

Abstract
Objective
Scleroderma, or systemic sclerosis (SSc), is characterized by progressive multi-organ fibrosis. We recently implicated lysophosphatidic acid (LPA) in the pathogenesis of pulmonary fibrosis. Here we investigated the roles of LPA and two of its receptors, LPA1 and LPA2, in dermal fibrosis in a mouse model of SSc.
Methods
Wild type (WT) and LPA1- and LPA2-deficient (LPA1 KO and LPA2 KO) mice were injected subcutaneously with bleomycin or PBS once daily for 28 doses. Dermal thickness, collagen content, and numbers of α-smooth muscle actin (αSMA)+ or phosphoSmad2+ cells were determined in bleomycin- and PBS-injected skin. In separate experiments, a novel selective LPA1 antagonist AM095, or vehicle alone, was administered by oral gavage to C57Bl/6 mice challenged with 28 daily injections of bleomycin or PBS. AM095 or vehicle treatments were initiated concurrently with, or 7 or 14 days after the onset of bleomycin and PBS injections, and continued to the end of the experiments. Dermal thickness and collagen content were determined in injected skin.
Results
LPA1 KO mice were markedly resistant to bleomycin-induced increases in dermal thickness and collagen, whereas LPA2 KO mice were as susceptible as WTs. Bleomycin-induced increases in dermal αSMA+ and phosphoSmad2+ cells were abrogated in LPA1 KO mice. Pharmacological antagonism of LPA1 with AM095 significantly attenuated bleomycin-induced dermal fibrosis when administered in either ‘preventive’ or ‘therapeutic’ regimens.
Conclusion
These results suggest that LPA-LPA1 pathway inhibition has the potential to be an effective new therapeutic strategy for scleroderma, and that LPA1 is a druggable target for dermal fibrosis.
Scleroderma, or systemic sclerosis (SSc), is a potentially fatal autoimmune disease of unknown etiology, characterized by progressive multi-organ fibrosis that is refractory to current therapies. Fibrogenesis in SSc is thought to result from tissue injury followed by dysregulated wound healing (1). Discovery of the mediators driving aberrant wound healing responses will hopefully identify new therapeutic targets for SSc. We hypothesize that one such target is LPA1, a receptor for lysophosphatidic acid (LPA).
LPA is a lipid mediator that signals through specific GPCRs. Five high-affinity LPA receptors have been definitively established and designated LPA1 to LPA5; P2Y5 is a lower affinity receptor that is likely to join the LPA receptor family as LPA6 (2). Our laboratory recently implicated LPA-LPA1 signaling in the pathogenesis of pulmonary fibrosis (3). We found that LPA1 KO mice were dramatically protected from bleomycin-induced pulmonary fibrosis and mortality, and that LPA-LPA1 signaling was responsible for the majority of fibroblast chemoattractant activity present in bronchoalveolar lavage (BAL) from patients with idiopathic pulmonary fibrosis (IPF). LPA-LPA2 signaling has also been implicated in pulmonary fibrosis. LPA-LPA2 signaling can induce αvβ6 integrin-mediated activation of latent TGF-β by lung epithelial cells in culture (4), and TGF-β activation by this integrin is critically required for the development of bleomycin-induced lung fibrosis (5).
LPA may also be involved in SSc pathogenesis, as suggested by the recent demonstration that arachidonoyl (20:4)-LPA levels are significantly higher in SSc patients’ serum versus controls (6). Injured human skin has been shown to contain increased amounts of both LPA and cells expressing LPA1 (7). We therefore investigated whether LPA signaling through either LPA1 or LPA2 is required for dermal fibrosis in the bleomycin model of scleroderma. In this model, repeated subcutaneous injections of the chemotherapeutic agent bleomycin results in dermal fibrosis resembling scleroderma (8), with collagen deposition and both fibroblast and myofibroblast accumulation (9). Lesional skin also shows increased Smad2 and Smad3 phosphorylation (10), indicating activation of the TGF-β pathway implicated in scleroderma (11, 12). We found that bleomycin-induced increases in dermal thickness, collagen content, myofibroblast accumulation and Smad2 phosphorylation were all markedly attenuated in LPA1 KO mice. Bleomycin-induced dermal fibrosis was also significantly reduced in WT mice treated with the novel, orally bioavailable, LPA1-selective antagonist AM095. In contrast, LPA2 KO mice were not protected from bleomycin-induced dermal fibrosis. These results indicate that LPA-LPA1 signaling importantly contributes to injury-induced dermal fibrosis.
MATERIALS AND METHODS
Animals
Experiments comparing LPA1 KO and WT mice used offspring of mice heterozygous for the LPA1 mutant allele, which were hybrids of the C57Bl/6 and 129Sv/J genetic backgrounds (13). LPA1 KO mice, generated by Dr. Jerold Chun’s laboratory (The Scripps Research Institute), demonstrate impaired suckling in neonatal pups due to defective olfaction, which leads to increased neonatal mortality, and reduced body size in survivors. Survivors also demonstrate craniofacial dysmorphism characterized by shorter snouts and more widely spaced eyes (13), but we have not noted any skin abnormalities at baseline. Experiments comparing LPA2 KO and WT mice used offspring of mice homozygous for the mutant LPA2 allele in the BALB/c genetic background (14), and WT BALB/c mice (Charles River Laboratories). LPA2 KO mice, also generated by Dr. Chun’s laboratory, are born at the expected frequency and display no obvious phenotypic abnormalities (14). Experiments measuring plasma AM095 concentrations and comparing AM095- and vehicle-treated mice used WT C57Bl/6 mice from Harlan Laboratories and the NCI-Frederick Mouse Repository, respectively. All experiments used sex- and age-matched mice at 6-8 weeks of age that were maintained in specific pathogen–free environments. All experiments were performed in accordance with NIH guidelines, and protocols approved by the Massachusetts General Hospital or the Amira Pharmaceuticals IACUC.
