

Summary
20-Hydroxyeicosatetraenoic acid (20-HETE) is a potent vasoconstrictor involved in vascular dysfunction and blood pressure regulation. Studies have revealed strong associations between 20-HETE and endothelial dysfunction however the signalling mechanisms are largely unknown. Therefore we sought to investigate the effect of 20-HETE on endothelial nitric oxide synthase (eNOS) and heat shock protein 90 (Hsp90) association.
20-HETE significantly enhanced the constriction and inhibited the relaxation of mouse aortic rings in response to phenylephrine and acetylcholine, respectively (p=0.05 versus control ring). In mice with chronic AMP-activated protein kinase (AMPK) activation this protected against the negative effects of 20-HETE (p<0.05). Immunoprecipitation of eNOS in cells treated with 20-HETE revealed a decrease in basal and vascular endothelial growth factor (VEGF) stimulated Hsp90 association with eNOS (p<0.05). Pre-treatment of the cells with AICAR (a chronic activator of AMPK) prevented the loss of Hsp90 association with eNOS following 20-HETE treatment. Treatment with 20-HETE for 24h induces an increase in eNOS phosphorylation, not observed following acute treatment (30mins). This was accompanied by transient changes in Akt phosphorylation.
20-HETE impairs eNOS-Hsp90 association which can be reversed via chronic activation of AMPK. This provides a mechanism for reduced NO bioactivity and endothelial dysfunction in diseases with elevated 20-HETE levels, such as hypertension.
Keywords: 20-HETE, endothelial dysfunction, eNOS-Hsp90 association, AMPK
Introduction
Endothelium-derived nitric oxide (NO) is an important regulator of vascular function. Produced by endothelial nitric oxide synthase (eNOS), under normal conditions, its vasodilatory actions regulate the diameter of blood vessels and maintain an anti-proliferative and anti-inflammatory environment in the vessel wall.(1) Phosphorylation of eNOS by Akt/protein kinase B at serine-1177 increases eNOS activity, while Hsp90 promotes increased eNOS activity via direct interaction with the enzyme. Exposure of endothelial cells to stimulants such as vascular endothelial growth factor (VEGF) induces increased Hsp90 association and phosphorylation by Akt, which ultimately lead to increased NO production.(2) Reductions in NO bioavailability or bioactivity contribute to impairment in normal endothelial function. Endothelial dysfunction is frequently associated with several cardiovascular disease risk factors.(3) The precise mechanisms behind endothelial dysfunction are largely unknown, although a number of factors are thought to be involved. These include: reduction in NO bioavailability and bioactivity via reaction with superoxide (O2−•), eNOS uncoupling, leading to increased O2−• formation and reduced cGMP production, and loss of tetrahydrobiopterin (BH4), an essential cofactor for eNOS activity.
Arachidonic acid is a major membrane fatty acid that can be metabolised by the cytochrome P450 (CYP450) enzyme system to a number of bioactive metabolites. Within the vasculature and kidney, a major product of this metabolism is 20-hydroxyeicosatetraenoic acid (20-HETE), a potent vasoconstrictor involved in vascular dysfunction and blood pressure (BP) regulation.(4) 20-HETE has been demonstrated to play an inhibitory role in eNOS activity and both animal and human studies have revealed strong associations between 20-HETE and endothelial dysfunction.(5, 6) We have previously demonstrated a significant association between urinary 20-HETE excretion and endothelial dysfunction in normotensive and hypertensive humans(6) as well as positive associations between 20-HETE and BP(7) and 20-HETE and oxidative stress.(8) More recently, 20-HETE was demonstrated to cause eNOS uncoupling(9), however the signalling mechanisms underlying these associations are largely unknown.
