

Abstract
Chronic alcoholism is associated with impaired cognitive functioning. Over 75% of autopsied chronic alcoholics have significant brain damage and over 50% of detoxified alcoholics display some degree of learning and memory impairment. However, the relative contributions of different etiological factors to the development of alcohol-related neuropathology and cognitive impairment are questioned. One reason for this quandary is that both alcohol toxicity and thiamine deficiency result in brain damage and cognitive problems. Two alcohol-related neurological disorders, alcohol-associated dementia and Wernicke-Korsakoff syndrome have been modeled in rodents. These pre-clinical models have elucidated the relative contributions of ethanol toxicity and thiamine deficiency to the development of dementia and amnesia. What is observed in these models—from repeated and chronic ethanol exposure to thiamine deficiency—is a progression of both neural and cognitive dysregulation. Repeated binge exposure to ethanol leads to changes in neural plasticity by reducing GABAergic inhibition and facilitating glutamatergic excitation, long-term chronic ethanol exposure results in hippocampal and cortical cell loss as well as reduced hippocampal neurotrophin protein content critical for neural survival, and thiamine deficiency results in gross pathological lesions in the diencephalon, reduced neurotrophic protein levels, and neurotransmitters levels in the hippocampus and cortex. Behaviorally, after recovery from repeated or chronic ethanol exposure there is impairment in working or episodic memory that can recover with prolonged abstinence. In contrast, after thiamine deficiency there is severe and persistent spatial memory impairments and increased perseverative behavior. The interaction between ethanol and thiamine deficiency does not produce more behavioral or neural pathology, with the exception of reduction of white matter, than long-term thiamine deficiency alone.
Keywords: alcoholism, amnesia, dementia, memory, Wernicke-Korsakoff Syndrome
Introduction
Alcohol addiction is a severe disorder with many long-lasting health consequences—one of which can be impaired cognitive functioning. Numerous studies have reported that 50 – 75% of detoxified alcoholics have some type of cognitive or memory disturbance (for reviews, see Dufour, 1993; Parsons & Nixon, 1993; Smith & Atkinson, 1995). Furthermore, chronic alcohol consumption can lead to at least two long-lasting neurological disorders associated with severe cognitive dysfunction: alcohol-associated dementia (AAD) and Wernicke-Korsakoff syndrome (WKS). The neurotoxic effects of long-term heavy alcohol consumption are believed to produce AAD, whereas long-term heavy alcohol consumption in combination with dietary deficiencies—particularly thiamine—can lead to WKS. Although WKS is a nutrition deficiency disorder, it is most frequently reported in alcoholic patients (Knopelman, Thomson, Guerrini & Marshall, 2009). Indeed, it has been estimated that greater than 10% of alcoholic patients have symptoms of either AAD or WKS (Harper & Kril, 1990; Parson & Nixon, 1993).
To gain mechanistic insights to the consequences of ethanol neural toxicity and thiamine deficiency on learning and memory function, a number of animal models of alcohol-related disorders have been developed. Some models isolate the effects of long-term or repeated high levels of ethanol exposure whereas others assess the effects of thiamine deficiency. Other work has assessed the synergistic interactions between ethanol toxicity and thiamine deficiency. The amount of ethanol-induced neural and behavioral change seems to be dependent on length of ethanol exposure, volume of alcohol, and degree of withdrawal signs or number of binge bouts (Crews & Nixon, 2009). If animals are allowed to progress through a severe a bout of thiamine deficiency (Zhang et al., 1995), there are massive lesions in the anterior and midline thalamus as well as the mammillary bodies. Furthermore, recent evidence demonstrates that thiamine deficiency alters hippocampal- and frontal cortical-dependent behaviors and neurochemistry.
I. Human neurological disorders associated with chronic alcohol consumption
Autopsy evaluations and in vivo neuroimaging of the brains of diagnosed human alcoholics has revealed that 78% of this population exhibits some degree of brain pathology (Goldstein & Shelley, 1980; Harper, 1998). The clinical presentation of brain damage in alcoholics is heterogeneous and results in a range of cognitive abnormalities. This is likely due to that a multitude of factors present in the alcoholic lifestyle (head injury, liver disease, malnourishment) that can cause brain damage. However, key factors that are important for brain damage in chronic alcoholics are: amount of consumption, length of drinking history, and malnourishment.
In fact, the diagnosis of AAD requires a careful clinical examination as this disorder includes a wide range of disrupted cognitive capacities that overlap with other types of dementia. Thus, a key diagnostic feature for this type of dementia is a history of alcohol abuse. The DSM-IV-TR (American Psychiatric Association, 2000) defines AAD as including memory impairment in addition to one or more other cognitive symptoms. The cognitive disturbances can include: aphasia (inability to use or understand language), apraxia (failure to make purposeful movements), agnosia (difficulty in identify objects), or disturbance in executive functioning (deficits in planning, organizing, attention, and/or changing cognitive strategies). In addition, the diagnosis of AAD cannot be made when a patient is acutely intoxicated or in the process of alcohol withdrawal.
A heavy drinking history is a cardinal feature in the criteria for AAD: Specifically, more than 35 drinks/week for men or 28 drinks/week for women for a period of 5 years. An additional component is the impairment of both executive control and memory that persists after 60 days of abstinence (Olsin, Atkinson, Smith, & Hendrie, 1998; Schmidt et al, 2005). Alcoholic-associated dementia is estimated to make up about 10% of all dementia cases and heavy drinking history is a significant contributing factor to the development of other forms of dementia (Smith & Kiloh, 1981). Although the existence of AAD is widely acknowledged by health professionals, it is not often identified due to the diffuse criteria and overlapping symptomology common in other cognitive disorders (Gupta & Warner, 2008).
In contrast, the clinical diagnosis of WKS has distinct behavioral criteria. Wernicke’s encephalopathy, which is the acute phase of WKS, is diagnosed by a classic triad of symptoms: oculomotor disturbances, motor-ataxia abnormalities, and global confusion (Victor, Adams, & Collins, 1971). The primary diagnostic feature of WKS is profound amnesia, both retrograde and anterograde (Victor, Adams, & Collins, 1989), but there are also impairments of perceptual and abstract problem solving skills (see Butters & Cermack, 1983; Parsons & Nixon, 1993). Despite these clear diagnostic criteria, WE and WKS is diagnosed more commonly in alcoholics at post-mortem than when while the patients are alive (Harper, 2007; Torvik, Lindboe, & Rogde, 1982). Indeed, post-mortem prevalence rates of WKS are 1 – 2% in the general population and 12 – 14% in the alcoholic population (Harper, Kril, & Holloway, 1986; Harper, 1998).
A number of reviews in the human literature (Bowden, 1990; Joyce, 1994; Victor et al., 1989; Harper, 1998) suggest that a clear clinical distinction between AAD and WKS may be unwarranted. This argument is based on the fact that it is difficult to determine the dietary status of many alcoholics making WKS an under-diagnosed disorder (Harper, 1998; Joyce, 1994). By this explanation, mild subclinical episodes of thiamine deficiency can account for most cases of AAD. Proteomic studies have shown that thiamine status is also altered in non-WKS alcoholics (Alexander-Kaufman, Harper, & Wilce, 2007). This finding provides some evidence that mild-to-moderate thiamine deficiency plays a role in the neurodegeneration observed in chronic alcoholics.
The cognitive decline observed in AAD patients is more variable than that reported in WKS patients. Alcoholic-associated dementia is said to result in “global” cognitive decline across a wide range of skills involving perceptual motor, visual spatial, abstract problem solving, as well as learning and memory processes (see Oscar-Berman, Kirkley, Gansler, & Couture, 2004; Parsons & Nixon, 1993; Tarter, 1975). The key behavioral features of WKS are profound amnesia that impedes new episodic and declarative learning and recall as well as the appearance of confabulation (Kopelman et al., 2009). In the human patient there is much overlap between AAD and WKS in both neuropathology and behavioral symptoms (Bowden, 1990; Harper, 1998; Joyce, 1994). What is missing is a “gold standard” for the neurological (both brain and behavior) diagnosis of AAD as distinct entity from WKS. This quandary results in problems for the clinical diagnosis of both syndromes (Harper, 1998; Olsin et al., 1998). Others (Lishman, 1986, 1990; Smith & Atkinson, 1995) argue that AAD and WKS are distinct disorders with overlapping clinical symptoms. This “dual vulnerability hypothesis” states that the development of these disorders in certain individuals likely involves numerous factors. One key factor that has been hypothesized is a genetic pre-disposition for the development of WKS (alterations in thiamine metabolism) or AAD (susceptibility to alcohol neurotoxicity). However, in human patients it is difficult to distinguish primary alcohol-induced pathology from brain damage due to the alcoholic lifestyle (vitamin deficiency, malnutrition, head trauma, liver disease, diabetes). Accordingly, neuropathological data as subsequently described in this review lends further support towards a continuum on the effects of alcohol to thiamine deficiency on the brain and memory.
A. Neuropathology in alcohol-related disorders
The earliest reports of brain abnormalities in those with excessive alcohol consumption were non-specific findings from CT imaging and post-mortem data. For instance, CT studies in alcoholics described reductions in brain volume (Harper & Blumbergs, 1982) and clinical pathology reports revealed reduced brain weights in alcoholics compared to controls (Harding, Halliday, Caine, & Kril, 1996). The most commonly reported findings in the brains of alcoholics are sulcal widening and ventricular enlargement as well as cortical white matter shrinkage, hippocampal abnormalities, and cell loss in the septal region and cerebellum (Harper, 1998; Lishman, 1990; Parson & Nixon, 1994; Pfefferbaum et al., 1992).
It has been well documented that there is neuronal loss in specific regions of the cerebral cortex, hypothalamus, and cerebellum of the alcoholic brain (Harper, Kril, & Daly, 1987). However, a stereology study in humans (Harding, Wong, Syoboda, Kril, & Halliday, 1997) found that there was no hippocampal neuronal loss in alcoholics compared to non-alcoholics. It was discovered that the hippocampal volume decrease observed in human alcoholics occurs from white matter loss and not from gray matter. Furthermore, both pathological (Harper & Kril, 1990) and imaging studies (Pfefferbaum, Lim, Desmond, & Sullivan, 1996) demonstrated reduced corpus callosum volume and white matter degeneration in the cerebellum of alcoholics (Sullivan, Deshmukh, & Desmond, 1998). The mechanism for white matter loss appears to be the occurrence of both myelin loss and the degradation of axonal circuitry (Pfefferbaum & Sullivan, 2005).
The frontal lobes appear to be more vulnerable to alcohol-related brain damage than other cerebral regions (Dirksen et al., 2006; Oscar-Berman et al., 2004; Pfefferbaum, Sullivan, Mathalon, & Lim, 1997; Ratti, Bo, Giarini, & Soragna, 2002). Examination of post-mortem tissue from the Australian brain bank has revealed decreased neuron density (15–23%) in the frontal cortex of alcoholics (Harper & Matsumoto 2005). In vivo methods have shown frontal cortex abnormalities in alcoholics, such as decreased frontal lobe volume detected by MRI (Pfefferbaum et al., 1997), abridged regional blood flow measurements that can recover with abstinence (Gansler et al., 2000), and decreased amplitude of event-related potentials during the processing of visual targets (Chen et al., 2007). In addition, hypermetabolism in white matter and hypometabolism in diencephalic-limbic gray matter (frontal cortex, temporal cortex, retrosplenial cortex) has been found in WKS patients using FDG-PET (Reed et al., 2003).
Numerous studies have revealed some significant neuropathological differences between WKS and non-WKS alcoholics. While non-WKS alcoholics typically evidence some degree of structural abnormalities in the same regions as WKS alcoholics, it is often less severe. For example, WKS patients have wider third ventricles, larger lateral ventricles, and wider interhemispheric fissures than non-WKS chronic alcoholics (Sullivan & Pfefferbaum, 2009). However, sulcal and Sylvian fissure widths are equivalent in WKS and non-WKS alcoholics (Jacobson & Lishman, 1990). Adding further credence to the notion of a continuum of pathology, a graded pattern of volume deficits were observed, from mild in non-WKS alcoholics to moderate and severe in WKS, in the frontal cortex, mammillary bodies, hippocampus, thalamus, cerebellum, and pons (Sullivan & Pfefferbaum, 2009).