Bleomycin injections and skin harvests
Bleomycin (Gensia Sicor) was dissolved in PBS at 10 μg/ml and sterilized by filtration. Bleomycin or PBS (100 μl) was injected subcutaneously into two locations on the shaved backs of LPA1 KO, LPA2 KO or WT mice, once per day for 28 doses. Mice were then sacrificed and full thickness 6 mm punch biopsies were obtained from each injection site. One skin sample was fixed in 10% formalin and embedded in paraffin for histology and immunohistochemistry studies; the other was frozen immediately at −80°C for hydroxyproline analysis.
Histology and dermal thickness measurement
Multiple 5 μm sections of paraffin-embedded skin samples were de-paraffinized, rehydrated and stained with H&E or Masson’s trichrome stains according to the standard protocols of our laboratory (3). Dermal thickness was determined using 100x magnification photomicrographs of H&E-stained sections, by measuring the distance between the epidermal-dermal junction and the dermal-fat junction at five randomly selected sites/high power field (HPF), for ten HPF per section.
Immunohistochemical analyses of α-SMA and phosphoSmad2
Multiple 5 μm sections of paraffin-embedded skin samples were cut onto ProbeOn Plus slides (Fisher Scientific), de-paraffinized and rehydrated. Immunolabeling of α-SMA and phosphoSmad2 was performed with primary rabbit anti-mouse α-SMA antibody (Abcam) and primary rabbit anti-mouse phosphoSmad2 antibody (Cell Signaling), respectively, using the MicroProbe Staining system (Fisher) per manufacturer instructions. Appropriate biotinylated secondary antibodies were used, followed by detection with an ABC Development Kit (Vector Laboratories) and color development with AEC (Dako). α-SMA+ and phosphoSmad2+ cells were then counted in 10 randomly selected non-overlapping HPF in dermal sections of WT and LPA1 KO mice.
Hydroxyproline Assay
Hydroxyproline content was determined as a measure of skin collagen using the standard protocol of our laboratory (15). Briefly, skin samples were homogenized in PBS and hydrolyzed overnight in 6N HCl at 120°C. A 25 μl aliquot was desiccated, resuspended in 25 μl H2O and added to 0.5 ml of 1.4% chloramine T (Sigma), 10% n-propranolol, and 0.5 M sodium acetate, pH 6.0. After 20-minute incubation at room temperature (RT), 0.5 ml of Erlich’s solution (1M p-dimethylaminobenzaldehyde (Sigma) in 70% n-propranolol, 20% perchloric acid) was added. After 15 minute incubation at 65°C, absorbance was measured at 550 nm and hydroxyproline concentration determined against a standard curve. Assay results were expressed as μg hydroxyproline/6 mm punch biopsy of skin.
Cell lines and culture
Human and mouse LPA1 and human LPA3 receptors were stably expressed in CHO cells (Invitrogen) and cultured in F12 media with 10% FBS and 1 mg/ml hygromycin B. Mouse LPA3 was stably expressed in HEK cells (Invitrogen) and cultured in DMEM with 10% FBS and 200 μg/ml hygromycin B. Human and mouse LPA2 and LPA5 and human LPA4 were transiently expressed in rat neuroblastoma B103 cells using Lipofectamine™ 2000 (Invitrogen) as per the manufacturer instructions.
Calcium Flux Assay
LPA receptor-transfected cells were plated in 96-well Poly-D-Lysine-coated black-wall clear-bottom plates (BD BioCoat) at 20,000-40,000 cells/well and cultured overnight in complete media. Cells were then washed with PBS and cultured in serum-free media either overnight (for stably expressing cells) or 4 hours (for transient transfectants) prior to dye loading. On the day of the assay, cells were loaded for 1 hour at 37°C with 100 μl FLIPR Calcium 4 dye (Molecular Devices) in HBSS supplemented with 20 mM HEPES, 2 mM probenecid and 0.3% FFA-HSA. Test compounds (25 μl 1% DMSO) were added to each well and incubated at RT for 30 minutes. LPA (50 μl of 5X stock solutions prepared in HBSS with 20 mM HEPES and 0.3% fatty acid-free HSA) was added after 15 seconds of baseline measurement. Final concentrations of LPA used were dependent on the receptor expressed: LPA1 and LPA3 assays used 10 nM LPA, LPA2 and LPA5 assays used 30 nM LPA and LPA4 assay used 300 nM LPA. Intracellular calcium mobilization was measured using the FLEXstation III (Molecular Devices). Inhibition curves were generated by plotting percentage inhibition of calcium flux versus log10 of the concentration of compound. IC50 values were calculated by nonlinear regression using the sigmoidal dose-response (variable slope) equation in Prism 5 (GraphPad Software).
Determination of AM095 concentrations in mouse plasma
C57Bl/6 mice were administered the selective LPA1 antagonist AM095 by oral gavage (30 mg/kg) at time 0 and 8h, and blood was collected by cardiac puncture under anesthesia in sodium EDTA tubes at 0, 4, 8, 9, 12 and 24h. Plasma samples were stored at −40°C prior to analysis of AM095 concentrations by liquid chromatography/mass spectrometry (LC–MS/MS). Known amounts of AM095 were added to thawed mouse plasma to yield a concentration range from 0.8 to 4,000 ng/ml. Plasma samples were precipitated using acetonitrile containing the internal standard buspirone. The analyte mixture (10 μl) was injected using a Leap PAL autosampler. Calibration curves were constructed by plotting the peak-area ratio of analyzed peaks against known concentrations. The lower limit of quantitation was 1 ng/ml. The data were subjected to linear regression analysis with 1/x2 weighting. The pharmacokinetic parameters of AM095 were calculated by non-compartmental analysis using WinNonlin Professional (Pharsight). Cmax and time to Cmax (Tmax) were obtained directly from the measured data.
AM095 administration in the bleomycin model
The selective LPA1 antagonist AM095 was dissolved in sterile water, and a dose of 30 mg/kg per mouse, or sterile water alone (vehicle), was administered by oral gavage to C57Bl/6 mice, twice daily on weekdays and once daily on weekends. AM095 was administered from the onset of bleomycin challenge in a ‘preventive’ regimen, or beginning either 7 or 14 days after the onset of bleomycin challenge in two ‘therapeutic’ regimens. For all AM095 regimens, bleomycin or PBS was injected subcutaneously for 28 consecutive days, and skin samples were obtained at the completion of the experiment as described above.