Previous studies have demonstrated that AMP-activated protein kinase (AMPK) activity is involved in the Hsp90-eNOS association.(10) AMPK is a serine/threonine protein kinase that is activated by physiological stimuli and oxidants. It is thought that the cardio-protective effects of several drugs such as metformin and statins may operate through AMPK activation.(11) Furthermore, a study in bovine endothelial cells revealed that metformin dose-dependently increased eNOS phosphorylation, eNOS-Hsp90 association and cGMP, and this effect was reversed in the presence of kinase-inactive AMPK.(12)
We hypothesised that 20-HETE would impair normal endothelial function and that this would be in part mediated through disruption to eNOS activation and function. The aims of the project were to investigate the functional consequences of 20-HETE on eNOS function in mouse aortic rings and whether this was improved with AMPK activation. We also sought to investigate the effect of 20-HETE on eNOS and Hsp90 association, and whether this was mediated by AMPK.
Methods
Materials
Cell culture reagents were obtained from Lonza (Allendale, USA) (human umbilical vein endothelial cells, HUVECs) and were maintained in commercially available Lonza media. All experiments were performed with confluent cells between passages 3 and 9. 20-HETE was purchased from Cayman Chemical (Ann Arbor MI), AICAR from Toronto Research Chemicals (Ontario, Canada), Compound C from Calbiochem (Gibbstown, NJ) and 2',7'-dichlorofluorescin diacetate (DCF) from Sigma (St Louis, MO). Polyclonal antibodies against Akt, phospho-Akt (serine-473) and phospho-eNOS (serine-1177 or threonine-495) were purchased from Cell Signaling (Danvers, MA). Monoclonal eNOS antibody and Hsp90 antibody were purchased from BD Biosciences (Franklin Lakes, NJ). All other chemicals were purchased from Sigma (St Louis MO).
ROS production
Intracellular ROS levels were determined using a fluorescence assay. For acute treatment, cells were washed with PBS and media was replaced with Hanks Buffered Saline Solution (HBSS, Gibco Carlsbad, CA) containing glucose and 10μM of DCF for 15mins. The HBSS was removed and replaced with fresh HBSS and the desired treatment. Cells were then washed quickly with ice cold PBS and 300μL of cold PBS was added to the cells on ice. The cells were collected into a black 96 well plate and DCF fluorescence was assessed with a fluorescence plate reader (Molecular Devices, Sunnyvale CA) with excitation at 485nm and emission at 535nm. For chronic treatments, cells were incubated with desired treatment, the media replaced with HBSS and cells were loaded with 10μM of DCF for 15mins before collection as described above. In both experiments, treatment with 100μM of H2O2 for 15 mins was used as a positive control.
Extracellular ROS production was determined using the Amplex-Red assay (Molecular Probes, Carlsbad, CA). After desired treatments, media was removed and replaced with HBSS containing glucose. Cells were incubated with 100μM of Amplex and 1U/mL of horseradish peroxidise and incubated at 37°C for 15mins, before 100μL of supernatant was collected in a black 96 well plate and the fluorescence determined (excitation=544nm, emission=590nm). Cells treated with 100μM of H2O2 for 15mins was used as a positive control.
Immunoprecipitation
After various treatments, cells were quickly washed with ice-cold PBS, lysed with 500μL of cell lysis buffer (Cell Signaling, Danvers MA) and placed on ice for approximately 3 mins. Cell lysates were collected on ice, briefly sonicated and spun at 14000 rpm, 4°C for 10 mins to pellet cell membrane. The supernatant was collected and incubated with 5μL of eNOS antibody with gentle rocking overnight at 4°C. The supernatant was then incubated with 20μL of 50% slurry of Immobilized Protein A/G Beads (Pierce, Rockford IL) for 2hrs with gentle rocking at 4°C. The sample was spun at 14000rpm, 4°C for 10mins, the beads washed once with 500μL of cold cell lysis buffer on ice and the pellet resuspended in 20μL of Laemmli's SDS-Sample Buffer (Boston Bioproducts, Boston MA). Proteins were immunoblotted as described below.
Immunoblotting
After various treatments, cells were washed with ice cold PBS and lysed with 250μL of Laemmli's SDS-Sample Buffer (Boston Bioproducts, Boston MA). Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis on 7.5% to 12% gels, transferred to nitrocellulose membrane and incubated with the primary antibody overnight (1:100 – 1:1000). After washing and addition of the secondary antibody (1:2000), protein bands were visualised with ECL reagent (GE Healthcare, Piscataway NJ) using a FluroChem HD2 (Alpha Tech, Burlington MA).