Studies by Harper and colleagues (1998 Harper and colleagues (2007) further suggest that thiamine deficiency is the foremost feature causing severe brain damage and structural changes in alcoholics. In WKS patients, regardless of etiology of thiamine deficiency (different diseases or conditions that result in restrict eating or limit adequate vitamin absorption), a prominent neuropathological consequence is lesions of the anterior and midline thalamic nuclei as well as the mammillary bodies (Mair et al., 1979; Mayes et al., 1988) and this extent of pathology is not evident in alcoholics without WKS. Recent studies suggest that the amnesic syndrome in WKS is due to damage in the diencephalic-hippocampal circuitry including thalamic nuclei and mammillary bodies (Sullivan & Marsh, 2003). Furthermore, Harding et al. (2000) found that neuronal loss in the anterior thalamic nuclei was critical to the production of amnesia in alcoholic-WKS patients.
B. Behavioral consequences of chronic alcoholism
In regards to a continuum of behavioral impairment, Butters (1985) suggested that while problem solving and visuoperceptual deficits develop slowly after many years of alcoholism, the amnesic symptoms associated with WKS appear more acutely when severe malnutrition and alcoholism are combined. The cognitive decline seen in AAD patients is more variable than that reported in WKS patients. Cognitive deficits on frontal cortical-dependent tasks has been observed in alcoholics with and without WKS (Dirksen, Howard, Cronin-Golomb, & Oscar-Berman, 2006; Gansler et al., 2000; Oscar-Berman & Evert, 1997; Oscar-Berman et al., 2004). Beyond tests of memory, WKS patients were impaired on tests of fluency, cognitive flexibility, and perseverative responding. Non-WKS alcoholics showed some frontal system deficits as well, but these generally were mild compared to the WKS patients.
The key clinical features of WKS are both anterograde amnesia, or the inability to create new memories following the event that caused the amnesia, and retrograde amnesia, or the inability to recall events prior to the amnesic state (Leng & Parkin, 1988). The retrograde component of WKS can extend as far back 20–30 years and there is a temporal gradient such that earlier memories are more intact (Kopelman, 1989). Studies that assessed anterograde memory functions have demonstrated that WKS patients have impaired declarative memory as assessed by both episodic (recall of passages) and semantic (letter and category fluency) tasks (Butters et al., 1987; Gold & Squire, 2006). Such results have been replicated many times (see Kopelman et al., 2009), demonstrating that WKS patients have difficulty establishing new explicit memories and poor long-term memories for personal experiences and the contexts in which they occur. Whether there is sparing of implicit or nondeclarative memory functions in WKS patients appears to be dependent on task demands. Studies that assess perceptual priming and perceptual motor performance reveal that implicit memory is relatively spared in WKS patients (d’Ydewalle & Van Damme, 2007; Fama, Pfefferbaum, & Sullivan, 2006; Gold & Squire, 2006). However, delayed eyeblink conditioning, a basic form of implicit memory, is impaired in both WKS patients and recovered alcoholics (McGlinchey-Berroth et al., 1995). This unique impairment of implicit memory is likely related to cerebellar deterioration that occurs with long-term alcohol consumption.
C. Recovery of impairment
While non-WKS alcoholic patients display some significant recovery of function over time following long-term detoxification of alcohol, WKS patients do not show the same trend. A number of longitudinal studies report that abstinent alcoholics show improvements of working memory and visuospatial abilities (Rosenbloom, Pfefferbaum, & Sullivan, 2004; Sullivan, Rosenbloom, Lim, & Pfefferbaum, 2000). Abstinence up to 7 years resolves many neurocognitive deficits associated with alcoholism, except for the suggestion of lingering deficits in spatial processing (Fein, Torres, & Prince, 2006). Gender appears to influence the recovery of motoric and postural capacities: Male alcoholics displaying significant recovery of these behaviors, whereas women alcoholics do not. Most of the human studies that have examined the development of alcohol-related disorders have used male participants. However, evidence suggests that women are more susceptible to alcohol-induced brain damage (Acker, 1986; Jacobson, 1986; Mann et al., 1992; Nixon, 1994; Wagner-Glen, 1993). This hypothesis arose from the observation that women alcoholics often drink less alcohol and have shorter drinking histories, but have brain damage comparable to male alcoholics (Nixon, 1994; Mann, Batra, Gunthner, & Schroth, 1992). Furthermore, the cognitive impairments seen in alcoholic females tend to be more severe than those seen in male alcoholics (Wagner-Glen, 1993). In addition, a disproportionately high number of women alcoholics exhibit AAD (Wagner-Glen, 1993) and WKS (Torvick et al., 1982; Victor et al., 1971). Whether this increase in “vulnerability” of women to the neurobehavioral and neuropathological consequences associated with alcoholism is related to the neurotoxic effects of alcohol and/or thiamine deficiency is unknown.
In neuropsychological tests, abstinent WKS patients of two years still show significant impairment in visual and verbal short- and long-term memory, working memory, basic executive functions, language, general knowledge, and visual-spatial abilities compared to controls (Fujiwara, Brand, Borsutzky, Steingass, & Markowitsch, 2008). Thus, it appears there is a continuum of functional recovery. Uncomplicated alcoholics, or those without features of WE or WKS, recover some of the behavioral and memory functioning albeit across years of abstinence. However, in WKS there is no significant recovery of functioning after abstinence. This suggests that the neurotoxic effects of alcohol on the brain may be temporary (Fein et al., 2006), while the neurological effects of thiamine deficiency are more severe and permanent. Such questions can be answered directly by animal models.
II. Animal Models
Animal models serve a heuristic function by allowing for the precise manipulation of variables in an experimentally testable situation. It is not possible to control for duration or amount of alcohol exposure, malnutrition, or the age at which such exposure occurs in humans. Animal models provide a framework for the integration of these variables, which in turn allows for the direct testing of specific hypotheses about how gender, diet and ethanol exposure interact. Thus, animal models may allow us to answer some questions about the role different factors play in the development of alcohol-related disorders.
Three etiological protocols (chronic ethanol intake, chronic intermittent [binge] ethanol intake, pyrithiamine-induced thiamine deficiency) have been used extensively to model alcohol-related neurological disorders in rodents. This body of research suggests that the neuropathology produced independently by thiamine deficiency differs in location and extent from that produced by chronic ethanol consumption (see Figure 1). Consequently, the extent of cognitive impairment is reflective of the degree and location of neuropathology induced by varying degrees of ethanol exposure and thiamine deficiency. Thus, in the review to follow, literature concerning learning and memory impairment will be discussed in relation to both ethanol-induced and thiamine deficiency-induced alterations in brain and behavior.
Figure 1.

A schematic outlining the similarities and differences of the chronic ethanol exposure and thiamine deficiency affects on brain structure and behavior in animal models. Bulleted points represent factors that can modify the degree of dysfunction. Although there is considerable overlap between the two etiological variables, thiamine deficiency is unique in producing lesions within the diencephalon.
A. Types of Chronic Ethanol Exposure
A prominent feature of chronic ethanol intake in rodents is impairment of hippocampal-dependent learning and memory (e.g., Cagetti, Pinna, Guidotti, Baicy, & Olsen, 2004; Lukoyanov, Pereira, Paula-Barbosa, & Cadete-Leite, 2003), which occurs as a consequence of the vulnerability of the hippocampus to ethanol-induced neurodegeneration (Garcia-Moreno et al., 2001; Paula-Barbosa, Brandao, Madeira, & Cadete-Leite, 1993). However, the degree of cognitive impairment is dependent upon the duration of ethanol exposure, the degree of resultant malnutrition, the animal strain, and number of withdrawal episodes. Consequently, with regards to animal models of human alcohol consumption, there is a continuum of ethanol exposure that produces neurological effects ranging from alternations in brain plasticity to cellular death and overt lesion. In the sections to follow, the literature concerning the rodent continuum of ethanol exposure and neuropathology will be reviewed in relation to the etiology of hippocampal learning and memory impairments.
i. Chronic Intermittent Ethanol
The chronic intermittent ethanol (CIE) exposure paradigm, which models human binge drinking and alcohol dependence, consists of repeated episodes of ethanol intoxication and withdrawal (Olsen, Liang, Cagetti, & Spigelman, 2005). Despite a lack of gross neuropathology in this model, it does appear to alter hippocampal function by modulating inhibition and excitation at the level of the synapse (Nelson, Ur, & Gruol, 2005; Olsen et al., 2005; Ward, Lallemand, & de Witte, 2009). Intermittent ethanol exposure and withdrawal hinders neural inhibition by reducing GABA(A) receptor function (Kang, Spigelman, Sapp, & Olsen, 1996; Mahmoudi, Kang, Tillakaratne, Tobin, & Olsen, 1997; Olsen et al., 2005) and modifying the expression of specific GABA(A) receptor subunits at the mRNA and protein level (Cagetti, Liang, Spigelman, & Olsen, 2003; Mahmoudi et al., 1997; Olsen et al., 2005). In addition, repeated episodes of intoxication and withdrawal decrease seizure threshold (Becker, Veatch, & Diaz-Granados, 1998; Kokka, Sapp, Taylor, & Olsen, 1993), which is presumed to occur as a consequence of kindling induction (Kang et al., 1996; Mahmoudi et al., 1997). Thus, downstream consequences of altered GABAergic function might include changes in synaptic transmission, diminished neuroplasticity and increased neurotoxicity.
The CIE paradigm also alters hippocampal and cortical glutamatergic synaptic function by elevating the expression of NMDA receptor subunits (NR2A and NR2B) after cessation of, but not during, CIE exposure (Nelson et al., 2005; Qiang, Denny, & Ticku, 2007; Rani & Ticku, 2006). The increase in NMDA receptor expression might also reflect altered glutamatergic synaptic transmission following CIE treatment (Nelson, Ur, & Gruol, 1999). A potential consequence of chronically elevated NMDA receptor activity might be reduced levels of the survival-promoting neurotrophins (i.e., brain-derived neurotrophic factor [BDNF; see Hardingham & Bading, 2003]) leading to excitotoxicity and cell death. However, CIE exposure has been shown to increase both septal and hippocampal BDNF levels (Miller, 2004) indicating that the effect of CIE on neurotrophins is structurally dependent. Another consequence of CIE is temporary suppression of long-term potentiation (LTP), a process thought to underlie neuroplasticity and hippocampal learning and memory (Malenka & Nicoll, 1999; Roberto, Nelson, Ur, & Gruol, 2002). Long-term potentiation is suppressed immediately following CIE exposure, but is recovered following as little as 5 days of withdrawal from this paradigm. This recovery of LTP coincides with the increased NMDA receptor up-regulation indicating that the alteration of NMDA-dependent receptor function might be involved in the recovery of LTP (Roberto et al., 2002). Subsequent experimentation revealed decreased levels of phosphorylated mitogen activated protein kinase (MAPK), which is critically involved in LTP formation, immediately following withdrawal of CIE (English & Sweatt, 1997; Roberto et al., 2003). The recovery of LTP and subsequent increase in NMDA receptor expression was accompanied by recovered MAPK signaling (Roberto et al., 2003). Thus, the reduction of LTP immediately following cessation of CIE is likely due to altered MAPK activation, whereas up-regulation of NMDA receptors appears to might contribute to the recovery of LTP.