Statistical analysis
Differences in dermal thickness, hydroxyproline content, and the numbers of α-SMA+ and phosphoSmad2+ cells between WT and LPA1 or LPA2 KO mice, and between AM095- and vehicle-treated mice, were tested for statistical significance by Student’s two tailed t-test, using Microsoft Excel software. P-values of <0.05 were considered statistically significant.
RESULTS
Bleomycin-induced dermal fibrosis is dependent on LPA1
Examination of H&E-stained skin sections of bleomycin- and PBS-challenged WT and LPA1 KO mice demonstrated that LPA1 KO mice were strikingly protected from bleomycin-induced dermal fibrosis (Figure 1A, upper panels). Compared with PBS-challenged mice, bleomycin-challenged WT mice demonstrated substantial thickening of the dermis with densely packed connective tissue replacing the subcutaneous fat. These changes were markedly reduced in bleomycin-challenged LPA1 KO mice. The substantial increase in dermal collagen induced by bleomycin in WT mice was also markedly attenuated in LPA1 KO mice, as visualized by Masson’s trichrome staining (Figure 1A, lower panels).
Figure 1.
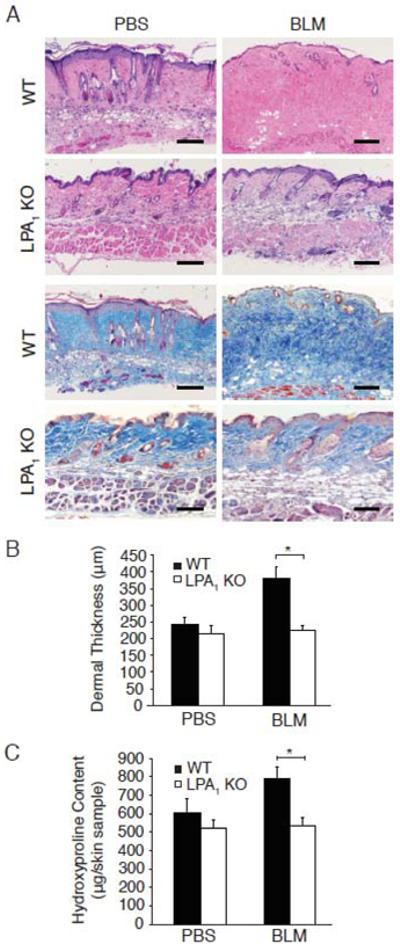
LPA1 KO mice are protected from bleomycin-induced dermal fibrosis. (A) H&E (upper panels) and Masson’s trichrome staining (lower panels) of the skin of WT and LPA1 KO mice following PBS or BLM challenge. Magnification x100; bar, 100 μm. (B) Dermal thickness measured in 5 locations/HPF, in 10 HPF/skin sample, *p<0.001, BLM-challenged WT vs. LPA1 KO mice; p<0.02, WT-BLM vs. WT-PBS mice. (C) Skin hydroxyproline content, *p<0.001, BLM-challenged WT vs. LPA1 KO mice; p<0.01, WT-BLM vs. WT-PBS mice. Data are from one of two experiments with similar results, each with n ≥ 5 WT and LPA1 KO mice for both treatment groups. Data are expressed as mean dermal thickness ± SEM or mean hydroxyproline content/6 mm punch biopsy skin sample ± SEM, respectively.
To quantify the protection of LPA1 KO mice, we first assessed the dermal thickness of bleomycin- and PBS-challenged WT and LPA1 KO mice. The dermal thickness of bleomycin-challenged LPA1 KO mice was significantly reduced compared with WT mice (Figure 1B). Whereas bleomycin challenge increased the dermal thickness of WT mice by 56%, the dermal thickness of bleomycin-challenged LPA1 KO mice was only 5% greater than PBS-challenged LPA1 KO mice (Figure 1B). Genetic deletion of LPA1 therefore attenuated the increase in bleomycin-induced dermal thickness by 91%. Biochemical assessment of skin collagen, by measuring skin hydroxyproline content, confirmed the significant protection of LPA1 KO mice. Bleomycin challenge increased the amount of skin hydroxyproline by 31% in WT mice, but only by 3% in LPA1 KO mice (Figure 1C). Genetic deletion of LPA1 therefore attenuated the bleomycin-induced increase in hydroxyproline by 90%. This dramatic protection of LPA1 KO mice suggests that the LPA-LPA1 pathway importantly contributes to dermal fibrosis.
Bleomycin-induced dermal fibrosis does not require LPA2
In contrast to LPA1 KO mice, LPA2 KO mice were not protected from bleomycin-induced dermal fibrosis. Bleomycin induced similar thickening of the dermis with densely packed connective tissue in WT and LPA2 KO mice, and similar increases in dermal collagen, as demonstrated by skin sections stained with H&E (Figure 2A, upper panels) and Masson’s trichrome (Figure 2A, lower panels), respectively. Dermal thickness and hydroxyproline content measurements confirmed that LPA2 deletion did not confer protection from bleomycin-induced fibrosis. Compared to PBS-challenged mice, bleomycin increased dermal thickness by 46% in WT mice and by 50% in LPA2 KO mice (Figure 2B). Similarly, bleomycin challenge increased skin hydroxyproline content by 67% in WT mice and by 66% in LPA2 KO mice (Figure 2C). The lack of protection of LPA2 KO mice suggests that LPA2 signaling is not required for dermal fibrosis.
Figure 2.