Vascular Activity
Male C57BL/6J mice, approximately 9 weeks old, were randomly assigned to receive a single intraperitoneal injection of either 200μL of PBS or 200μL of AICAR (1mM). Twenty-four hours later, the mice were anesthetised by Ketamine and perfused with 0.9% NaCl. The aortic rings were then harvested, cleaned of excess tissue and cut into equal segments, approximately 2mm long. All aortas were incubated for approx 30 mins in Physiological Saline Solution (PSS; 119mM NaCl, 4.69mM KCl, 1.17mM MgSO4, 1.18mM KH2PO4, 2.5mM CaCl2, 25mM NaHCO3, 0.03mM EDTA and 5.5mM Glucose), aerated at 95% O2/5% CO2 at 37°C under constant passive force (~1mM). The rings were primed with KCl (40mM) alone and KCl (40mM) and phenylephrine (10−5 M) to reactivate the mechanical and functional properties of the vessel. Rings were then allowed to equilibrate before addition of either 20-HETE (1μM) or vehicle (ethanol). Contraction response of the rings was determined with increasing doses of phenylephrine (10−9 to 10−5 M). Separate experiments were conducted to determine relaxation response where rings were pre-constricted with phenylephrine (~3 × 10−7 M) and then relaxed with increasing doses of acetylcholine (10−9 to 10−5 M). Relaxation was calculated as a % of the starting point for each ring. Aortic activity experiments were recorded using a Multiwire Myograph System (DMT-USA Inc, Model 610M, Version 2.2, Atlanta GA). All experiments were approved by the University of Massachusetts Animal Ethics Committee.
Statistical analysis
All immunoblots are representative of 3 to 5 independent experiments. Numerical data are presented as mean ± SD. Comparisons between treatment groups were performed using 1-way ANOVA with post-hoc Dunnett comparison or repeated measures 2-way ANOVA as appropriate, using the Statistical Packages for the Social Sciences (SPSS Version 15) program.
Results
20-HETE effects on ex vivo aortic ring function
To determine the functional consequence of 20-HETE on endothelial function in intact blood vessels, we investigated the effects of 20-HETE on aortic ring constriction and relaxation in the presence or absence of AMPK activation. To achieve this, male C57BL/6J mice, approximately 9 weeks of age, were randomly assigned to receive a single intraperitoneal injection of 200μL PBS or 200μL AICAR (1mM). 24hr later, the mice aortic rings were harvested and mounted in ring baths to determine their constriction and relaxation responses in the presence or absence of 1μM of 20-HETE. As shown, rings treated with 20-HETE alone resulted in a significant increase in the constriction response to increasing doses of phenylephrine when compared to both control rings (p = 0.045) and AICAR+20-HETE rings (p = 0.025) (figure 1a). When rings were pre-constricted with phenylephrine and then relaxed using increasing doses of acetylcholine, the 20-HETE treated ring relaxed significantly less when compared to the control ring (p = 0.05), the AICAR treated ring (p = 0.044) or the AICAR+20-HETE treated ring (p = 0.027) (figure 1b). Addition of Indomethacin to the organ bath did not alter the results (data not shown). This data suggests that 20-HETE has a negative impact on normal vessel function, supporting our previous findings in humans, and that this may be prevented via AMPK activation following pre-treatment with AICAR.
Figure 1.

Mouse aortic ring (a) constriction with increasing doses of phenylephrine. 2-way ANOVA with repeated measures analysis, p = 0.05 versus control and p = 0.025 versus AICAR+20-HETE (n=6–9) and (b) relaxation in response to increasing doses of acetylcholine, following pre-constriction with phenylephrine. 2-way ANOVA with repeated measures analysis, p = 0.05 versus control, p = 0.04 versus AICAR and p = 0.03 versus AICAR+20-HETE (n=5).
