decrease in brain mass or atrophy [Fig. 10].
Figure 10. In vivo molecular resonance imaging [MRI] of WT and Tg mice.
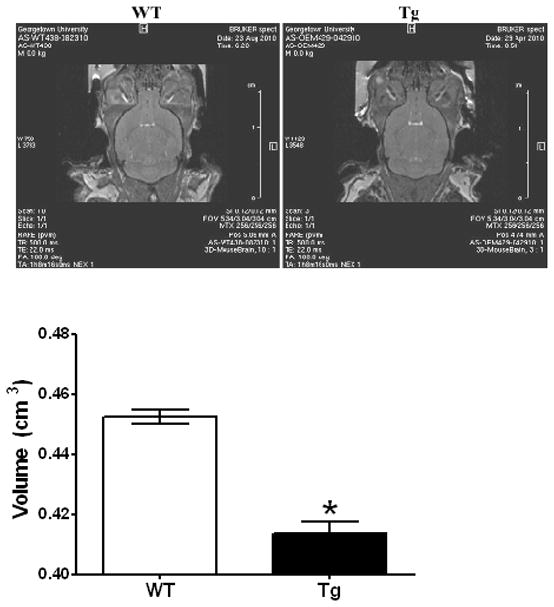
Volumetric measurements after in vivo MRI were performed as described in Methods with the use of Paravision 4.0 software by defining threshold values that segmented brain mass from structures such as the ventricles and determining the pixels and area corresponding to brain on each slice. Data are the averages from two mice in each group [18–22 months of age], while the image is representative from each animal species. [*, P < 0.05] compared to age-matched WT animals.
Discussion
We describe here novel findings demonstrating the spontaneous development of tauopathy in striata of a mouse model of Parkinsonism, which overexpresses human α-Syn under the PDGF promoter. In these animals and in the striatum of PD patients, Tau is hyperphosphorylated at specific serine residues reminiscent of Alzheimer’s-like neuropathology. This finding provides additional support for our previous studies [Duka et al, 2006; Duka & Sidhu, 2006; Kozikowski et al, 2006; Duka et al, 2009], suggesting that Parkinson’s disease, and probably other synucleinopathies, are also tauopathic diseases. In this regard, it should be noted that in the MPTP model of PD and in human postmortem striata, we found hyperphosphorylation of Tau at the same sites demonstrated here, namely, Ser202, Ser262 and Ser396/404 [Duka et al, 2006; Duka & Sidhu, 2006; Kozikowski et al, 2006;Duka et al, 2009], suggesting that the PDGF-α-Syn overexpressing mice may be a good model system to study tauopathy in the context of PD. Only limited neurochemical studies have been conducted demonstrating and identifying the epitopes of hyperphosphorylated Tau in postmortem tissues in PD brains or in animal models of PD. A recent study found increased levels of Tau hyperphosphorylated at Ser396 in synaptic-enriched fractions isolated from the frontal cortex of PD patients [Muntané et al, 2008]. In animal models of PD, tauopathic changes have been observed in only very few studies, such as in the brainstem of symptomatic A30P α-Syn mutant mice at pSer202 and pSer396/404 [Frasier et al, 2005]. In these studies, striatum was not analyzed and it remains unknown whether these animals also develop tauopathy in this region. Another study using the neurotoxin rotenone to model Parkinsonism in rats found immunoreactive Tau hyperphosphorylated at Ser202/Thr205, Thr212/Ser214 and Ser396/404 [Höglinger et al, 2005]. Using the A53T mice, we have recently obtained evidence to indicate the development of tauopathy in striatum of these mice [Wills & Sidhu, Unpublished Observations]. Thus, the existence of tauopathy in several transgenic α-Syn mouse models of PD, as well as our findings in postmortem PD and PD with dementia brains [Wills et al, 2010], along with several clinical findings mentioned in the Introduction, supports our contention that tauopathy is a central feature of PD. Conversely, emerging studies now describe an active role for the participation of α-Syn in Alzheimer’s disease [for a review see Crews et al, 2009]. While further studies are necessary, it is likely that amyloidogenic proteins such as α-Syn contribute to the tauopathic processes seen in both Alzheimer’s disease and Parkinson’s disease. A distinguishing feature between Parkinson’s disease and Alzheimer’s disease, however, is the limited nature of tauopathy seen in the former, where tauopathy is restricted primarily to dopaminergic neurons [Jellinger, 2010; Wills et al, 2010], whereas in Alzheimer’s disease, tauopathy is seen throughout the brain.