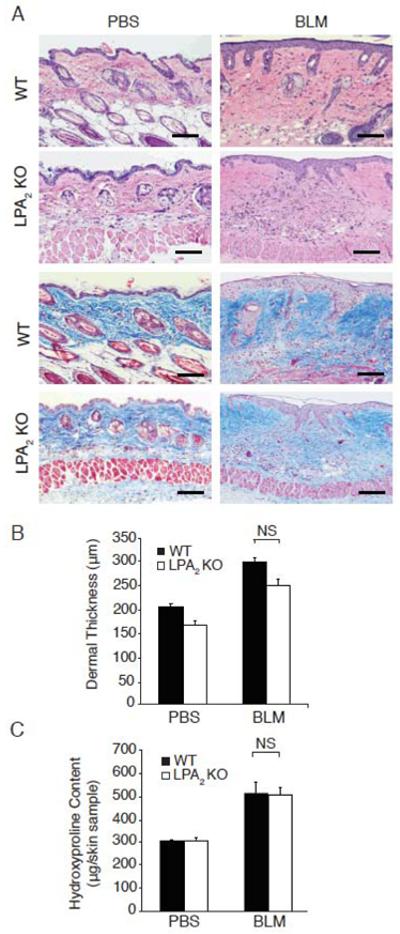
LPA2 KO mice are not protected from bleomycin-induced dermal fibrosis. (A) H&E (upper panels) and Masson’s trichrome staining (lower panels) of the skin of WT and LPA2 KO mice following PBS or BLM challenge. Magnification x100; bar, 100 μm. (B) Dermal thickness measured in 5 locations/HPF, in 10 HPF/skin sample. BLM-challenged WT and LPA2 KO mice were not significantly different (NS); p<0.002, WT-BLM vs. WT-PBS; p<0.001, LPA2 KO-BLM vs. LPA2 KO-PBS mice. (C) Skin hydroxyproline content, BLM-challenged WT vs. LPA2 KO mice were not significantly different (NS); p<0.003, WT-BLM vs. WT-PBS, p<0.002, LPA2 KO-BLM vs. LPA2 KO-PBS mice. Data are from one of two experiments with similar results, each with n ≥ 5 WT and LPA2 KO mice for both treatment groups. Data expressed as mean dermal thickness ± SEM or mean hydroxyproline content/6 mm punch biopsy skin sample ± SEM, respectively.
Bleomycin-induced dermal myofibroblast accumulation is dependent on LPA1
To begin to investigate the mechanism(s) through which LPA and LPA1 contribute to dermal fibrosis, we assessed two processes implicated in scleroderma, accumulation of myofibroblasts and activation of the TGF-β–Smad signaling pathway, in bleomycin-challenged WT and LPA1 KO mice. SSc fibrogenesis is associated with fibroblast differentiation into myofibroblasts, which secrete increased amounts of extracellular matrix components, including collagen (16). Myofibroblast differentiation is characterized by the acquisition of smooth muscle cell features, including α-SMA expression (17). To determine whether LPA-LPA1 signaling contributes to bleomycin-induced myofibroblast accumulation, we compared the number of α-SMA+ cells in the dermis of bleomycin- and PBS-challenged WT and LPA1 KO mice. Bleomycin challenge substantially increased the number of α-SMA+ cells in the dermis of WT but not LPA1 KO mice (Figure 3A). The number of α-SMA+ cells increased by 70% in bleomycin-challenged WT mice, but only by 5% in LPA1 KO mice (Figure 3B), suggesting that LPA and LPA1 importantly contribute to myofibroblast accumulation in dermal fibrosis.
Figure 3.
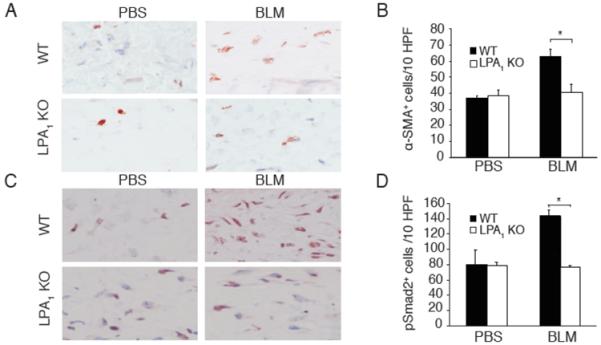
Bleomycin-induced accumulation of α-SMA+ myofibroblasts and phosphoSmad2+ cells is diminished in LPA1 KO mice. (A) Skin of WT and LPA1 KO mice following PBS- or BLM-challenge stained with anti-α-SMA antibody (magnification x400). (B) Quantification of α-SMA+ cells in dermis of WT and LPA1 KO mice following PBS or BLM challenge. Data are from one of two independent experiments with similar results, each with n = 6 WT and LPA1 KO mice for both treatment groups, and are expressed as mean number of α-SMA+ cells per 10 HPF ± SEM (*p<0.01, WT-BLM vs. LPA1 KO-BLM; p<0.001, WT-BLM vs. WT-PBS; LPA1 KO-BLM vs. LPA1 KO-PBS was not significant). (C) Skin of WT and LPA1 KO mice following PBS or BLM challenge, stained with anti-phosphoSmad2 (pSmad2) antibody (x400). (D) Quantification of pSmad2+ cells in dermis of WT and LPA1 KO mice following PBS or BLM challenge. Data are from one of two independent experiments with similar results, each with n = 3 WT and LPA1 KO mice for both treatment groups, and are expressed as mean numbers of pSmad+ cells per 10 HPF ± SEM (*p<0.002, WT-BLM vs. LPA1 KO-BLM; p<0.03, WT-BLM vs. WT-PBS mice, LPA1 KO-BLM vs. LPA1 KO-PBS mice was not significant).