20-HETE effects on eNOS-Hsp90 association
To investigate the mechanistic effects of 20-HETE treatment on Hsp90 association with eNOS, we treated HUVECs with 20-HETE, immunoprecipitated with the eNOS antibody and then immunoblotted with eNOS serine-1177 and Hsp90 antibodies. Under basal conditions, acute and chronic treatment with 20-HETE reduced Hsp90 association with eNOS. Following stimulation with VEGF (10ng for 5 mins), only chronic 20-HETE treatment reduced Hsp90 association with eNOS (figure 2). These results were only achieved when 10μM of 20-HETE was used. The lack of response at 1μM concentration may be due to poor uptake by the cell or sequestration of 20-HETE by the media.
Figure 2.
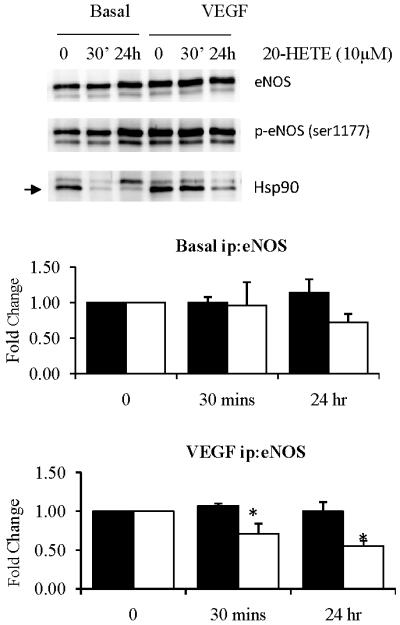
eNOS immunprecipitation of HUVECs treated with 20-HETE (10μM), under basal and VEGF stimulated conditions (n=3). Fold change where black bars are p-eNOS/eNOS and white bars are Hsp90/eNOS. ANOVA with Dunnett's post-hoc analysis *p<0.05 versus control.
In vitro activation of AMPK protects against the effects of 20-HETE
To further determine the role of AMPK in 20-HETE mediated eNOS-Hsp90 disassociation, we treated cells with AICAR (1mM for 1hr) prior to treatment with 20-HETE. Separate experiments confirmed phosphorylation of AMPK and ACC following treatment with AICAR (data not shown). Following immunoprecipitation with the eNOS antibody, we immunoblotted with phospho-eNOS serine-1177 and Hsp90 antibodies. Pre-treatment with AICAR to activate AMPK protected against 20-HETE mediated eNOS-Hsp90 disassociation both basally and following VEGF stimulation (figure 3a and b), supporting our ex vivo findings. Again, these results were only achieved using 10μM of 20-HETE.
Figure 3.

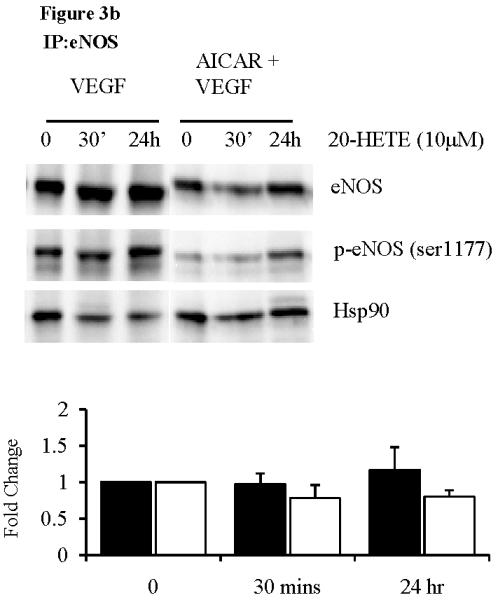
eNOS immunoprecipitation of HUVECs pre-treated with AICAR (1mM, 1hr), followed by 20-HETE (10μM) under (a) basal and (b) VEGF stimulated conditions (n=3). (c) eNOS immunoprecipitation of HUVECs pre-treated with AICAR and/or Compound C (20μM, 1 hr), followed by 20-HETE (10μM). Fold change where black bars are p-eNOS/eNOS and white bars are Hsp90/eNOS.