That α-Syn is central to observing tauopathy in PD models was demonstrated previously when we showed a failure of MPTP to provoke increases in p-Tau levels in vivo and in vitro in both α-Syn−/− mice, as well as in neuronal cells lacking α-Syn [Duka et al, 2006; Duka et al, 2009]. Moreover, in the absence of α-Syn, MPTP failed to cause an activation of p-GSK-3β [Duka et al, 2009]. In the current studies as well, our data indicate that tauopathy is seen only upon overexpression of α-Syn. Thus, whereas the 4 month old animal did not overexpress α-Syn, there was no tauopathy or increases in p-GSK-3β. However, in the 8 and 11 month old animal, where we found a progressive increase in accumulation of α-Syn, we also noted robust increases in p-Tau and p-GSK-3β levels. Indeed, at 11 months of age, where we saw elevated levels of α-Syn compared to the 8 month old animal, we also observed higher levels of p-Tau [pSer262 and pSer396/404] relative to 8 month old mice. Interestingly, pSer202 levels remained unchanged in the 8 and 11 month old mice, consistent with our earlier findings in cells and neurons, where we found activation of pSer202 to occur earlier than pSer262 or pSer396/404 [Duka et al, 2006]. Together, these studies further confirm the tight linkage between accumulation of α-Syn and development of tauopathy in Parkinson’s disease.
We earlier demonstrated that p-GSK-3β is central to the development of the tauopathic process in PD models, and that blockade of the kinase with specific inhibitors prevented tauopathic changes and cell death in mesencephalic neurons and cultured cells [Kozikowski et al, 2006], suggesting that inhibiting tauopathy was neuroprotective to dopaminergic neurons. In the present studies, GSK-3β, a kinase which targets Tau, was phosphorylated at Tyr216 resulting in its activation, with concurrent Tau hyperphosphorylation in both the PDGF-α-Syn mouse and PD patients, consistent with our previous findings that p-GSK-3β is necessary for the tauopathic process [Duka et al, 2006; Duka et al, 2009]. Although the precise mechanisms leading to the GSK-3β-dependent hyperphosphorylation of Tau are still elusive, studies have shown that amyloid proteins such as β-amyloid and prions may have a unique capability to activate p-GSK-3β [Perez et al, 2003; Hernandez et al, 2010]. As an amyloidogenic protein, α-Syn may be a new member of amyloidogenic proteins that can activate p-GSK-3β, initiating the series of reactions that results in hyperphosphorylation of Tau. Indeed, we have shown that α-Syn can cause the specific recruitment of p-GSK-3β via protein:protein interactions [Duka et al, 2009]. Moreover, time course studies in mesencephalic neurons show that accumulation of α-Syn and activation of p-GSK-3β precedes p-Tau formation [Duka et al, 2006]. In the current studies, it appears that α-Syn may actually recruit GSK-3β even prior to its hyperphosphorylation. Thus, increased levels of non-phosphorylated GSK-3β were found to be co-localized with α-Syn in Triton X-100 insoluble fractions of transgenic mice, along with p-GSK-3β, [see Fig. 3A]. We speculate that α-Syn may initiate the activation of GSK-3β by recruiting total GSK-3β, possibly by direct protein:protein interaction, which may in turn promote conformational changes in GSK-3β resulting in its autophosphorylation at Tyr216 leading to its activation. Once activated, p-GSK-3β causes the hyperphosphorylation of Tau.
Other studies have demonstrated a toxic synergistic interaction between α-Syn and Tau. Thus, α-Syn and Tau were shown to polymerize into amyloid fibrils, forming intraneuronal filamentous inclusions, and in vitro studies showed that co-incubation of Tau and α-Syn synergistically promoted the fibrilization of both proteins [Giasson et al, 2003], with the A53T mutation of α-Syn accelerating such fibrilization [Kotzbauer et al, 2004]. Our own studies have shown that α-Syn, p-GSK-3β and p-Tau interact with one another through protein:protein interactions [Duka et al, 2009], and such interactions between α-Syn and p-Tau may be necessary for their fibrilization and amyloid production. Moreover, Triton X-100 extractions in the present studies demonstrate the presence of aggregated α-Syn, along with aggregated p-Tau and p-GSK-3β.