Bleomycin-induced dermal Smad2 phosphorylation is dependent on LPA1
Myofibroblast differentiation and synthesis of matrix proteins are driven by TGF-β (18, 19). By directing these key pro-fibrotic processes, TGF-β is thought to play a central role in SSc fibrogenesis. When bound by active TGF-β, TGF-β receptors transmit signals through phosphorylation of cytoplasmic Smad proteins, which translocate to the nucleus and act as transcription factors (20). To determine whether LPA-LPA1 signaling contributes to the activation of the TGF-β signaling pathway following bleomycin challenge, we compared the number of cells with nuclear Smad2 phosphorylation in the dermis of bleomycin- and PBS-challenged WT and LPA1 KO mice. Bleomycin challenge increased the number of nuclear phosphoSmad2+ cells in the dermis of WT but not LPA1 KO mice (Figure 3C). The number of phosphoSmad2+ cells increased by 81% in bleomycin-challenged WT mice, but not at all in LPA1 KO mice (Figure 3D), suggesting that LPA and LPA1 may contribute to the activation of the TGF-β-Smad signaling pathway during the development of dermal fibrosis. Alternatively, the reduced number of nuclear phosphoSmad2+ cells in bleomycin-challenged LPA1 KO mice could be at least partially attributable to reductions in the numbers of fibroblasts and myofibroblasts accumulating in the dermis of these mice. Reductions in fibroblast and myofibroblast numbers would reduce the number of cells present in the dermis that are able to respond to TGF-β by Smad phosphorylation.
AM095 is a novel potent and selective LPA1 antagonist
To investigate LPA1’s potential as a therapeutic target for dermal fibrosis, we evaluated a potent new LPA1-selective antagonist, AM095 (Amira Pharmaceuticals, sodium{4′-[3-methyl-4-((R)-1-phenyl-ethoxycarbonylamino)-isoxazol-5-yl]-biphenyl-4-yl}-acetate, Figure 4A). AM095 inhibited the LPA-induced calcium flux of CHO cells stably transfected with human or mouse LPA1 (Figure 4B). The IC50 for AM095 antagonism of LPA-induced calcium flux of human or mouse LPA1-transfected CHO cells was 0.025 and 0.023 μM, respectively (Table 1). In contrast, the IC50 for AM095 antagonism of LPA-induced calcium flux was >5 μM for CHO, human embryonic kidney (HEK), or B103 rat neuroblastoma cells transfected with one of the other four established human or mouse LPA receptors, demonstrating AM095’s selectivity for LPA1 (Table 1). The average plasma AM095 concentrations produced in mice over a 24-hour period by the administration of two 30 mg/kg doses by oral gavage eight hours apart are shown in Figure 4C. The AM095 AUC value was 118.7 μg*hr/ml, with a maximum plasma concentration (Cmax) value of 6200 nM or 28 μg/ml and a Cmin value of 170 nM or 0.08 μg/ml. Twice daily 30 mg/kg dosing by oral gavage was therefore used in all subsequent experiments, as this produced plasma AM095 concentrations that were greater than the IC50 versus the LPA1 receptor throughout the treatment period.
Figure 4.

Structure and pharmacokinetics of AM095. (A) Chemical structure of the selective LPA1 antagonist, AM095 (Sodium{4′-[3-methyl-4-((R)-1-phenyl-ethoxycarbonylamino)-isoxazol-5-yl]-biphenyl-4-yl}-acetate). (B) AM095 inhibition of LPA-induced calcium flux in CHO cells recombinantly expressing human or mouse LPA1. (C) Average plasma concentrations of AM095 in C57Bl/6 mice receiving 30 mg/kg by oral gavage at 0 and 8h (n = 2 per time point analyzed).
Table 1.
Inhibition by AM095 of LPA-stimulated intracellular calcium release from cells recombinantly expressing LPA1-5
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| LPA1 | LPA2 | LPA3 | LPA4 | LPA5 | |
| Human | 0.025 (n=5) |
>10 (n=3) |
>10 (n=5) |
8.5 (n=2) |
>10 (n=3) |
| Mouse | 0.023 (n=3) |
>10 (n=3) |
5.4 (n=4) |
ND | >10 (n=3) |
Preventive or therapeutic pharmacological antagonism of LPA1 with AM095 attenuates bleomycin-induced dermal fibrosis
Administration of AM095 from the onset of bleomycin challenge in a ‘preventive’ regimen attenuated bleomycin-induced dermal fibrosis, substantially mitigating bleomycin-induced increases in dermal thickness and dermal collagen, as demonstrated by skin sections stained with H&E (Figure 5A, left panels) and Masson’s trichrome (Figure 5A, right panels) respectively. Delayed administration of AM095 beginning after the onset of bleomycin challenge in two ‘therapeutic’ regimens also attenuated bleomycin-induced dermal fibrosis. Dermal thickness and hydroxyproline content measurements indicated the protective efficacy of treatment in all three regimens studied, designated preventive AM095, therapeutic AM095 #1 in which treatment began on day 7 after the onset of bleomycin challenge, and therapeutic AM095 #2 in which treatment began on day 14. Bleomycin challenge increased the dermal thickness of vehicle-treated mice by 82%, but by only 25%, 12% or 32% in the preventive, therapeutic #1 or therapeutic #2 AM095-treated mice, respectively (Figure 5B). Preventive pharmacological inhibition of LPA1 therefore attenuated the bleomycin-induced increase in dermal thickness by 70%, while therapeutic pharmacological inhibition of LPA1 begun on day 7 or 14 attenuated the bleomycin-induced increase in dermal thickness by 85% or 61%, respectively. Similarly, bleomycin challenge increased the skin hydroxyproline content of vehicle-treated mice by 117%, but by only 56%, 79% or 86% in the preventive, therapeutic #1 or therapeutic #2 AM095-treated mice, respectively (Figure 5C). Preventive inhibition of LPA1 therefore attenuated the bleomycin-induced increase in hydroxyproline by 52%, while therapeutic inhibition of LPA1 begun on day 7 or 14 attenuated the bleomycin-induced increase in hydroxyproline by 32% and 26%, respectively. These data suggest an ongoing requirement for the LPA-LPA1 pathway in the maintenance of pathological dermal fibrosis, suggesting that this pathway is a viable target for therapeutic intervention in fibrotic diseases of the skin.
Figure 5.