To investigate the role of AMPK further, we then treated cells with both AICAR to activate AMPK and Compound C to inhibit AMPK, prior to 20-HETE treatment. As before, treatment with AICAR in the presence of 20-HETE, rescued the eNOS-Hsp90 association. Co-treatment with Compound C (20μM) however, resulted in a reduction in Hsp90-eNOS association following chronic 20-HETE treatment (figure 3c). Taken together these results further strengthen a role for AMPK, suggesting that chronic activation of AMPK can protect against 20-HETE induced eNOS-Hsp90 disassociation and subsequent impaired vascular function.
20-HETE effects on eNOS and Akt phosphorylation
Phosphorylation of eNOS and Akt are both important steps in the activation of the eNOS system. Treatment of HUVECs with 20-HETE resulted in transient increases in the phosphorylation of Akt at serine-473 residue (figure 4a). Chronic 20-HETE treatment also led to increased eNOS phosphorylation at both the serine-1177 and threonine-495 residues (figure 4b). There appeared to be no effect of 20-HETE on eNOS phosphorylation at the serine-633 residue (data not shown). Again, these results were only achieved at 10μM concentrations. This data indicates that chronic exposure to 20-HETE effects both eNOS and Akt phosphorylation in HUVECs.
Figure 4.

Phosphorylation of (a) Akt and (b) eNOS in HUVECs following acute and chronic treatment with 20-HETE (10μM). ANOVA with Dunnett's post-hoc analysis *p<0.05 versus control.
20-HETE induced ROS production
We also determined whether these effects may have been mediated through increases in ROS production. Intracellular ROS production was assessed via the DCF assay, which measures predominately intracellular superoxide production. Acute treatment with 20-HETE (1–30 min) produced a marked increase in ROS production compared to untreated cells (figure 5a). However, following chronic treatment with 20-HETE (2–24 hr) we observed that ROS production was similar to control cells (figure 5b). We also investigated extracellular ROS production via the Amplex-Red assay. Following both acute and chronic 20-HETE treatment, the level of ROS production was similar to that seen in control cells (figure 5c). This data suggests that 20-HETE produces acute increases in intracellular ROS, which is expected to be predominately superoxide.
Figure 5.

Intracellular ROS production following (a) acute 20-HETE treatment. ANOVA with Dunnett's post-hoc analysis, p < 0.05 versus control (n=4) and (b) chronic 20-HETE treatment. ANOVA with Dunnett's post-hoc analysis (n=4). Extracellular ROS production (c) following acute and chronic 20-HETE treatment. ANOVA with Dunnett's post-hoc analysis (n=4).
To determine if our findings were due to 20-HETE toxicity, we investigated its effect on release of lactate dehydrogenase (a marker of cell death) and formation of formazan (a marker of cell viability). At doses up to 10μM and time points up to 24hr, there was no effect on either marker of cell viability (data not shown).
Discussion
The major finding of the present study is that exposure of endothelial cells to 20-HETE leads to a disruption of the eNOS-Hsp90 association, which is at least in part, mediated through AMPK. Chronic activation of AMPK protects against the effects of 20-HETE on both the disassociation of Hsp90 and eNOS. Chronic activation of AMPK also protects ex vivo endothelial function however it remains unclear whether this is solely due to an NO-dependent pathway. This finding provides mechanistic evidence for disrupted eNOS function and endothelial dysfunction in diseases associated with elevated 20-HETE levels, such as hypertension.(6, 7)
We investigated the functional effects of 20-HETE by using a mouse aortic ring system. In this model, we investigated if 20-HETE could impair vasorelaxation and whether chronic activation of AMPK could protect the vessel function from the deleterious effects of 20-HETE. In response to both constrictive and relaxing stimuli, AMPK activation by AICAR alone had no effect, while 20-HETE significantly enhanced the constriction response and inhibited the relaxation response. However, when AMPK was chronically activated through AICAR pre-treatment, this protected against the effects of 20-HETE. This suggests that chronic AMPK activation via AICAR treatment results in a “healthier” vessel that is able to protect against the deleterious effects of 20-HETE.