Interestingly, high levels of p-Tau were also seen in the Triton X-100 soluble fraction and is consistent with the fact that hyperphosphorylation of Tau favors its dissociation from the microtubules, disrupting axonal transport, compromising the function and viability of neurons, and has been suggested to represent an early disease-associated change in AD [Gong & Iqbal, 2008]. In vitro studies have shown that hyperphosphorylation of Tau at Ser202 inhibits its binding to microtubules by 35% [Alonso et al, 2004], while hyperphosphorylation at Ser202, Ser262 and Ser396, among other sites, are key in converting Tau into an inhibitory molecule that sequesters normal microtubule-associated proteins from microtubules [Abraha et al, 2000; Weaver et al, 2000]. Moreover, hyperphosphorylation at Ser396 promotes self-aggregation of Tau into filaments [Abraha et al, 2000]. Dissociation of p-Tau from microtubules was also demonstrated by our current studies. Thus, in PIPES-extracted, cytoskeleton-free fractions, large increases in pSer202 Tau and pSer396/404 Tau were observed in α-Syn Tg mice. This was accompanied by increased levels of MAP1, total Tau, tubulin and actin, suggestive of cytoskeleton destabilization and remodeling.
The current study also demonstrates in vivo a conformational change in p-Tau, defined by antibody MC1, representing the transition of p-Tau from soluble to aggregated filamentous tau. The folding of Tau into a paperclip-like structure, recognized by MC1 antibodies, occurs when the amino acids at residues 7–9 interact with residues 312–342, and is one of the earliest pathological alterations of Tau in Alzheimer disease, preceding the formation of neurofibrillary tangles in AD [Jeganathan et al, 2008]. Interestingly, the MC1 conformation is induced by hyperphosphorylation of Tau at Ser199, Ser202, Thr205 and the PHF-1 sites [Ser396/404], causing compaction of the paperclip structure [Jeganathan et al, 2008]. Moreover, it has been shown that paperclip conformation of p-Tau enhances its ability to aggregate to paired helical filaments [Jeganathan et al, 2008].
From immunohistochemistry of TH-positive neurons and fibers in striatum and SN, our data shows loss of TH-positive fibers in the striatum, without significant changes in SN. This is consistent with our previous findings where loss of TH-positive fibers was shown in SN [Masliah et al, 2000]. Moreover, in vivo volumetric MRI studies showing decreased brain volume in Tg mice are suggestive of atrophy in these animals. In vivo anatomical evaluation of PD and PDD in humans by MRI has been associated with whole brain as well regional atrophy (medial or lateral atrophy involving structures such as the caudate nucleus, globus pallidum or putamen) which appear to correlate closely with neuronal loss [Schulz et al, 1999; Piccini et al, 2006]. Immunohistochemical studies in Tg mice also demonstrated the presence of peri-nuclear accumulation and co-localization of α-Syn, p-GSK-3β and p-Tau as opposed to its usual location being enriched in the soma and axon terminals. Such inclusion bodies are highly reminiscent of Lewy bodies, a common visual pathology of PD.
In conclusion, the present findings suggest that changes in Tau metabolism may indeed be a common denominator in neurodegenerative diseases such as PD and AD. Despite differences in their topographic distributions and phenotypic manifestations, these diseases are linked by the progressive accumulation of hyperphosphorylated Tau, activated GSK-3β and elevated levels of α-Syn. Taken together, these findings highlight the potential importance of targeting the tauopathic pathway in the therapeutic management of diverse human neurodegenerative diseases, including PD.
Acknowledgments
We thank Ciaran Smolinsky and Tiffany Kaul for technical assistance, to Peter Davies for the CP13, MC1 and PHF-1 antibodies and to Eliezer Masliah for the PDGF-alpha-synuclein overexpressing mouse colony. We are grateful to the families of the many patients for their generosity in donating the organs that made this study possible and to Sun Health Research Institute Brain Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain Donation Program is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium) and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson’s Research. This study was supported in part by grants from NIA, AG28108 [A.S.].