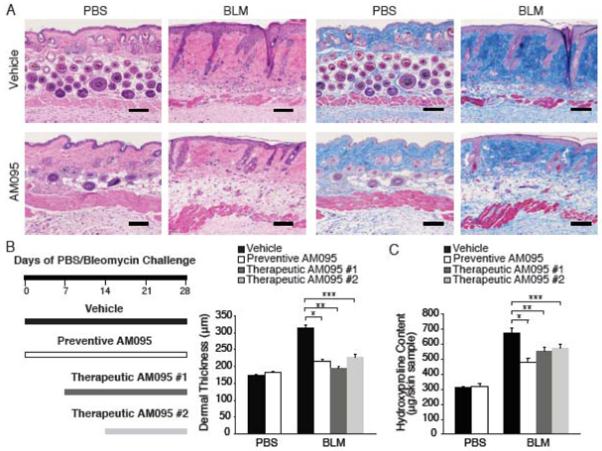
Pharmacological LPA1 receptor antagonism attenuates bleomycin-induced fibrosis. (A) H&E (left panels) and Masson’s trichrome staining (right panels) of the skin of PBS- or BLM-challenged C57Bl/6 mice treated with vehicle or AM095 in a ‘preventive’ regimen. Magnification x100; bar, 100μm. (B) Dermal thickness of PBS- or BLM-challenged C57Bl/6 mice treated with vehicle, ‘preventive’ AM095, or ‘therapeutic’ AM095 begun 7 (AM095 #1) or 14 days (AM095 #2) after PBS or BLM. N = 3 mice/group, data expressed as mean dermal thickness ± SEM (*p<0.0008, preventive AM095- vs. vehicle-BLM-mice; **p<0.002 and ***p<0.01, AM095 #1- and AM095 #2- vs. vehicle-BLM-mice, respectively). (C) Skin hydroxyproline content of the same groups of mice in (A). N = 5 mice/group, data expressed as mean hydroxyproline/skin sample ± SEM (*p<0.0005, **p<0.02 and ***p<0.02, vehicle-treated vs. preventive AM095-, therapeutic AM095 #1- and therapeutic AM095 #2-treated BLM-challenged mice, respectively). In (B) and (C), PBS, vehicle-BLM, and preventive AM095-BLM data are combined from 2 experiments; therapeutic AM095-BLM data are from one experiment.
DISCUSSION
Our results demonstrate that LPA1 is required for the development of bleomycin-induced dermal fibrosis. Genetic deletion of this receptor, or its pharmacological antagonism with a new orally bioavailable selective inhibitor, protected mice from the increases in dermal thickness and collagen content produced in this model. In contrast, genetic deletion of LPA2 did not confer protection against dermal fibrosis. Taken together, these data suggest that LPA signaling specifically through LPA1 is critical for the development of skin fibrosis induced by tissue injury.
Although its precise cellular origin in biological fluids and tissues has yet to be established, LPA production has been demonstrated in response to injury and promotes wound healing in multiple tissues, including the skin (7, 21, 22). Recurrent tissue injury and aberrant wound healing responses appear to contribute to the pathogenesis of multiple fibrotic diseases, including scleroderma (1, 23), and arachidonoyl (20:4)-LPA levels were recently noted to be significantly higher in SSc patients’ serum versus controls (6). We would therefore expect levels of LPA to be increased in the skin during the development of injury-induced dermal fibrosis in both bleomycin-challenged mice and SSc patients, though we have yet to investigate this.
Our initial investigations of the mechanism(s) through which LPA-LPA1 signaling contributes to dermal fibrosis revealed that in contrast to WT mice, bleomycin-challenged LPA1 KO mice failed to demonstrate two hallmarks of scleroderma-associated skin fibrosis: increased numbers of dermal myofibroblasts and of dermal cells with nuclear Smad2 phosphorylation. These results suggest that LPA-LPA1 signaling is required for two central processes in scleroderma fibrogenesis: (1) myofibroblast accumulation, and (2) TGF-β-Smad signaling pathway activation. We believe that attenuation of both of these interconnected fibrogenic processes in the absence or inhibition of LPA1 accounts for the dramatic protection of LPA1 KO and AM095-treated mice from bleomycin-induced dermal fibrosis.
Myofibroblasts predominate in areas of increased collagen deposition in scleroderma lesional skin (24, 25), where the number of myofibroblasts correlates with fibrosis severity (25). Promotion of myofibroblast accumulation by LPA-LPA1 signaling therefore would be expected to promote dermal fibrosis. LPA mediates multiple fibroblast activities that lead to the accumulation of these cells, including their recruitment and proliferation, and prevention of their apoptosis (3, 22, 26-31). We hypothesize that reduced fibroblast accumulation in the absence of LPA-LPA1 signaling contributes to reduced myofibroblast accumulation in bleomycin-challenged LPA1 KO mice, by reducing the pool of cells from which myofibroblasts differentiate. Our ability to evaluate this hypothesis in our immunohistochemical studies, however, was limited by difficulties enumerating tissue fibroblasts in situ, as opposed to myofibroblasts, by immunostaining, due to the lack of antigens specifically expressed by these cells.