Chronic bouts of intoxication and withdrawal alter neural plasticity in the hippocampus that result in cognitive deficits as evidenced by impairment on the spatial Morris water maze (MWM). On this task, a rat is trained to learn the location of a submerged platform set in opaque water. After several training sessions, the subject is then released from random quadrants following a temporal delay within the circular apparatus and the latency and swim path provides a reliable measure of spatial memory. Chronic intermittent ethanol exposure impairs spatial recall of the location of an escape platform, but does not hinder spatial learning during the training session (Cagetti et al., 2004). The altered plasticity and memory impairments might be, in part, a corollary of diminished neurogenesis. Neurogenesis involves the generation, maturation, migration and functional integration of new neurons into the hippocampal dentate gyrus and has been implicated in hippocampal-mediated learning and memory (Gould, Beylin, Tanapat, Reeves, & Shors, 1999; Kempermann, Wiskott, & Gage, 2004; Shors et al., 2001). Proper GABAergic (Ge et al., 2006) and glutamatergic functioning at the NMDA receptor (Suzuki et al., 2006) is critical for successful integration of newly generated cells. Altered inhibition and excitation, coupled with reduced pro-survival neurotrophic support in the hippocampus, might lead to reduced neurogenesis in the CIE model. Indeed, brief binge ethanol exposure reduces both proliferation and survival of pluripotent cells (Crews et al., 2006; Nixon & Crews, 2002). This is of relevance to learning and memory as reductions in neurogenesis are associated with decreased hippocampal learning and memory (Madsen, Kristjansen, Bolwig, & Wortwein, 2003; Winocur, Wojtowicz, Sekeres, Snyder, & Wang, 2006). However, further research is necessary to determine if similar effects occur following chronic exposure.
ii. Chronic Ethanol Treatment
Chronic ethanol treatment (CET) models long-term drinking behavior and consists of prolonged daily ethanol administration (10% – 20% v/v) that lasts weeks to months (Franke, Kittner, Berger, Wirkner, & Schramek, 1997; Savage, Candon, & Hohmann, 2000). This is accomplished either by exposure to successive increases in ethanol concentration added to drinking water or atmosphere (Robles & Sabria, 2008; Zahr et al., 2009), or ethanol paired with an isocaloric liquid diet, which is thought to control for nutritional deficiencies common to other CET models (Santucci, Cortes, Bettica, & Cortes, 2008; Thinschmidt, Walker, & King, 2003). The result of these treatments is selective neural damage and cognitive impairment that persists following a period of abstinence (Farr, Scherrer, Banks, Flood, & Morley, 2005; Tremwel & Hunter, 1994). Interestingly, CET does not differentially affect the progression of neuropathology in male and female rats (Savage et al., 2000).
The cortex and hippocampus, two regions intimately involved in learning and memory, are atrophied following chronic ethanol ingestion. Chronic ethanol treatment culminates in shrinkage of both gray and white matter in the frontal cortex (Savage et al., 2000) and decreased thickness of cortical layers I–III in the prelimbic cortex (Cadete-Leite, Alves, Tavares, & Paula-Barbosa, 1990). Within the hippocampus, CET results in a 10 – 40% reduction in neuronal populations (pyramidal cells, granule cells, and interneurons) with a concomitant 20 – 60% decrease in the density of dendritic processes (Walker, King, & Hunter, 1993). Interestingly, hippocampal pyramidal cells appear more vulnerable to the neurotoxic effects of CET than granule cells (Bengoechea & Gonzalo, 1991). In addition, the loss of hippocampal granule (16%) and pyramidal cell (20%) populations (Franke et al., 1997; Paula-Barbosa et al., 1993) is evident after a 2-month abstinence period (Riley & Walker, 1978; Walker, Barnes, Zornetzer, Hunter, & Kubanis, 1980). Thus, long-term ethanol intake is neurotoxic to neurons in the hippocampal formation and cortex, and the cellular death incurred likely contributes to learning and memory dysfunction.
Compounding CET-induced neuroanatomical damage is dysregulation of the basal forebrain cholinergic system. This region is important for proper hippocampal function as it modulates learning and memory in this region (for review, see Gold, 2003). Specifically, chronic ethanol exposure reduces the populations of choline acetyltransferase (ChAT)- and acetylcholinesterase (AChE)-positive cells in the medial septum/diagonal band (MS/DB [Floyd et al., 1997; Hodges et al., 1991; Savage et al., 2000]), a structure that provides the majority of acetylcholine (ACh) input to the hippocampus. The result of blunted cholinergic input to the hippocampus is decreased ChAT and AChE activity and function (Casamenti, Scali, Vannucchi, Bartolini, & Pepeu, 1993; Hodges et al., 1991; Lukoyanov et al., 2003; Pires et al., 2001, 2005) as well as ACh receptor down-regulation within this region (Robles & Sabria, 2008). However, there is no evidence of cortical cholinergic dysfunction following CET (Pereira, Menezes, Franco, Costa, & Ribeiro, 1998; Pires, Pereira, Oliveira-Silva, Franco, & Ribeiro, 2001; Pires et al., 2005). This culminates in downstream reductions in hippocampal ACh efflux (~57% decrease) and concomitant behavioral dysfunction (Casamenti et al., 1993; Hodges et al., 1991; Melis, Stancampiano, Imperato, Carta, & Fadda, 1996), suggestive of behaviorally-elicited hippocampal hypofunction.
A family of proteins that modulate neural plasticity underlying hippocampal learning and memory are the neurotrophins (Chao, 2000; Lu & Chow, 1999). Chronic ethanol exposure decreases expression of several of these proteins, including nerve growth factor (NGF) and BDNF in the hippocampus (Hellmann, Rommelspacher, & Wernicke, 2009; Miller & Mooney, 2004; Tapia-Arancibia et al., 2001), which might contribute to the behavioral impairments (Davis, 2009). Indeed, a downstream consequence of reduced neurotrophin levels in the hippocampus is diminished neurogenesis as these proteins are important regulators of cell survival (Choi, Li, Parada, & Sisodia, 2009). Thus, diminished hippocampal neurogenesis maybe contributing to the cognitive impairments observed in the rodent CET model—but this has not been directly tested.
Long-term potentiation (LTP) is a form of activity-dependent synaptic plasticity induced by high frequency stimulation that may underlie hippocampal-dependent learning and memory (Bliss & Lomo, 1973; Bliss & Gardner, 1973). Chronic exposure to ethanol diminishes the magnitude of LTP in the hippocampus (Peris et al., 1997). Unlike the dual role of GABA and glutamate in decreasing LTP in the CIE paradigm, LTP in the CET model is altered due to changes in GABAergic inhibition and acetylcholine transmission (Hu, Walker, Vickroy, & Peris, 1999; Peris et al., 1997). Although the precise mechanism of CET-induced potentiation of hippocampal GABAergic inhibition remains to be determined, it may involve muscarinic receptor stimulation of GABAergic interneurons within the hippocampus (Hu et al., 1999; Peris et al., 1997). Another form of synaptic plasticity, long-term depression (LTD), involves low-frequency stimulation and is also thought to be involved in learning and memory processes (for review, see Collingridge, Peineau, Howland, & Wang, 2010). Similar to LTP, chronic ingestion of ethanol impairs LTD (Thinschmidt et al., 2003). However, further research is necessary to delineate the precise relationship between alterations in LTP and LTD as they relate to CET-induced learning and memory impairment.
As was discussed in the preceding section, a hallmark feature of chronic alcohol intake in humans is learning and memory deficits. Similarly in rodents, a prominent feature of CET is deficits in hippocampal-dependent learning and memory function. Chronic ethanol treatment-induced neuropathology and cholinergic biochemical dysfunction culminate in behavioral impairments on the spatial MWM. Rodents exposed to long-term ethanol (20% v/v for 6 months) demonstrate both impaired learning of the spatial MWM task as well as decreased spatial memory of the location of a submerged platform relative to control subjects (Lukoyanov et al., 2003). However, performance on the radial arm maze (RAM), which assesses reference memory and working memory, has yielded contradictory results following CET. The spatial RAM task targets both forms of memory by assessing the rats’ ability to discriminate between baited and unbaited arms and memory of arms from which it as already retrieved food. Exposure to a moderate dose of ethanol (6.7 v/v for 7 months) does not impair reference or working memory on the spatial RAM task (Steigerwald & Miller, 1997). Interestingly, the concentration of ethanol the subject is exposed to appears to be critical as higher doses (20% v/v) for similar durations severely impairs working and reference memory on the spatial RAM task (Arendt et al., 1988; Arendt et al., 1989; Hodges et al., 1991). These latter spatial deficits are congruent with reduced ChAT activity in both the medial septum and nucleus basalis magnocellularis of the basal forebrain (Hodges et al., 1991). There are also disparate findings regarding the effects of higher doses (20% v/v) of ethanol on working and reference memory performance on the RAM task. However, these differences appear to be due to the animal strain as Sprague-Dawley rats were impaired (Arendt et al., 1988; Arendt et al., 1989; Hodges et al., 1991), but similar doses did not impair performance of Fisher rats (see Pereira et al., 1998).
Although there does not appear to be any differences in CET-induced neuropathological progression across gender, it does differentially affect cognitive processes in male and female rats. Indeed, on delayed matching- and delayed non-matching-to-position working memory tasks, male rats that were exposed to CET (20% v/v) for 20 weeks evidenced impairment on the non-matching version, whereas female rats displayed matching-to-position impairment (Savage et al., 2000). The factors underlying these gender differences are unclear, but are presumed to involve a complex interplay between hormonal and neurobiological processes.
Further research is warranted to determine the precise nature of the spatial deficits associated with CET, but pharmacological studies suggest that cholinergic hypofunction might be of particular importance. This cholinergic deficit hypothesis is supported by the reversal of maze performance deficits in CET rats treated with cholinergic agonists. Specifically, administration of arecoline and nicotine, two ACh agonists, enhances working memory on the spatial RAM task rats exposed to CET (20% v/v) for 28 weeks (Hodges et al., 1991). Furthermore, transplantation of ACh-rich, but not ACh-poor, foetal neural tissue from the basal forebrain into the hippocampus improved spatial RAM performance following CET (Arendt et al., 1989; Hodges et al., 1991). Thus, the forebrain cholinergic system holds promise as a therapeutic target for the treatment of cognitive deficits associated with CET.
B. Models of Thiamine Deficiency
Experimental thiamine deficiency paradigms model the dementia and neurodegeneration that typify nutritional deficiencies associated with chronic alcohol-induced Wernicke-Korsakoff syndrome (WKS).
i. Thiamine Deficiency
In its simplest form, thiamine deficiency (TD) is induced via exposure to a thiamine-depleted diet for an extended period of time (3–4 wks). As such, exposure to a thiamine-deficient diet for as little as 9 days in mice has been demonstrated to reduce hippocampal neurogenesis without negatively affecting the anatomy or physiology of the hippocampus (Zhao et al., 2008). As thiamine deficiency progresses to 30 days, levels of AChE are reduced in the cortex and hippocampus (Pires et al., 2001, 2005), which is associated with deficits in spatial memory on the MWM (Pires et al., 2005) and impairments on passive avoidance (Nakagawasai et al., 2000; 2001). Emotional behavior changes, including impairment on forced swim and increased muricide (Nakagawasai et al., 2001). The cognitive/memory deficits observed during the later phases of acute TD are improved by administration of acetylcholinesterase inhibitors (AChEIs), muscarinic agonists or herbal compounds that increase ACh release (Nakagawasai, 2005). After prolonged TD there is also a loss of cholinergic neurons within the forebrain (Zhao et al., 2008) and a loss of cholinergic fibers that innervate the hippocampus (Nakagawasai et al., 2000) that could contribute to some of the cognitive/memory abnormalities seen in the late stages of TD.
ii. Pyrithiamine-Induced Thiamine Deficiency
The pyrithiamine-induced thiamine deficiency (PTD) model has been used with rodents for the past 40 years to study both the molecular and behavioral consequences of thiamine deficiency-induced WKS (e.g., Holowach, Kauffman, Ikossi, Thomas, & McDougal, Jr., 1968; Langlais, Zhang, & Savage, 1996; Watanabe, 1978; Witt, 1985). It involves co-administration of pyrithiamine (an inhibitor of thiamine absorption and metabolism that also affects thiamine pryophosphokinase and thiamine triphosphate [Butterworth & Heroux, 1989]) and feeding of a thiamine-deficient diet, which reduces complications and mortality rates that tend to accompany dietary thiamine deficiency alone (Freeman, Nielsen, & Gibson, 1987). Furthermore, dietary restriction alone typically produces lesion restricted to brainstem nuclei and fails to produce the pattern of diencephalic lesion observed in WKS (Troncoso, Johnston, Hess, Griffin, & Price, 1981; Witt, 1985). In contrast, the PTD model replicates both the acute neurological signs of Wernicke’s Encephalopathy (WE), and the chronic neuropathological and cognitive deficits that define alcohol-induced WKS (Langlais et al., 1996; Troncoso et al., 1981; Witt, 1985).