Abbreviations
- PD
Parkinson’s disease
- AD
Alzheimer’s disease
- p-Tau
hyperphosphorylated Tau
- PDGF
platelet derived growth factor
- Tg
transgenic
- α-Syn
α-synuclein
- LBs
Lewy bodies
- p-Tau
hyperphosphorylated Tau
- GSK-3β
glycogen synthase kinase 3β
- p-GSK-3β
GSK-3β phosphorylated at Tyr216
- SN
Substantia nigra
References
- 1.Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LIJ. C-terminal inhibition of Tau assembly in vitro and in Alzheimer's disease. Cell Sci. 2000;113(21):3737–3745. doi: 10.1242/jcs.113.21.3737. [DOI] [PubMed] [Google Scholar]
- 2.Alonso AD, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of Tau isoforms with Alzheimer's disease abnormally hyperphosphorylated Tau and in vitro phosphorylation into the disease-like protein. J Biol Chem. 2001;276(41):37967–73. doi: 10.1074/jbc.M105365200. [DOI] [PubMed] [Google Scholar]
- 3.Corti O, Hampe C, Darios F, Ibanez P, Ruberg M, Brice A. Parkinson's disease: from causes to mechanisms. C R Biol. 2005;328(2):131–42. doi: 10.1016/j.crvi.2004.10.009. Review. [DOI] [PubMed] [Google Scholar]
- 4.Crews L, Tsigelny I, Hashimoto M, Masliah E. Role of Synucleins in Alzheimer's Disease. Neurotox Res. 2009;16(3):306–317. doi: 10.1007/s12640-009-9073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–46. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 6.Duka T, Duka V, Joyce JN, Sidhu A. α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J. 2009;23(9):2820–30. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, Sidhu A. Alpha-synuclein induces hyper-phosphorylation of Tau in the MPTP model of Parkinsonism. FASEB J. 2006;20:2302–2312. doi: 10.1096/fj.06-6092com. [DOI] [PubMed] [Google Scholar]
- 8.Duka T, Sidhu A. The Neurotoxin MPP induces hyperphosphorylation of Tau in the presence of alpha-synuclein in SHSY-5Y neuroblastoma cells. Neurotox Res. 2006;10(1):1–10. doi: 10.1007/BF03033329. [DOI] [PubMed] [Google Scholar]
- 9.El-Agnaf OMA, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson’s disease. FEBS Letts. 1998;440(1):67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 10.Esper CD, Weiner WJ, Factor SA. Progressive supranuclear palsy. Rev Neurol Dis. 2007;4(4):209–216. [PubMed] [Google Scholar]
- 11.Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55(3):259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C, Kahle P, Wolozin B. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol. 2005 Apr;192(2):274–287. doi: 10.1016/j.expneurol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 13.Fricke ST, Rodriguez O, VanMeter J, Dettin LE, Casimiro M, Chien CD, Newell T, Johnson K, Ileva L, Ojeifo J, Johnson MD, Albanese C. In vivo magnetic resonance volumetric and spectroscopic analysis of mouse prostate cancer models. The Prostate. 2006 May 15;66:708–717. doi: 10.1002/pros.20392. [DOI] [PubMed] [Google Scholar]
- 14.Gasparini L, Terni B, Spillantini MG. Frontotemporal dementia with Tau pathology. Neurodegener Dis. 2007;4(2–3):236–253. doi: 10.1159/000101848. [DOI] [PubMed] [Google Scholar]
- 15.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of Tau and alpha-synuclein. Science. 2003;300(5619):636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 16.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein Tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15(23):2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusions. J Biol Chem. 2002;277(50):48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez F, Gomez de Barreda E, Fuster-Matanzo A, Lucas J, Avila J. GSK3: A possible link between beta amyloid peptide and Tau protein. Experimental Neurology. 2010;223(2):322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Höglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Féger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral Tauopathy. J Neurochem. 2005;95(4):930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- 20.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Letts. 1994;354(1):27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 21.Jeannotte AM, Sidhu A. Regulated interactions of the norepinephrine transporter by actin and microtubule cytoskeletons. J Neurochem. 2008;105(5):1668–82. doi: 10.1111/j.1471-4159.2008.05258.x. [DOI] [PubMed] [Google Scholar]
- 22.Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283(46):32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- 23.Jellinger KA. Interaction between α-synuclein and tau in Parkinson's disease. Exp Neurol. 2010 Oct 18; doi: 10.1016/j.expneurol.2010.10.006. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Joyce JN, Ryoo HL, Beach TB, Caviness JN, Stacy M, Gurevich EV, Reiser M, Adler CH. Loss of response to levodopa in Parkinson's disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 2002;955(1–2):138–152. doi: 10.1016/s0006-8993(02)03396-6. [DOI] [PubMed] [Google Scholar]