Evidence from both mouse models and SSc patients indicates that TGF-β plays a central role in scleroderma fibrogenesis. Fibroblast-specific expression of a constitutively-active TGF-β receptor is sufficient to recapitulate many features of scleroderma in mice, including dermal fibrosis (32), while inhibition of TGF-β signaling protects against dermal fibrosis in commonly used mouse models of scleroderma, including the bleomycin model (33). Gene expression profiling of lesional skin from scleroderma patients demonstrates increased expression of many TGF-β targets, similar to gene expression induced by treating normal fibroblasts with TGF-β (10, 34, 35). In prior studies, we found that LPA1 expression was not required for TGF-β downstream signaling in fibroblasts, since increases in procollagen type I α1, fibronectin and α-SMA expression induced by TGF-β were similar in WT and LPA1-deficient mouse lung fibroblasts (3). TGF-β activity, however, is primarily regulated through the post-translational activation of latent TGF-β complexes (36, 37). The failure of phosphoSmad2+ cells to increase in bleomycin-challenged LPA1 KO mice therefore raises the possibility that LPA-LPA1 signaling may mediate TGF-β activation during the development of dermal fibrosis. Although decreased fibroblast and myofibroblast accumulation in bleomycin-challenged LPA1 KO mice could also have contributed to the reduced number of nuclear phosphoSmad2+ cells observed, by decreasing the number of TGF-β-responsive cells present in the dermis, LPA has been reported to mediate TGF-β activation. LPA treatment of keratinocytes, as well as lung epithelial cells, has been shown to induce active TGF-β (4, 38). While LPA signaling through LPA2 has been shown to induce αvβ6 integrin-dependent activation of latent TGF-β by lung epithelial cells in culture (4), our results suggest that LPA1 would be the receptor likely to mediate LPA-induced TGF-β activation in the skin. Activation of TGF-β by the epithelial cell-restricted αvβ6 integrin is required for the development of lung fibrosis in several animal models, including the bleomycin model of pulmonary fibrosis (5). In the lung, TGF-β driven fibroblast activation and differentiation to myofibroblasts therefore is dependent on the activation of TGF-β by adjacent epithelial cells in a paracrine fashion. In the skin of scleroderma patients however, activation of TGF-β by fibroblasts themselves contributes to fibroblast activation and differentiation into myofibroblasts in an autocrine fashion (39). The ability of scleroderma fibroblasts to activate TGF-β has been shown to result from their over-expression of two other αν-containing integrins that are capable of activating latent TGF-β, ανβ5 and ανβ3 (40, 41). We therefore hypothesize that LPA signaling through LPA1 mediates TGF-β activation during the development of dermal fibrosis through the αvβ5 and αvβ3 integrins expressed by skin fibroblasts.
As noted, our laboratory recently implicated LPA-LPA1 signaling in the pathogenesis of pulmonary fibrosis (3). In addition, LPA has been implicated in renal and hepatic fibrogenesis. LPA1 KO mice were significantly protected in the unilateral ureteral obstruction model of renal tubulointerstitial fibrosis (42), and circulating LPA concentrations correlated with the extent of hepatic fibrosis in the carbon tetrachloride rodent model of liver fibrosis (43). Including our results in this study, data now implicate the LPA-LPA1 pathway in the development of lung, kidney, liver and skin fibrosis, suggesting that this pathway is of fundamental importance in the pathogenesis of fibrotic diseases associated with tissue injury. Additionally, the efficacy of an LPA1-selective antagonist in our dermal fibrosis model provides pre-clinical support for targeting LPA1 in fibrotic diseases such as scleroderma.
In summary, we have shown that LPA signaling through LPA1 but not LPA2 is critically required for the development of bleomycin-induced dermal fibrosis, and for both myofibroblast accumulation and TGF-β-Smad signaling in this model. In addition to genetic deletion of LPA1, we found that both preventive and therapeutic pharmacological inhibition of this receptor protected mice from dermal fibrosis. The ability of AM095 to attenuate dermal fibrosis when initiated after tissue injury onset in a therapeutic regimen suggests that antagonism of LPA1 may be effective in the treatment of patients with already existing fibrosis, as would be needed for clinically useful anti-fibrotic drugs (40). Our results therefore indicate that LPA-LPA1 inhibition has the potential to be an effective new therapeutic strategy for scleroderma, and that LPA1 is a druggable target for fibrosis.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge that this work was supported by a Scleroderma Research Foundation Grant and NIH R01-HL095732 to A.M.T, an American College of Rheumatology REF Physician Scientist Development Award to F.V.C, NIH T32-AR007258-32 to A.D.L., and NIH R01-DA019674, R01-NS048478, and R01-DC009505 to J.C. The authors also thank C.P. Leary for her expert assistance.
Footnotes
Conflict of interest: Drs. Seiders, Bain, King, Swaney and Lorrain receive income and have ownership of equity in Amira Pharmaceuticals, Drs. Chun and Tager receive consulting fees from Amira Pharmaceuticals, and Drs. Luster and Tager have filed a patent cooperation treaty application on “Lysophosphatidic Acid Receptor Targeting for Lung Disease.”
REFERENCES
- 1.Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends in Immunology. 2005;26(11):587–595. doi: 10.1016/j.it.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 50:157–86. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 3.Tager AM, Lacamera P, Shea BS, Campanella GS, Selman M, Zhao Z, et al. The lysophosphatidic acid receptor LPA(1) links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 4.Xu MY, Porte J, Knox AJ, Weinreb PH, Maher TM, Violette SM, et al. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q) Am J Pathol. 2009;174(4):1264–79. doi: 10.2353/ajpath.2009.080160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 6.Tokumura A, Carbone LD, Yoshioka Y, Morishige J, Kikuchi M, Postlethwaite A, et al. Elevated serum levels of arachidonoyl-lysophosphatidic acid and sphingosine 1-phosphate in systemic sclerosis. Int J Med Sci. 2009;6(4):168–76. doi: 10.7150/ijms.6.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazereeuw-Hautier J, Gres S, Fanguin M, Cariven C, Fauvel J, Perret B, et al. Production of lysophosphatidic acid in blister fluid: involvement of a lysophospholipase D activity. J Invest Dermatol. 2005;125(3):421–7. doi: 10.1111/j.0022-202X.2005.23855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto T, Takagawa S, Katayama I, Yamazaki K, Hamazaki Y, Shinkai H, et al. Animal Model of Sclerotic Skin. I: Local Injections of Bleomycin Induce Sclerotic Skin Mimicking Scleroderma. J Invest Dermatol. 1999;112(4):456–462. doi: 10.1046/j.1523-1747.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu M, Varga J. In perspective: murine models of scleroderma. Curr Rheumatol Rep. 2008;10(3):173–82. doi: 10.1007/s11926-008-0030-9. [DOI] [PubMed] [Google Scholar]