Glutamate excitotoxicity is the primary mechanism of cell death the PTD model (Hazel Butterworth, & Hakim, 1993; Langlais & Zhang, 1993; Todd & Butterworth, 1998). Recent studies further confirm the role of oxidative stress in neuropathology associated with TD as elevated levels of the oxidative stress indicators hemeoxygenase-1 and intracellular cell adhesion molecule-1, have been reported in the brains of TD rats (Calingasan & Gibson, 2000b; Gibson & Zhang, 2002). During the early WE stage of PTD treatment, anatomical damage is localized primarily to the gelatinosus and anteroventral ventrolateral thalamic nuclei (Zhang et al., 1995). As thiamine deficiency progresses into the later WE stage, cell death extends throughout several nuclei within the posterior thalamus (Langlais & Mair, 1990; Zhang et al., 1995). As PTD treatment progresses to model WKS, this pathological profile expands to encompass damage to numerous midline intralaminar thalamic nuclei and the medial mammillary bodies (Langlais & Savage, 1995; Langlais et al., 1996). Figure 2 displays the lesions and cell loss in the diencephalon in the PTD model using the immunocytochemical marker NeuN that stains the nucleus of neurons. In addition to the neuronal loss, there is degeneration of key limbic system fiber tracts (i.e., fimbria/fornix and mammillothalamic tract [Langlais & Zhang, 1997; Markowitsch, 1988]). Interestingly, there is little indication of gross hippocampal damage (Langlais, Mandel, & Mair, 1992) despite evidence of functional impairment (Savage, Chang, & Gold, 2003; Vetreno, Anzalone, & Savage, 2008). This has led to the hypothesis that learning and memory impairments in PTD model is a product of a disconnect between the diencephalon and the extended hippocampal system (see Markowitsch & Pritzel, 1985; Warrington & Weiskrantz, 1982 for a description of the disconnection syndrome theory).
Figure 2.
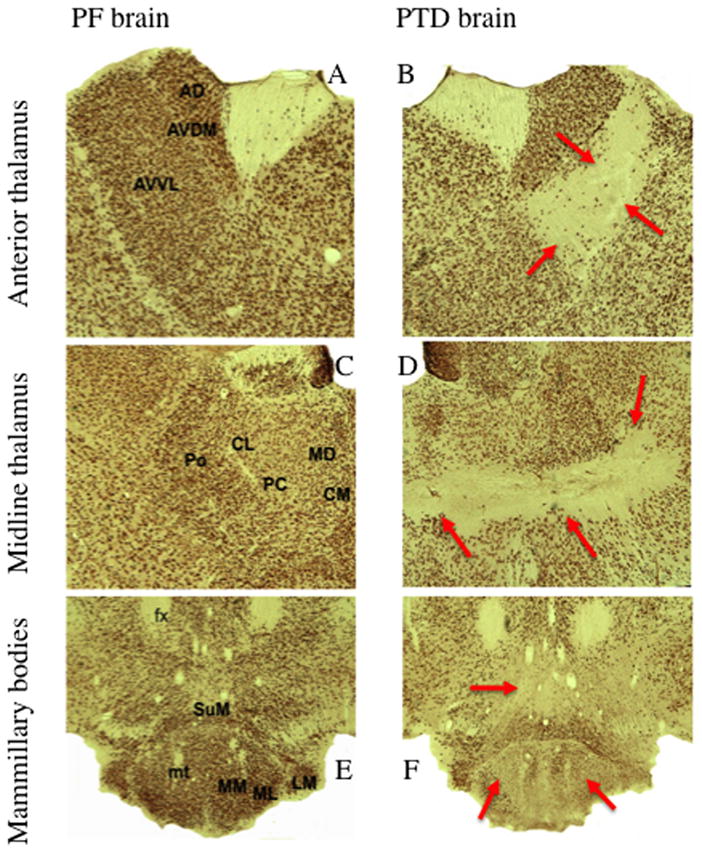
Neuronal specific nuclear protein (NeuN) stained slides comparing the thalamus and mammillary bodies of PF and PTD rats (from Anzalone et al, 2010). The top row (A and B) reveal the anterior thalamic nuclei in both PF and PTD rats. In PTD rats there is selective cell loss in the anteroventral ventrolateral (AVVL) with relative sparing of the anteroventral dorsal medial (AVDM) and anterodorsal nuclei (AD). The middle row (C and D) are images of midline and intralaminar thalamic structures. There is significant cell loss in the intralaminar nuclei (central medial (CM), paracentral (PC) and centrolateral (CL) nuclei) as well as the posterior thalamic nucleus (Po). However, the medial dorsal (MD) nucleus is spared—with the exception of the most ventral tip. The bottom row (E and F) are examples of the hypothalamus including the mammillothalamic tract (mt), medial mammillary nucleus (MM), lateral medial mammillary nucleus (ML) lateral mammillary nucleus (LM) and the supramammillary nucleus (SuM). There is significant cell loss medial mammillary ncuclei (MM, ML) and within the SuM.
Alterations in basal forebrain cholinergic input to the hippocampus is another factor likely contributing to learning and memory impairment in the PTD model. Indeed, memory deficits during the WE stage of PTD are reversed via administration of AChEIs (Nakagawasai, 2005) indicative of cholinergic dysfunction. Furthermore, our laboratory consistently demonstrated a ~30% reduction in ChAT-positive cells in the MS/DB during the WKS stage (Pitkin & Savage, 2001, 2004; Roland & Savage, 2009b), which correlates with behavioral impairment on spatial tasks (e.g., Roland & Savage, 2009b). This effect is congruent with thiamine deficiency as thiamine is a necessary co-factor for ACh synthesis (Todd & Butterworth, 1999) and ACh-synthesizing cells in the basal forebrain are especially liable to thiamine deficiency-induced cell death (Szutowicz et al., 2007). Loss of cholinergic cells, in turn, reduces ACh fiber densities within the hippocampus and cortex (Anzalone, Vetreno, Ramos, & Savage, 2010) that is correlated to blunted hippocampal cholinergic efflux during behavioral testing (Roland & Savage, 2007; Roland, Mark, Vetreno, & Savage, 2008; Savage et al., 2003; Savage, Roland, & Klintsova, 2007; Vetreno et al., 2008). This neuroanatomical and neurochemical data supports the “disconnection syndrome” hypothesis. Furthermore, as shown in Figure 3, not only is the hippocampus functionally down regulated after PTD treatment, but so are the limbic (prefrontal, frontal and retrosplenial) cortices. It is important to note that not all memory-related structures are affected: ACh efflux in the amygdala and striatum are normal when rats are tested on hippocampal-dependent tasks (Savage et al., 2007; Vetreno et al., 2008).
Figure 3.

Mean (± SEM) changes in ACh efflux (relative to baseline) before (B1-3), during (M1-3) and after (A1-3) training on hippocampal-dependent tasks in different memory-related brain structures in both PF (squares) and PTD (circles) rats. The greatest reductions were seen in the medial frontal cortex (A) prefrontal cortex (B), and hippocampus (C) followed by a moderate decrease in the retrosplenial [area 29ab] cortex (D). In contrast, no impairments in ACh efflux were observed in the amygdala (F) or dorsal striatum (F).
Additional support of the disconnection syndrome hypothesis is provided by the demonstration of recovery of function following administration of AChEIs, either systemically or intrahippocampally (see Figure 4, Roland et al., 2008; Roland, Levinson, Vetreno, & Savage, 2010), or intraseptal administration of bicuculline (Roland & Savage, 2009a). Thus, the extended hippocampal system atrophy, coupled with reduced hippocampal cholinergic innervation, culminates in severe and long-lasting learning and memory impairments. The precise nature of this interaction remains unclear, but the decrease in cholinergic innervation to the hippocampus might reduce neurogenesis, which would produce impaired learning and memory of hippocampal-mediated tasks. Although no studies have assessed this to date, this seems likely given the recent finding from our laboratory of reduced BDNF expression in the hippocampus of PTD-treated rats (see Figure 5).
Figure 4.

Acetylcholinesterase inhibors (AChEIs) acutely infused into the hippocampus (A: 40 ng, ●=0.97), medial septum (B: 5 μg,●=0.99) and medial frontal cortex (C: 1 μg,●=3.14) all significantly increased spontaneous alternation performance in PTD rats. NOTE: Dose response curves for all AChEIs were conducted for behavior and ACh efflux. Only the maximum effective dose is shown for each drug condition. *= significant difference between control and treatment conditions in PTD rats; ^ =Drug recovery in PTD rats is still reduced relative to PF saline score; ns= Recovery in PTD rats is equal to PF control score. For comparisons across studies, Cohen’s effect size (●) was reported.
Figure 5.

(A) Basal brain derived neurotropic factor (BDNF) levels in PF (open bars) and PTD rats (black bars). (B) There is a strong positive correlation between BDNF levels in the hippocampus and frontal cortex.
Several long-term behavioral deficits remain after administration of thiamine replacement therapy to resolve PTD treatment. Working memory deficits, as assessed on non-matching-to-position and non-matching-to-sample tasks (NMTP/NMTS) as well as matching-to-position and matching-to-sample tasks (MTP/MTS), have been consistently demonstrated in PTD rats (Langlais & Savage, 1995; Mumby, Cameli, & Glenn, 1999). These tasks require the subject to remember, for varying latencies, the order of stimulus presentation and use this information to guide behavior on future trials. Most studies implicate damage to the midline thalamic nuclei as causal to the working memory impairments (Mair, Otto, Knoth, Rabchenuk, & Langlais, 1991; Langlais et al., 1992; Langlais & Savage, 1995). Spatial learning and memory is also impaired in the PTD model. Using the spontaneous alternation task, which provides a good measure of hippocampal-dependent spatial memory (McIntyre, Pal, Marriott, & Gold, 2002; McIntyre, Marriott, & Gold, 2003), our laboratory has consistently demonstrated impaired alternation behavior in PTD rats (Roland et al., 2008; Savage et al., 2003, 2007; Vetreno et al., 2008). Pyrithiamine-treated rats are also impaired on the spatial MWM as evidenced by longer latencies to locate the hidden platform and increased thigmotaxic behavior (Langlais et al., 1992; Pires et al., 2005).