- 25.Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, Lee VM. Fibrillization of alpha-synuclein and Tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp Neurol. 2004;187(2):279–288. doi: 10.1016/j.expneurol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Kozikowski A, Petukhov P, Sridhar J, King L, Blond S, Duka T, Rusnak M, Sidhu A. Highly potent and specific GSK-3β inhibitors that block Tau phosphorylation and decrease α-synuclein protein expression in a cellular model of Parkinson's disease. ChemMedChem. 2006;1(2):256–266. doi: 10.1002/cmdc.200500039. [DOI] [PubMed] [Google Scholar]
- 27.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 28.Ling H, O'Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Paviour DC, Lees AJ. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(7):2045–57. doi: 10.1093/brain/awq123. [DOI] [PubMed] [Google Scholar]
- 29.Ludolph AC, Kassubek J, Landwehrmeyer BG, Mandelkow E, Mandelkow EM, Burn DJ, Caparros-Lefebvre D, Frey KA, de Yebenes JG, Gasser T, Heutink P, Höglinger G, Jamrozik Z, Jellinger KA, Kazantsev A, Kretzschmar H, Lang AE, Litvan I, Lucas JJ, McGeer PL, Melquist S, Oertel W, Otto M, Paviour D, Reum T, Saint-Raymond A, Steele JC, Tolnay M, Tumani H, van Swieten JC, Vanier MT, Vonsattel JP, Wagner S, Wszolek ZK. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur J Neurol. 2009;16(3):297–309. doi: 10.1111/j.1468-1331.2008.02513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masliah E, Rockenstein E, Veinbergs J, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 31.Muntané G, Dalfó E, Martinez A, Ferrer I. Phosphorylation of Tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer's disease, and in Parkinson's disease and related alpha-synucleinopathies. Neuroscience. 2008;152(4):913–923. doi: 10.1016/j.neuroscience.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 32.Murray B, Lynch T, Farrell M. Clinicopathological features of the Tauopathies. Biochemical Society Transactions. 2005;33(4):595–599. doi: 10.1042/BST0330595. [DOI] [PubMed] [Google Scholar]
- 33.Perez M, Rojo AI, Wandosell F, Diaz-Nido J, Avila J. Prion peptide induces neuronal cell de3th through a pathway involving glycogen synthase kinase 3. Biochem J. 2003;372(1):129–136. doi: 10.1042/BJ20021596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piccini P, Brooks DJ. New developments of brain imaging for Parkinson’s disease and related disorders. Mov Disord. 2006;21(12):2035–2041. doi: 10.1002/mds.20845. [DOI] [PubMed] [Google Scholar]
- 35.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52(3):183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Polymeropoulos HM, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzasini AM, Duvoisin RC, Di Iorio G, Golbe IL, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 37.Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68(5):568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- 38.Schulz JB, Skalej M, Wedekind D, Luft AR, Abele M, Voigt K, Dichgans J, Klockgether T. Magnetic resonance imaging-based volumetry differentiates idiopathic Parkinson’s syndrome from multiple system atrophy and progressive supranuclear palsy. Ann Neurol. 1999;45(1):65–74. [PubMed] [Google Scholar]
- 39.Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357(2):299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- 40.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 41.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95(11):6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord. 2007;13(3):336–340. doi: 10.1016/S1353-8020(08)70027-0. [DOI] [PubMed] [Google Scholar]
- 43.Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of Tau in Alzheimer's disease. Neurobiol Aging. 2000;21(5):719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 44.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–9. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 45.Wills J, Jones J, Haggerty T, Valeriy D, Joyce J, Sidhu A. Elevated Tauopathy and alpha synuclein pathology in postmortem Parkinson’s disease brain with and without dementia. Exp Neurol. Sep;225(1):210–8. doi: 10.1016/j.expneurol.2010.06.017. Epub 2010 Jun 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004 Feb;55(2):164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Freed CR. Tyrosine-to-cysteine modification of human alpha-synuclein enhances protein aggregation and cellular toxicity. J Biol Chem. 2004;279(11):10128–10135. doi: 10.1074/jbc.M307563200. [DOI] [PubMed] [Google Scholar]