- 10.Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100(21):12319–24. doi: 10.1073/pnas.1635114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takagawa S, Lakos G, Mori Y, Yamamoto T, Nishioka K, Varga J. Sustained activation of fibroblast transforming growth factor-beta/Smad signaling in a murine model of scleroderma. J Invest Dermatol. 2003;121(1):41–50. doi: 10.1046/j.1523-1747.2003.12308.x. [DOI] [PubMed] [Google Scholar]
- 12.Mori Y, Hinchcliff M, Wu M, Warner-Blankenship M, K ML, Varga J. Connective tissue growth factor/CCN2-null mouse embryonic fibroblasts retain intact transforming growth factor-beta responsiveness. Exp Cell Res. 2008;314(5):1094–104. doi: 10.1016/j.yexcr.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97(24):13384–9. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2) Mol Cell Biol. 2002;22(19):6921–9. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31(4):395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 16.Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13(1):7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- 17.Abraham DJ, Eckes B, Rajkumar V, Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr Rheumatol Rep. 2007;9(2):136–43. doi: 10.1007/s11926-007-0008-z. [DOI] [PubMed] [Google Scholar]
- 18.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 19.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83(3):835–70. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 20.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 21.Demoyer JS, Skalak TC, Durieux ME. Lysophosphatidic acid enhances healing of acute cutaneous wounds in the mouse. Wound Repair Regen. 2000;8(6):530–7. doi: 10.1046/j.1524-475x.2000.00530.x. [DOI] [PubMed] [Google Scholar]
- 22.Balazs L, Okolicany J, Ferrebee M, Tolley B, Tigyi G. Topical application of the phospholipid growth factor lysophosphatidic acid promotes wound healing in vivo. Am J Physiol Regul Integr Comp Physiol. 2001;280(2):R466–72. doi: 10.1152/ajpregu.2001.280.2.R466. [DOI] [PubMed] [Google Scholar]
- 23.Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9(Suppl 2):S2. doi: 10.1186/ar2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sappino AP, Masouye I, Saurat JH, Gabbiani G. Smooth muscle differentiation in scleroderma fibroblastic cells. Am J Pathol. 1990;137(3):585–91. [PMC free article] [PubMed] [Google Scholar]
- 25.Kissin EY, Merkel PA, Lafyatis R. Myofibroblasts and hyalinized collagen as markers of skin disease in systemic sclerosis. Arthritis Rheum. 2006;54(11):3655–60. doi: 10.1002/art.22186. [DOI] [PubMed] [Google Scholar]
- 26.Kundra V, Anand-Apte B, Feig LA, Zetter BR. The chemotactic response to PDGF-BB: evidence of a role for Ras. J Cell Biol. 1995;130(3):725–31. doi: 10.1083/jcb.130.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietruck F, Busch S, Virchow S, Brockmeyer N, Siffert W. Signalling properties of lysophosphatidic acid in primary human skin fibroblasts: role of pertussis toxin-sensitive GTP-binding proteins. Naunyn Schmiedebergs Arch Pharmacol. 1997;355(1):1–7. doi: 10.1007/pl00004906. [DOI] [PubMed] [Google Scholar]
- 28.Cerutis DR, Dreyer A, Cordini F, McVaney TP, Mattson JS, Parrish LC, et al. Lysophosphatidic acid modulates the regenerative responses of human gingival fibroblasts and enhances the actions of platelet-derived growth factor. J Periodontol. 2004;75(2):297–305. doi: 10.1902/jop.2004.75.2.297. [DOI] [PubMed] [Google Scholar]
- 29.Tangkijvanich P, Melton AC, Chitapanarux T, Han J, Yee HF. Platelet-derived growth factor-BB and lysophosphatidic acid distinctly regulate hepatic myofibroblast migration through focal adhesion kinase. Exp Cell Res. 2002;281(1):140–7. doi: 10.1006/excr.2002.5657. [DOI] [PubMed] [Google Scholar]
- 30.Fang X, Yu S, LaPushin R, Lu Y, Furui T, Penn LZ, et al. Lysophosphatidic acid prevents apoptosis in fibroblasts via G(i)-protein-mediated activation of mitogen-activated protein kinase. Biochem J. 2000;352(1):43. [PMC free article] [PubMed] [Google Scholar]
- 31.Song J, Clair T, Noh JH, Eun JW, Ryu SY, Lee SN, et al. Autotaxin (lysoPLD/NPP2) protects fibroblasts from apoptosis through its enzymatic product, lysophosphatidic acid, utilizing albumin-bound substrate. Biochem Biophys Res Commun. 2005;337(3):967–75. doi: 10.1016/j.bbrc.2005.09.140. [DOI] [PubMed] [Google Scholar]
- 32.Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56(1):334–44. doi: 10.1002/art.22328. [DOI] [PubMed] [Google Scholar]
- 33.Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, et al. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165(1):203–17. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardner H, Shearstone JR, Bandaru R, Crowell T, Lynes M, Trojanowska M, et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum. 2006;54(6):1961–73. doi: 10.1002/art.21894. [DOI] [PubMed] [Google Scholar]
- 35.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3(7):e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51(5):1376–82. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 37.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 38.Piazza GA, Ritter JL, Baracka CA. Lysophosphatidic acid induction of transforming growth factors alpha and beta: modulation of proliferation and differentiation in cultured human keratinocytes and mouse skin. Exp Cell Res. 1995;216(1):51–64. doi: 10.1006/excr.1995.1007. [DOI] [PubMed] [Google Scholar]
- 39.Ihn H, Yamane K, Kubo M, Tamaki K. Blockade of endogenous transforming growth factor beta signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor beta receptors. Arthritis Rheum. 2001;44(2):474–80. doi: 10.1002/1529-0131(200102)44:2<474::AID-ANR67>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Increased expression levels of integrin alphavbeta5 on scleroderma fibroblasts. Am J Pathol. 2004;164(4):1275–92. doi: 10.1016/s0002-9440(10)63215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol. 2005;175(11):7708–18. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 42.Pradere JP, Klein J, Gres S, Guigne C, Neau E, Valet P, et al. LPA1 receptor activation promotes renal interstitial fibrosis. J Am Soc Nephrol. 2007;18(12):3110–8. doi: 10.1681/ASN.2007020196. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Tomiya T, et al. Plasma lysophosphatidic acid level and serum autotaxin activity are increased in liver injury in rats in relation to its severity. Life Sci. 2007;81(12):1009–15. doi: 10.1016/j.lfs.2007.08.013. [DOI] [PubMed] [Google Scholar]