Despite all of the learning and memory deficits associated with PTD treatment, some learning and memory ability are preserved. Learning of simple discrimination tasks, such as light-dark discrimination (Mair, Anderson, Langlais, & McEntee, 1988), is unaffected in PTD-treated subjects. Similarly, these subjects are unimpaired on spatial tasks that do not require the hippocampus, such as an egocentric, striatal-based t-maze task (Vetreno et al., 2008) and an amygdala-based MTP task (Langlais & Savage, 1995).
iii. Thiamine Deficiency & Ethanol Exposure
Exposing rodents to ethanol in combination with bouts of thiamine deficiency is used to assess the combined effects of these processes on brain physiology and function (Ciccia & Langlais, 2000; He, Sullivan, Stankovic, Harper, & Pfefferbaum, 2007; Pires et al., 2001, 2005). It is thought that thiamine deficiency might exacerbate ethanol-induced neurotoxicity (Lotfi & Meyer, 1989; Zimitat et al., 1990). The onset of the clinical stages associated with thiamine deficiency do occur earlier and progress at a faster rate in rats that received ethanol than in those that did not (Zimitat, Kril, Harper, & Nixon, 1990). However, assessment of the effects of thiamine deficiency and ethanol in combination failed to demonstrate consistent synergistic effects in accelerating cognitive impairments (Ciccia & Langlais, 2000): Chronic ethanol alone increased preservative errors on spontaneous alternation and reduced delayed nonmatching- and matching-to-sample scores, whereas thiamine deficiency alone or in combination with CET only impaired delayed matching-to-sample scores. Combined CET and 1 week of exposure to a thiamine deficient diet was reported to reduce cortical and hippocampal AChE activity to a greater extent than ethanol alone (Pires et al., 2001). However, white matter atrophy appears to be more sensitive to the combined effects of thiamine deficiency and ethanol exposure: Rodents exposed to PTD + ethanol had greater thinning of myelin in the corpus callosum than did subjects that received only PTD treatment (He et al, 2007). The timing of thiamine deficiency during chronic ethanol exposure appears to affect the degree of frontal cortical cell loss. If thiamine deficiency occurs at the beginning, but not end, of the protracted ethanol exposure there is a loss of neurons in the FR1 region of the rat cortex (Kril & Homewood, 1993). Although these combined paradigms provide some insights into the contributions of the interactions between ethanol and thiamine deficiency, they do not encompass the entire spectrum of anatomical and behavioral pathology that characterize human WKS.
Summary
The neurological disorders of AAD and WKS are difficult to distinguish by their clinical or neuropathological features. There appears to be a continuum of brain and behavioral dysfunction in alcoholics with and without WKS. Numerous imaging studies have revealed the cortical and subcortical shrinkage, as well as ventricular enlargement, is greater in WKS alcoholics than uncomplicated alcoholics (Sullivan & Pfefferbaum, 2009). Despite relatively clear diagnostic criteria for WKS it is still under diagnosed in living alcoholics (see Harper, 2007). This occurs because the early clinical features of thiamine deficiency (the WE phase) can be missed or masked by alcohol intoxication. There is clear evidence that if patients are treated by high doses of thiamine in the WE phase, the amnestic Korsakoff phase can be modified or eliminated. However, if left untreated, the declarative-type of amnesia persists with little recovery across time. The WKS patient population was pivotal in the development of the theoretical dual memory dissociation between declarative (hippocampal-based memory) and nondeclarative (nonhippocampal memory) memory processes (see Squire, 1982).
In chronic alcoholics without WKS, some of the brain damage and cognitive impairment appears to be reversible if abstinence is achieved. The pattern of cognitive deficits seen in abstinent uncomplicated alcoholics is ‘frontal’ in nature, but hippocampal-dependent memory also seems to be affected. White matter loss within the cortex appears to be a major contributor to cognitive dysfunction (Sullivan & Pfefferbaum, 2009). However, neuronal loss has also been documented in specific regions of the frontal cortex (Harper et al., 1987). The frontal lobes regulate what has been termed “executive functions”: complex cognitive skills such as working memory, sequential ordering and reversal learning. These are the types of tasks that chronic alcoholics perform poorly on. However, binge drinking (most alcoholics have multiple episodes of heavy drinking followed by abstinence) that leads to repeated withdrawal syndromes appears to create hippocampal dysfunction that includes impaired neurogenesis and memory (see Crews & Nixon, 2009).
Animal models have shown us that both thiamine deficiency and chronic ethanol exposure adversely affect several brain regions and produce a range of behavioral impairment (see Figure 1). Both etiological factors produce some similar brain effects: loss of cholinergic cells in the basal forebrain, hippocampal ACh hypofunction, as well as shrinkage of frontal cortical grey and white matter. Thiamine deficiency has additional lesions and significant cell loss in the diencephalon. Numerous studies have demonstrated in both WKS patients and animal models that anterior and midline thalamic damage is critical in the production of anterograde amnesic state associated with thiamine deficiency. However, hippocampal and frontal cortical cholinergic dysfunctions are emerging as critical variables in the extent of amnesia and recovery of function. If ACh levels within the septohippocampal system are increased in PTD rats, then spatial memory can be recovered (see Figure 4).
Studies that have directly compared the two treatment protocols are few—as are the number of studies that have examined the interactive effects of ethanol toxicity and thiamine deficiency. To date, although ethanol may hasten the progression of thiamine deficiency, no additive behavioral impairment is found when ethanol exposure and thiamine deficiency is combined. These results suggest that severe neuropathology produced by thiamine deficiency decreases spatial memory to such an extreme dysfunctional level that is not further disrupted by additional brain damage. However, a recent study demonstrated that the corpus callosum appears to be affected to a greater extent when chronic ethanol exposure is combined with thiamine deficiency (He et al,. 2007).
Although there has been some effort to distinguish differences in neuropathology between the two models, there has been minimal effort to characterize the models on behavioral signs. Understanding the behavioral impairments produced by different etiological factors may lead to greater understanding of the human clinical conditions of AAD and WKS, and contribute to our knowledge of behavioral-brain relationships. This approach will result in a more valid model system for use in exploring potential therapeutic interventions for disorders of cognition and memory.
Acknowledgments
This research was funded by grant NINDS 054272 to LMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acker C. Neuropsychological deficits in alcoholics: the relative contributions of gender and drinking history. British Journal of Addiction. 1986;81:395–403. doi: 10.1111/j.1360-0443.1986.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Alexander-Kaufmann K, Harper C, Wilce P. Cerebellar vermis proteome of chronic alcoholic individuals. Alcoholism: Clinical and Experimental Research. 2007;31:1286–1296. doi: 10.1111/j.1530-0277.2007.00437.x. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington, DC: American Psychiatric Association; 2000. pp. 199–205. Revised. [Google Scholar]
- Anzalone S, Vetreno RP, Ramos RL, Savage LM. Cortical cholinergic abnormalities contribute to the amnesia state induced by pyrithiamine-induced thiamine deficiency in the rat. European Journal of Neuroscience. 2010;32:847–858. doi: 10.1111/j.1460-9568.2010.07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Marchbanks RM, Schugens MM, Sinden J, Lantos PL, Gray JA. Cholinergic system and memory in the rat: Effects of chronic ethanol, embryonic basal forebrain brain transplants and excitotoxic lesions of cholinergic basal forebrain projection system. Neuroscience. 1989;33:435–462. doi: 10.1016/0306-4522(89)90397-7. [DOI] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Sinden J, Schugens MM, Marchbanks RM, Lantos PL, Gray JA. Cholinergic-rich brain transplant reverse alcohol-induced memory deficits. Nature. 1988;332:448–450. doi: 10.1038/332448a0. [DOI] [PubMed] [Google Scholar]
- Becker HC, Veatch LM, Diaz-Granados JL. Repeated ethanol withdrawal experience selectively alters sensitivity to different chemoconvulsant drugs in mice. Psychopharmacology. 1998;139:145–153. doi: 10.1007/s002130050699. [DOI] [PubMed] [Google Scholar]
- Bengoechea O, Gonzalo LM. Effects of alcoholization on the rat hippocampus. Neuroscience Letters. 1991;123:112–114. doi: 10.1016/0304-3940(91)90170-x. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmisstion in the dentate area of the anesthetized rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anesthetized rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden SC. Separating cognitive impairment in neurologically asymptomatic alcoholism from Wernicke-Korsakoff syndrome: Is the neuropsychological distinction justified? Psychological Bulletin. 1990;107:355–366. doi: 10.1037/0033-2909.107.3.355. [DOI] [PubMed] [Google Scholar]
- Butters N. Alcoholic Korsakoff’s syndrome: Some unresolved issues concerning etiology, neuropathology, and cognitive deficits. Journal of Clinical and Experimental Neuropsychology. 1985;7:181–210. doi: 10.1080/01688638508401252. [DOI] [PubMed] [Google Scholar]
- Butters N, Granholm E, Salmon DP, Grant I, Wolfe J. Episodic and semantic memory: A comparison of amnesic and demented patients. Journal of Clinical and Experimental Neuropsychology. 1987;9:479–497. doi: 10.1080/01688638708410764. [DOI] [PubMed] [Google Scholar]
- Butterworth RF, Heroux M. Effect f pyrithiamine treatment and subsequent thiamine rehabilitation on regional cerebral amino acids and thiamine-dependent enzymes. Journal of Neurochemistry. 1989;52:1079–1084. doi: 10.1111/j.1471-4159.1989.tb01850.x. [DOI] [PubMed] [Google Scholar]
- Cadete-Leite A, Alves MC, Tavares MA, Paula-Barbosa MM. Effects of chronic alcohol intake and withdrawal on the prefrontal neurons and synapses. Alcohol. 1990;7:145–152. doi: 10.1016/0741-8329(90)90076-o. [DOI] [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW. Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Molecular Pharmacology. 2003;63:53–64. doi: 10.1124/mol.63.1.53. [DOI] [PubMed] [Google Scholar]
- Cagetti E, Pinna G, Guidotti A, Baicy K, Olsen RW. Chronic intermittent ethanol (CIE) administration in rats decreases levels of neurosteroids in hippocampus, accompanied by altered behavioral responses to neurosteroids and memory function. Neuropharmacology. 2004;46:570–579. doi: 10.1016/j.neuropharm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Gibson GE. Dietary restriction attenuates the neuronal loss, induction of hemeoxygenase-1 and blood barrier breakdown induced by impaired oxidative metabolism. Brain Research. 2000;885:62–69. doi: 10.1016/s0006-8993(00)02933-4. [DOI] [PubMed] [Google Scholar]
- Casamenti F, Scali C, Vannucchi MG, Bartolini L, Pepeu G. Long-term ethanol consumption by rats: Effect on acetylcholine release in vivo, choline acetyltransferase activity, and behavior. Neuroscience. 1993;56:465–471. doi: 10.1016/0306-4522(93)90346-h. [DOI] [PubMed] [Google Scholar]
- Chao MV. Trophic factors: An evolutionary cul-de-sac or door into higher neuronal function? Journal of Neuroscience Research. 2000;59:353–355. [PubMed] [Google Scholar]
- Chen AC, Porjesz B, Rangaswamy M, Kamarajan C, Tang Y, Jones KA, et al. Reduced frontal lobe activity in subjects with high impulsivity and alcoholism. Alcoholism: Clinical and Experimental Research. 2007;31:156–165. doi: 10.1111/j.1530-0277.2006.00277.x. [DOI] [PubMed] [Google Scholar]
- Choi SH, Li Y, Parada LF, Sisodia SS. Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Molecular Degeneration. 2009;4:52. doi: 10.1186/1750-1326-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia RM, Langlais PJ. An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcoholism: Clinical and Experimental Research. 2000;24:622–634. [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nature Reviews: Neuroscience. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol & Alcoholism. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J. BHT blocks NF-kB activation and ethanol-induced brain damage. Alcoholism: Clinical and Experimental Research. 2006;30:1938–1949. doi: 10.1111/j.1530-0277.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- Davis MI. Ethanol-BDNF interactions: Still more questions than answers. Pharmacology & Therapeutics. 2008;118:36–57. doi: 10.1016/j.pharmthera.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen CL, Howard JA, Cronin-Golomb A, Oscar-Berman M. Patterns of prefrontal dysfunction in alcoholics with and without Korsakoff’s syndrome, patients with Parkinson’s disease, and patients with rupture and repair of the anterior communicating artery. Neuropsychiatric Disease and Treatment. 2006;2:327–339. doi: 10.2147/nedt.2006.2.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour MC. The epidemiology of alcohol-induced brain damage. In: Hunt WA, Nixon SJ, editors. Alcohol-induced Brain Damage. National Institute on Alcohol Abuse and Alcoholism; Rockville, MD: 1993. pp. 39–69. [Google Scholar]
- d’Ydewalle G, Van Damme I. Memory and the Korsakoff syndrome: Not remembering what is remembered. Neuropsychologia. 2007;45:905–920. doi: 10.1016/j.neuropsychologia.2006.08.025. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. The Journal of Biological Chemistry. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Fama R, Pfefferbaum A, Sullivan EV. Perceptual learning in detoxified alcoholic men: Contributions from explicit memory, executive function, and age. Alcoholism: Clinical and Experimental Research. 2004;28:1657–1665. doi: 10.1097/01.alc.0000145690.48510.da. [DOI] [PubMed] [Google Scholar]
- Fama R, Pfefferbaum A, Sullivan EV. Visuoperceptual learning and alcoholic Korsakoff syndrome. Alcoholism: Clinical and Experimental Research. 2006;30:680–687. doi: 10.1111/j.1530-0277.2006.00085.x. [DOI] [PubMed] [Google Scholar]
- Farr SA, Scherrer JF, Banks WA, Flood JF, Morley JE. Chronic ethanol consumption impairs learning and memory after cessation of ethanol. Alcoholism: Clinical and Experimental Research. 2005;29:971–982. doi: 10.1097/01.alc.0000171038.03371.56. [DOI] [PubMed] [Google Scholar]
- Fein G, Torres J, Price LJ, Di Sclafani V. Cognitive performance in long-term abstinent alcoholic individuals. Alcoholism: Clinical and Experimental Research. 2006;30:1538–1544. doi: 10.1111/j.1530-0277.2006.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd EA, Young-Seigler AC, Ford BD, Reasor JD, Moore EL, Townsel JG, Rucker HK. Chronic ingestion produces cholinergic hypofunction in rat brain. Alcohol. 1997;14:93–98. doi: 10.1016/s0741-8329(97)86147-2. [DOI] [PubMed] [Google Scholar]
- Franke H, Kittner H, Berger P, Wirkner K, Schramek J. The reaction of astrocytes and neurons in the hippocampus of adult rats during chronic ethanol treatment and correlations to behavioral impairments. Alcohol. 1997;14:445–454. doi: 10.1016/s0741-8329(96)00209-1. [DOI] [PubMed] [Google Scholar]
- Freeman GB, Nielsen PA, Gibson GE. Effect of age on behavioural and enzymatic changes during thiamine deficiency. Neurobiology of Aging. 1987;8:429–434. doi: 10.1016/0197-4580(87)90037-6. [DOI] [PubMed] [Google Scholar]
- Fujiwara E, Brand M, Borsutzky S, Steingass HP, Markowitsch HJ. Cognitive performance of detoxified alcoholic Korsakoff syndrome patients remains stable over two years. Journal of Clinical and Experimental Neuropsychology. 2008;30:576–587. doi: 10.1080/13803390701557271. [DOI] [PubMed] [Google Scholar]
- Gansler DA, Harris GJ, Oscar-Berman M, Streeter C, Lewis RF, Ahmed I, et al. Hypoperfusion of inferior frontal brain regions in abstinent alcoholics: A pilot SPECT study. Journal of Studies on Alcohol. 2000;61:32–37. doi: 10.15288/jsa.2000.61.32. [DOI] [PubMed] [Google Scholar]
- Garcia-Moreno LM, Conejo NM, Pardo HG, Gomez M, Martin FR, Alonso MJ, Arias JL. Hippocampal AgNOR activity after chronic alcohol consumption and alcohol deprivation in rats. Physiology & Behavior. 2001;72:115–121. doi: 10.1016/s0031-9384(00)00408-x. [DOI] [PubMed] [Google Scholar]
- Ge S, Goh ELK, Sailor KA, Kitabatake Y, Ming G, Song H. GABA regulates synaptic integraion of newly generated neurons in the adult brain. Nature Letters. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Zhang H. Interactions of oxidative stress with thiamine homeostasis promote neurodegeneration. Neurochemistry Internatinal. 2002;40:493–504. doi: 10.1016/s0197-0186(01)00120-6. [DOI] [PubMed] [Google Scholar]
- Gold PE. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiology of Learning and Memory. 2003;80:194–210. doi: 10.1016/j.nlm.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Gold JJ, Squire LR. The anatomy of amnesia: Neurohistological analysis of three new cases. Learning & Memory. 2006;13:699–710. doi: 10.1101/lm.357406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein G, Shelly C. Neuropsychological investigation of brain lesion localization in alcoholism. In: Begleiter H, editor. Biological Effect of Alcohol. Plenum Press; New York, NY: 1980. pp. 731–743. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nature Neuroscience. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Gupta S, Warner J. Alcohol–related dementia: A 21st-century silent epidemic? British Journal of Psychiatry. 2008;19:351–353. doi: 10.1192/bjp.bp.108.051425. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Halliday G, Caine D, Kril JJ. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123:141–154. doi: 10.1093/brain/123.1.141. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Wong A, Svoboda M, Kril JJ, Halliday GM. Chronic alcohol consumption does not cause hippocampal neuron loss in humans. Hippocampus. 1997;7:78–87. doi: 10.1002/(SICI)1098-1063(1997)7:1<78::AID-HIPO8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The yin and yang of NMDA receptor signaling. Trends in Neuroscience. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Harper CG. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? Journal of Neuropathology and Experimental Neurology. 1998;57:101–110. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- Harper CG. The neurotoxicity of alcohol. Human & Experimental Toxicology. 2007;26:251–257. doi: 10.1177/0960327107070499. [DOI] [PubMed] [Google Scholar]
- Harper CG, Blumbergs PC. Brain weights in alcoholics. Journal of Neurology, Neurosurgery & Psychiatry. 1982;45:838–840. doi: 10.1136/jnnp.45.9.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Neuropathology of alcoholism. Alcohol. 1990;25:207–216. doi: 10.1093/oxfordjournals.alcalc.a044994. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ, Daly J. Are we drinking our neurones away? British Medical Journal. 1987;294:534–536. doi: 10.1136/bmj.294.6571.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Kril JJ, Holloway RL. Brain shrinkage in chronic alcoholics: A pathological study. British Medical Journal. 1985;290:501–504. doi: 10.1136/bmj.290.6467.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Matsumoto I. Ethanol and brain damage. Current Opinion in Pharmacology. 2005;5:73–78. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, Hakim AM. Cerebral vulnerability is associated with selective increase in extracellulary glutamate concentration in experimental thiamine deficiency. Journal of Neurochemistry. 1993;61:1155–1158. doi: 10.1111/j.1471-4159.1993.tb03635.x. [DOI] [PubMed] [Google Scholar]
- He X, Sullivan EV, Stankovic RK, Harper CG, Pfefferbaum A. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology. 2007;32:2207–2016. doi: 10.1038/sj.npp.1301332. [DOI] [PubMed] [Google Scholar]
- Hellmann J, Rommelspacher H, Wernicke C. Long-term ethanol exposure impairs neuronal differentiation of human neuroblastoma cells involving neurotrophin-mediated intracellular signaling and in particular protein kinase C. Alcoholism: Clinical and Experimental Research. 2008;33:538–550. doi: 10.1111/j.1530-0277.2008.00867.x. [DOI] [PubMed] [Google Scholar]
- Hodges H, Allen Y, Sinden J, Mitchell SN, Arendt T, Lantos PL, Gray JA. The effects of cholinergic drugs and cholinergic-rich foetal neural transplants on alcohol-induced deficits in radial maze performance in rats. Behavioural Brain Research. 1991;43:7–28. doi: 10.1016/s0166-4328(05)80048-8. [DOI] [PubMed] [Google Scholar]
- Holowach J, Kauffman F, Ikossi MG, Thomas C, McDougal DB., Jr The effects of a thiamine antagonist, pyrithiamine, on levels of selected metabolic intermediates and on activities of thiamine-dependent enzymes in brain and liver. Journal of Neurochemistry. 1968;15:621–631. doi: 10.1111/j.1471-4159.1968.tb08961.x. [DOI] [PubMed] [Google Scholar]
- Hu M, Walker DW, Vickroy TW, Peris J. Chronic ethanol exposure increases 3H-GABA release in rat hippocampus by presynaptic muscarinic receptor modulation. Alcoholism: Clinical and Experimental Research. 1999;23:1587–1595. [PubMed] [Google Scholar]
- Jacobson R. Female alcoholics: a controlled CT and brain scan and clinical study. British Journal of Addictions. 1986;81:661–669. doi: 10.1111/j.1360-0443.1986.tb00386.x. [DOI] [PubMed] [Google Scholar]
- Jacobson RR, Lishman WA. Cortical and diencephalic lesions in Korsakoff’s Syndrome: A clinical and scan study. Psychological Medicine. 1990;20:63–75. doi: 10.1017/s0033291700013234. [DOI] [PubMed] [Google Scholar]
- Joyce EM. Aetiology of alcoholic brain damage: Alcoholic neurotoxicity or thiamine malnutrition? British Medical Bulletin. 1994;50:99–114. doi: 10.1093/oxfordjournals.bmb.a072888. [DOI] [PubMed] [Google Scholar]
- Kang M, Spigelman I, Sapp DW, Olsen RW. Persistent reduction of GABA(A) receptor-mediated inhibition in rat hippocampus after chronic intermittent ethanol treatment. Brain Research. 1996;709:221–228. doi: 10.1016/0006-8993(95)01274-5. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Current Opinion in Neurobiology. 2004;14:186–191. doi: 10.1016/j.conb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Kopelman MD. Remote and autobiographical memory, temporal context memory and frontal atrophy in Korsakoff and Alzheimer patients. Neuropsychologia. 1989;27:437–460. doi: 10.1016/0028-3932(89)90050-x. [DOI] [PubMed] [Google Scholar]
- Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korsakoff syndrome: Clinical aspects, psychology and treatment. Alcohol & Alcoholism. 2009;44:148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- Kokka N, Sapp DW, Taylor AM, Olsen RW. The kindling model of alcohol dependence: Similar persistent reduction in seizure threshold to pentylenetetrazol in animals receiving chronic ethanol or chronic pentylenetetrazol. Alcoholism: Clinical and Experimental Research. 1993;17:525–531. doi: 10.1111/j.1530-0277.1993.tb00793.x. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Homewood J. Neuronal changes in the cerebral cortex of the rat following alcohol treatment and thiamine deficiency. Journal of Neuropathological and Experimental Neurology. 1993;52:586–593. doi: 10.1097/00005072-199311000-00005. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG. Protective effects of the glutamate antagonist MK-801 on pyrithiamine–induced lesions and amino acid changes in rat brain. The Journal of Neuroscience. 1990;10:1664–1674. doi: 10.1523/JNEUROSCI.10-05-01664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Mandel RJ, Mair RG. Diencephalic lesions, learning impairments, and intact retrograde memory following acute thiamine deficiency in the rat. Behavioural Brain Research. 1992;48:177–185. doi: 10.1016/s0166-4328(05)80155-x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behavioural Brain Research. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Extracellular glutamate is increased in thalamus during thiamine deficiency–induced lesions and is blocked by MK–801. Journal of Neurochemistry. 1993;61:2175–2182. doi: 10.1111/j.1471-4159.1993.tb07457.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang S. Cortical and subcortical white matter damage without Wernicke’s Encephalopathy after recovery from thiamine deficiency in the rat. Alcoholism: Clinical and Experimental Research. 1997;21:434–443. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang S, Savage LM. Neuropathology of thiamine deficiency: An update on the comparative analysis of human disorders and experimental models. Metabolic Brain Disease. 1996;11:19–37. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- Lukoyanov NV, Pereira PA, Paula-Barbosa MM, Cadete-Leite A. Nerve growth factor improves spatial learning and restores hippocampal cholinergic fibers in rats withdrawn from chronic treatment with ethanol. Experimental Brain Research. 2003;148:88–94. doi: 10.1007/s00221-002-1290-7. [DOI] [PubMed] [Google Scholar]
- Lishman WA. Alcoholic dementia: A hypothesis. The Lancet. 1986;327:1184–1186. doi: 10.1016/s0140-6736(86)91162-1. [DOI] [PubMed] [Google Scholar]
- Lishman WA. Alcohol and the Brain. British Journal of Psychiatry. 1990;156:635–644. doi: 10.1192/bjp.156.5.635. [DOI] [PubMed] [Google Scholar]
- Lotfi J, Meyer J. Cerebral hemodynamic and metabolic effects of chronic alcoholism. Cerebrovascular and Brain Metabolism Reviews. 1989;1:2–25. [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. The Journal of Neuroscience. 1990;10:1372–1379. doi: 10.1523/JNEUROSCI.10-04-01372.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. Journal of Neuroscience Research. 1999;58:76–87. [PubMed] [Google Scholar]
- Madsen TM, Kristjansen P, Bolwig TG, Wortwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience. 2003;119:635–642. doi: 10.1016/s0306-4522(03)00199-4. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M, Kang M, Tillakaratne N, Tobin AJ, Olsen RW. Chronic intermittent ethanol treatment in rats increases GABAA receptor a4-subunit expression: Possible relevance to alcohol dependence. Journal of Neurochemistry. 1997;68:2485–2492. doi: 10.1046/j.1471-4159.1997.68062485.x. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. Behavioral impairments, brain lesions, and monoaminergic activity in the rat following recovery from a bout of thiamine deficiency. Behavioural Brain Research. 1988;27:223–239. doi: 10.1016/0166-4328(88)90119-2. [DOI] [PubMed] [Google Scholar]
- Mair RG, Otto TA, Knoth RL, Rabchenuk SA, Langlais PJ. Analysis of aversively conditioned learning and memory in rats recovered from pyrithiamine-induced thiamine deficiency. Behavioral Neuroscience. 1991;105:351–359. [PubMed] [Google Scholar]
- Mair RG, Warrington EK, Weiskrantz L. Memory disorder in Korsakoff’s psychosis: A neuropathological and neuropsychological investigation of two cases. Brain. 1979;102:749–783. doi: 10.1093/brain/102.4.749. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation: A decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mann K, Batra A, Gunthner A, Schroth G. Do women develop alcoholic brain damage more readily than men? Alcoholism: Clinical and Experimental Research. 1992;16:1052–1056. doi: 10.1111/j.1530-0277.1992.tb00698.x. [DOI] [PubMed] [Google Scholar]
- Markowitsch HJ. Diencephalic amnesia: A reorientation towards tracts? Brain Research Reviews. 1988;13:351–370. doi: 10.1016/0006-8993(88)91226-7. [DOI] [PubMed] [Google Scholar]
- Markowitsch HJ, Pritzel M. The neuropathology of amnesia. Progress in Neurobiology. 1985;25:189–287. doi: 10.1016/0301-0082(85)90016-4. [DOI] [PubMed] [Google Scholar]
- Mayes AR, Meudell PR, Mann D, Pickering A. Location of lesions in Korsakoff’s syndrome: Neuropsychological and neuropathological data on two patients. Cortex. 1988;24:367–388. doi: 10.1016/s0010-9452(88)80001-7. [DOI] [PubMed] [Google Scholar]
- McGlinchey-Berroth R, Cermak LS, Carrillo MC, Armfield S, Gabrieli JD, Disterhoft JF. Impaired delay eyeblink conditioning in amnesic Korsakoff’s patients and recovered alcoholics. Alcoholism: Clinical and Experimental Research. 1995;19:1127–1132. doi: 10.1111/j.1530-0277.1995.tb01590.x. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Marriott LK, Gold PE. Patterns of brain acetylcholine release predict individual differences in preferred learning strategies in rats. Neurobiology of Learning and Memory. 2003;79:177–183. doi: 10.1016/s1074-7427(02)00014-x. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Pal SN, Marriott LK, Gold PE. Competition between memory systems: Acetylcholine release in the hippocampus correlates negatively with good performance on an amygdala-dependent task. Neuroscience. 2002;22:1171–1176. doi: 10.1523/JNEUROSCI.22-03-01171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis F, Stancampiano R, Imperato A, Carta G, Fadda F. Chronic ethanol consumption in rats: Correlation between memory performance and hippocampal acetylcholine release in vivo. Neuroscience. 1996;74:155–159. doi: 10.1016/0306-4522(96)00109-1. [DOI] [PubMed] [Google Scholar]
- Miller MW. Repeated episodic exposure to ethanol affects neurotrophin content in the forebrain of the mature rat. Experimental Neurology. 2004;189:173–181. doi: 10.1016/j.expneurol.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Miller MW, Mooney SM. Chronic exposure to ethanol alters neurotrophin content in the basal forebrain-cortex system in the mature rat: Effects on autocrine-paracrine mechanisms. Journal of Neurobiology. 2004;60:490–498. doi: 10.1002/neu.20059. [DOI] [PubMed] [Google Scholar]
- Mumby DG, Cameli L, Glenn MJ. Impaired allocentric spatial working memory and intact retrograde memory after thalamic damage cause by thiamine deficiency in rats. Behavioral Neuroscience. 1999;113:42–50. [PubMed] [Google Scholar]
- Nakagawasai O. Behavioral and neurochemical alterations following thiamine deficiency in rodents: Relationship to functions of cholinergic neurons. Yakugaku Zasshi. 2005;125:549–554. doi: 10.1248/yakushi.125.549. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Taniguchi R, Tan-No K, Esashi A, Niijima F, Kisara K. Characteristics of depressive behavior induced by feeding thiamine- deficiency diet in mice. Life Science. 2001;69:1181–1191. doi: 10.1016/s0024-3205(01)01206-1. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Tan-No K, Niijima F, Kisara K. Immunohistochemical estimation of brain choline acetyltransferase and somatostatin related to the impairment of avoidance learning induced by thiamine deficiency. Brain Research Bulletin. 2000;52:189–196. doi: 10.1016/s0361-9230(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Cruol DL. Chronic intermittent ethanol exposure alters CA1 synaptic transmisstion in rat hippocampal slices. Neuroscience. 1999;94:431–442. doi: 10.1016/s0306-4522(99)00336-x. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL. Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Research. 2005;1048:69–79. doi: 10.1016/j.brainres.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Nixon SJ. Cognitive deficits in alcoholic women. Alcohol World Health & Research. 1994;18:228–232. [PMC free article] [PubMed] [Google Scholar]
- Nixon K, Crews FT. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. Journal of Neurochemistry. 2002;83:1087–1093. doi: 10.1046/j.1471-4159.2002.01214.x. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Liang J, Cagetti E, Spigelman I. Plasticity of GABAA receptors in brains of rats treated with chronic intermittent ethanol. Neurochemical Research. 2005;30:1579–1588. doi: 10.1007/s11064-005-8836-6. [DOI] [PubMed] [Google Scholar]
- Oscar-Berman M, Evert DL. Alcoholic Korsakoff’s syndrome. In: Nussbaum PD, editor. Handbook of neuropsychology and aging. Plenum; New York, NY: 1997. pp. 201–215. [Google Scholar]
- Oscar-Berman M, Kirkley SM, Gansler DA, Couture A. Comparisons of Korsakoff and non-Korsakoff alcoholics on neuropsychological tests of prefrontal brain functioning. Alcoholism: Clinical and Experimental Research. 2004;28:667–675. doi: 10.1097/01.alc.0000122761.09179.b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons OA, Nixon SJ. Neurobehavioral sequelae of alcoholism. Neurologic Clinics. 1993;11:205–218. [PubMed] [Google Scholar]
- Paula-Barbosa MM, Brandao F, Madeira MD, Cadete-Leite A. Structural changes in the hippocampal formation after long-term alcohol consumption and withdrawal in the rat. Addiction. 1993;88:237–247. doi: 10.1111/j.1360-0443.1993.tb00807.x. [DOI] [PubMed] [Google Scholar]
- Pereira S, Menezes GA, Franco GC, Costa AE, Ribeiro AM. Chronic ethanol consumption impairs spatial remote memory in rats but does not affect cortical cholinergic parameters. Pharmacology, Biochemistry and Behavior. 1998;60:305–311. doi: 10.1016/s0091-3057(97)00472-3. [DOI] [PubMed] [Google Scholar]
- Peris J, Anderson KJ, Vickroy TW, King MA, Hunter BE, Walker DW. Neurochemical basis of disruption of hippocampal long-term potentiation by chronic alcohol exposure. Frontiers in Bioscience. 1997;2:309–316. doi: 10.2741/a193. [DOI] [PubMed] [Google Scholar]
- Peris J, Eppler B, Hu M, Walker DW, Hunter BE, Mason K, Anderson KJ. Effects of chronic ethanol exposure on GABA receptors and GABAB receptor modulation of 3H-GABA release in the hippocampus. Alcoholism: Clinical and Experimental Research. 1997;21:1047–1052. [PubMed] [Google Scholar]
- Pfefferbaum A, Lim KO, Desmond JE, Sullivan EV. Thinning of the corpus callosum in older alcoholic men. A magnetic resonance imaging study. Alcoholism: Clinical and Experimental Research. 1996;20:752–757. doi: 10.1111/j.1530-0277.1996.tb01682.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Lim K, Zipursky R, Mathalon D, Rosenbloom M, Lane B, Ha CN, Sullivan E. Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: A quantitative MRI study. Alcoholism: Clinical and Experimental Research. 1992;16:1078–1089. doi: 10.1111/j.1530-0277.1992.tb00702.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV. Disruption of brain white matte microstructure by excessive intracellular and extracellular fluid in alcoholism: Evidence from diffusion tensor imaging. Neuropsychopharmacology. 2005;30:423–432. doi: 10.1038/sj.npp.1300623. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, Lim KO. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcoholism: Clinical and Experimental Research. 1997;21:521–529. doi: 10.1111/j.1530-0277.1997.tb03798.x. [DOI] [PubMed] [Google Scholar]
- Pires R, Pereira S, Oliveira-Silva IF, Franco GC, Ribeiro AM. Cholinergic parameters and the retrieval of learned and re-learned spatial information: A study using a model of Wernicke-Korsakoff syndrome. Behavioural Brain Research. 2005;162:11–21. doi: 10.1016/j.bbr.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Pires R, Pereira S, Pittella J, Franco GC, Ferreira C, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacology, Biochemistry and Behavior. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behavioural Brain Research. 2001;119:167–177. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: The role of time of insult. Behavioural Brain Research. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny AD, Ticku MK. Chronic intermittent ethanol treatment selectively alters N-Methyl-D-aspartate receptor subunit surface expression in cultured cortical neurons. Molecular Pharmacology. 2007;72:95–102. doi: 10.1124/mol.106.033043. [DOI] [PubMed] [Google Scholar]
- Rani CS, Ticku MK. Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABAA and NMDA receptor subunits. Alcohol. 2006;38:89–97. doi: 10.1016/j.alcohol.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Ratti MT, Bo P, Giardini A, Soragna D. Chronic alcoholism and the frontal lobe: Which executive functions are impaired? Acta Neurologica Scandinavica. 2002;105:276–281. doi: 10.1034/j.1600-0404.2002.0o315.x. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Lasserson D, Marsden P, Stanhope N, Stevens T, Bello F, Kingsley D, Colchester A, Kopelman MD. FDG-PET findings in the Wernicke-Korsakoff syndrome. Cortex. 2003;39:1027–1045. doi: 10.1016/s0010-9452(08)70876-1. [DOI] [PubMed] [Google Scholar]
- Riley JN, Walker DW. Morphological alternations in hippocampus after long-term alcohol consumption in mice. Science. 1978;201:646–648. doi: 10.1126/science.566953. [DOI] [PubMed] [Google Scholar]
- Roberto M, Nelson TE, Ur CL, Brunelli M, Sanna PP, Gruol DL. The transient depression of hippocampal CA1 LTP induced by chronic intermittent ethanol exposure is associated with an inhibition of the MAP kinase pathway. European Journal of Neuroscience. 2003;17:1646–1654. doi: 10.1046/j.1460-9568.2003.02614.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Nelson TE, Ur CL, Gruol DL. Long-term potentiation in the rat hippocampus is reversibly depressed by chronic intermittent ethanol exposure. Journal of Neurophysiology. 2002;87:2385–2397. doi: 10.1152/jn.2002.87.5.2385. [DOI] [PubMed] [Google Scholar]
- Robles N, Sabria J. Effects of moderate chronic ethanol consumption on hippocampal nicotinic receptors and associative learning. Neurobiology of Learning and Memory. 2008;89:497–503. doi: 10.1016/j.nlm.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Roland JJ, Levinson M, Vetreno RP, Savage LM. Differential effects of systemic and intraseptal administration of the acetylcholinesterase inhibitor tacrine on the recovery of spatial behavior in an animal model of diencephalic amnesia. European Journal of Pharmacology. 2010;629:31–39. doi: 10.1016/j.ejphar.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Mark K, Vetreno RP, Savage LM. Increasing hippocampal acetylcholine levels enhance behavioral performance in an animal model of diencephalic amnesia. Brain Research. 2008;1234:116–127. doi: 10.1016/j.brainres.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blunted hippocampal, but not striatal, acetylcholine efflux parallels learning impairment in diencephalic-lesioned rats. Neurobiology of Learning and Memory. 2007;87:123–132. doi: 10.1016/j.nlm.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blocking GABA-A receptors in the medial septum enhances hippocampal acetylcholine release and behavior in a rat model of diencephalic amnesia. Pharmacology, Biochemistry and Behavior. 2009a;92:480–487. doi: 10.1016/j.pbb.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. The role of cholinergic and GABAergic medial septal/diagonal band cell populations in the emergence of diencephalic amnesia. Neuroscience. 2009b;160:32–41. doi: 10.1016/j.neuroscience.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom MJ, Pfefferbaum A, Sullivan EV. Recovery of short-term memory and psychomotor speed but not postural stability with long-term sobriety in alcoholic women. Neuropsychology. 2004;18:589–597. doi: 10.1037/0894-4105.18.3.589. [DOI] [PubMed] [Google Scholar]
- Santucci AC, Cortes C, Bettica A, Cortes F. Chronic ethanol consumption in rats produces residual increases in anxiety 4 months after withdrawal. Behavioural Brain Research. 2008;188:24–31. doi: 10.1016/j.bbr.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Savage LM, Candon PM, Hohmann HL. Alcohol-induced brain pathology and behavioral dysfunction: Using an animal model to examine sex differences. Alcoholism: Clinical and Experimental Research. 2000;24:465–475. [PubMed] [Google Scholar]
- Savage LM, Chang Q, Gold PE. Diencephalic damage decreases hippocampal acetylcholine release during spontaneous alternation testing. Learning & Memory. 2003;10:242–246. doi: 10.1101/lm.60003. [DOI] [PubMed] [Google Scholar]
- Savage LM, Roland JJ, Klintsova A. Selective septohippocampal-but not forebrain amygdalar–cholinergic dysfunction in diencephalic amnesia. Brain Research. 2007;1139:210–219. doi: 10.1016/j.brainres.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Sweet AJ, Castillo R, Langlais PJ. The effects of lesions to thalamic lateral internal medullary lamina and posterior nuclei on learning, memory and habituation in the rat. Behavioural Brain Research. 1997;82:133–147. doi: 10.1016/s0166-4328(97)80983-7. [DOI] [PubMed] [Google Scholar]
- Schmidt KS, Gallo JL, Ferri C, Giovannetti T, Sestito N, Libon DJ, Schmidt PS. The neuropsychological profile of alcohol-related dementia suggests cortical and subcortical pathology. Dementia and Geriatric Cognitive Disorders. 2005;20:286–291. doi: 10.1159/000088306. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Smith DM, Atkinson RM. Alcoholism and dementia. The International Journal of the Addictions. 1995;30:1843–1869. doi: 10.3109/10826089509071058. [DOI] [PubMed] [Google Scholar]
- Smith JS, Kiloh LG. The investigation of dementia: Results in 200 consecutive admissions. The Lancet. 1981;1:824–827. doi: 10.1016/s0140-6736(81)92692-1. [DOI] [PubMed] [Google Scholar]
- Squire LR. The neuropsychology of human memory. Annual Reviews in Neuroscience. 1982;5:241–273. doi: 10.1146/annurev.ne.05.030182.001325. [DOI] [PubMed] [Google Scholar]
- Steigerwald ES, Miller MW. Performance by adult rats in sensory-mediated radial arm maze is not impaired and may be transiently enhanced by chronic exposure to ethanol. Alcoholism: Clinical and Experimental Research. 1997;21:1553–1559. [PubMed] [Google Scholar]
- Sullivan EV, Deshmukh A, Desmond JE. Cerebellar volume deficits and neuropsychological functions in alcoholics. Alcoholism: Clinical and Experimental Research. 1998;22:63A. [PubMed] [Google Scholar]
- Sullivan EV, Marsh L. Hippocampal volume deficits in alcoholic Korsakoff’s Syndrome. Neurology. 2003;61:1716–1719. doi: 10.1212/01.wnl.0000098940.31882.bb. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Lim KO, Pfefferbaum A. Longitudinal changes in cognition, gait, and balance in abstinent and relapsed alcoholic men: Relationships to changes in brain structure. Neuropsychology. 2000;14:178–188. [PubMed] [Google Scholar]
- Suzuki M, Nelson AD, Eickstaedt JB, Wallace K, Wright LS, Svendsen CN. Glutamate enhances proliferation and neurogenesis in human neural progenitor cell cultures derived from the fetal cortex. The European Journal of Neuroscience. 2006;24:645–653. doi: 10.1111/j.1460-9568.2006.04957.x. [DOI] [PubMed] [Google Scholar]
- Szutowicz A, Bielarczyk H, Gul S, Ronowska A, Pawelczyk T, Jankowska-Kulawy A. Phenotype-dependent susceptibility of cholinergic neuroblastoma cells to neurotoxic inputs. Metabolic Brain Disease. 2007;21:149–161. doi: 10.1007/s11011-006-9007-4. [DOI] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Rage F, Givalois L, Dingeon P, Arancibia S, Beauge R. Effects of alcohol on brain-derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. Journal of Neuroscience Research. 2001;63:200–208. doi: 10.1002/1097-4547(20010115)63:2<200::AID-JNR1012>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Tartar RE. Psychological deficit in chronic alcoholics: A review. International Journal of Addiction. 1975;10:327–368. [Google Scholar]
- Tarvares MA, Paula-Barbosa MM. Alcohol-induced granule cell loss in the cerebellar cortex of the adult rat. Experimental Neurology. 1984;78:574–582. doi: 10.1016/0014-4886(82)90075-9. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, Walker DW, King MA. Chronic ethanol treatment reduces the magnitude of hippocampal LTD in the adult rat. Synapse. 2003;48:189–197. doi: 10.1002/syn.10203. [DOI] [PubMed] [Google Scholar]
- Todd KG, Butterworth RF. Evaluation of the role of NMDA-mediated excitotoxicity in the selective neuronal loss in experimental Wernicke’s encephalopathy. Experimental Neurology. 1998;149:130–138. doi: 10.1006/exnr.1997.6677. [DOI] [PubMed] [Google Scholar]
- Todd KG, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Annals of the New York Academy of Sciences. 1999;893:404–411. doi: 10.1111/j.1749-6632.1999.tb07866.x. [DOI] [PubMed] [Google Scholar]
- Torvik A. Brain lesions in alcoholics: A neuropathological study with clinical correlations. Journal of the Neurological Sciences. 1982;56:233–248. doi: 10.1016/0022-510x(82)90145-9. [DOI] [PubMed] [Google Scholar]
- Tremwel MF, Hunter BE. Effects of chronic ethanol ingestion on long-term potentiation remain even after a prolonged recovery from ethanol exposure. Synapse. 1994;17:141–148. doi: 10.1002/syn.890170210. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Johnston MV, Hess KM, Griffin JW, Price DL. Model of Wernicke’s Encephalopathy. Archives of Neurology. 1981;38:350–354. doi: 10.1001/archneur.1981.00510060052007. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Anzalone SJ, Savage LM. Impaired, spared, and enhanved ACh efflux across the hippocampus and striatum in diencephalic amnesia is dependent on task demands. Neurobiology of Learning and Memory. 2008;90:237–244. doi: 10.1016/j.nlm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome. F.A. Davis; Philadelphia, PA: 1971. [PubMed] [Google Scholar]
- Victor M, Adams RC, Collins GH. The Wernicke-Korsakoff syndrome and related neurological disorders due to alcoholism and malnutrition. 2. Philadelphia: F.A. Davis. Philadelphia, PA; 1989. [Google Scholar]
- Wagner-Glenn S. Sex differences in alcohol-induced brain damage. In: Hunt WA, Nixon SJ, editors. Alcohol-Induced Brain Damage. U.S. Department of Health and Human Services; Rockville, MD: 1993. pp. 195–212. [Google Scholar]
- Walker DW, Barnes DE, Zornetzer SF, Hunter BE, Kubanis P. Neuronal loss in hippocampus induced by prolonged ethanol consumption in rats. Science. 1980;209:711–713. doi: 10.1126/science.7394532. [DOI] [PubMed] [Google Scholar]
- Walker DW, King MA, Hunter BE. Alternations in the structure of the hippocampus after long-term ethanol consumption. In: Hunt WA, Nixon SJ, editors. Alcohol-induced Brain Damage. National Institute on Alcohol Abuse and Alcoholism; Rockville, MD: 1993. pp. 231–248. [Google Scholar]
- Ward RJ, Lallemand F, de Witte P. Biochemical and neurotransmitter changes implicated in alcohol-induced brain damage in chronic or ‘binge drinking’ alcohol abuse. Alcohol & Alcoholism. 2009;44:128–135. doi: 10.1093/alcalc/agn100. [DOI] [PubMed] [Google Scholar]
- Warrington EK, Weiskrantz L. Amnesia: a disconnection syndrome? Neuropsychologia. 1982;20:233–248. doi: 10.1016/0028-3932(82)90099-9. [DOI] [PubMed] [Google Scholar]
- Watanabe I. Pyrithiamine-induced acute thiamine-deficient encephalopathy in the mouse. Experimental and Molecular Pathology. 1978;28:381–394. doi: 10.1016/0014-4800(78)90012-6. [DOI] [PubMed] [Google Scholar]
- Winocur G, Wojtowicz J, Sekeres M, Snyder JS, Wang S. Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus. 2006;16:296–304. doi: 10.1002/hipo.20163. [DOI] [PubMed] [Google Scholar]
- Witt ED. Neuroanatomical consequences of thiamine deficiency: A comparative analysis. Alcohol and Alcoholism. 1985;2:201–221. [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Vinco S, Orduna J, Luong R, Sullivan EV, Pfefferbaum A. In vivo evidence for alcohol-induced neurochemical changes in rat brain without protracted withdrawal, pronounced thiamine deficiency, or severe liver damage. Neuropsychopharmacology. 2009;34:1427–1442. doi: 10.1038/npp.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Weilersbacher GS, Henderson SW, Corso T, Olney JW, Langlais PJ. Excitotoxic cytopathology, progression, and reversibility of thiamine deficiency-induced diencephalic lesions. Journal of Neuropathology and Experimental Neurology. 1995;54:255–267. doi: 10.1097/00005072-199503000-00012. [DOI] [PubMed] [Google Scholar]
- Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou C, Xu T, Hong Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiology of Disease. 2008;29:176–185. doi: 10.1016/j.nbd.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Zimitat C, Kril J, Harper C, Nixon P. Progression of neurological disease in thiamine-deficient rats is enhanced by ethanol. Alcohol. 1990;7:493–501. doi: 10.1016/0741-8329(90)90038-e. [DOI] [PubMed] [Google Scholar]