Hsp90 is an important chaperone molecule that binds both Akt and eNOS, facilitating phosphorylation at the serine-1177 residue of the eNOS complex by Akt. This then leads to activation of the eNOS enzyme and subsequent production of NO. Preventing eNOS from binding to the Hsp90 chaperone leads to disassociation of the enzyme, increased ROS production and decreased NO bioavailability. Previous work has shown that AMPK may be required for Hsp90-eNOS binding,(10) and more recently that AMPK is required for eNOS phosphorylation.(13) A recent study has demonstrated that treatment with 20-HETE leads to eNOS-Hsp90 uncoupling following stimulation with a calcium ionophore,(9) although the exact mechanism remains unclear. In the present study we demonstrate that under both basal and VEGF stimulated conditions, 20-HETE is able to disrupt the association of the eNOS-Hsp90 complex following both acute and chronic treatment. Pre-treatment of the cells with AICAR, a compound which activates the AMPK complex is able to prevent this disassociation. Strengthening this argument is evidence that Compound C, an inhibitor of AMPK reverses the protective effects of AICAR on the eNOS-Hsp90 complex following 20-HETE treatment.
Both CYP4A and 4F enzyme activity and 20-HETE itself are able to increase ROS production in endothelial cells.(14) Excessive endothelial ROS production may result in depletion of NO, via rapid reaction with superoxide, resulting in endothelial dysfunction. We have previously shown a significant association between increased urinary 20-HETE excretion and increased oxidative stress in hypertensive individuals.(8) Alternatively, ROS when produced in controlled amounts can function as signalling molecules in cells.(15) During the acute phase, 20-HETE caused an increase in intracellular ROS production, which had returned to control levels by 2 hr.
There was no change in extracellular ROS production. This suggests that direct application of 20-HETE to the cultured endothelial cells led to an increase in intracellular ROS production, presumably due to some cellular uptake of 20-HETE. This was not caused by 20-HETE toxicity as evidenced by the cell death and cell viability studies.
Treatment with 20-HETE resulted in transient increases in Akt phosphorylation, which is required for subsequent eNOS phosphorylation. Interestingly, eNOS phosphorylation at the serine-1177 residue increased with longer exposure to 20-HETE. The serine-1177 residue is typically seen as an activation site that is required for normal eNOS function.(13) There were no changes observed at the serine-633 residue, however the threonine-495 site also had increasing phosphorylation over time. The threonine-495 site is known as an inactivation site with phosphorylation of the site being associated with decreased eNOS activity. Indeed previous evidence has shown that eNOS phosphorylation at both the threonine-495 residue and serine-1177 residue facilitates eNOS uncoupling and an increased production of superoxide.(16) These coordinated eNOS phosphorylation events following treatment with 20-HETE, may contribute to disruption of eNOS-Hsp90 complex.
The present work suggests that 20-HETE effects endothelial function through eNOS-Hsp90 disassociation. Other possibilities include inhibition of the formation of the eNOS dimer, also required for normal function or possibly direct interaction between the Hsp90 chaperone and 20-HETE, preventing the eNOS from binding. Investigation of these other pathways of action is warranted. Furthermore, we acknowledge that the dose of 20-HETE used in the cell culture experiments was higher than that used in the ex vivo ring studies. At 1μM concentrations, the effects of 20-HETE on cultured endothelial cells were not observed. This may be partly due to insufficient uptake by the cell either via sequestration in the media or reaction with oxygen. Given no receptor for 20-HETE has been identified, it remains unknown as to how the cells take up 20-HETE and in what form. Further work to elucidate this pathway is also warranted. In summary, the present study has demonstrated that 20-HETE gives rise to eNOS-Hsp90 disassociation and this results in reduced endothelial function as measured via response to phenylephrine and acetylcholine. Treatment with AICAR, an AMPK activator is able to prevent the adverse effects of 20-HETE both in an endothelial cell culture system and ex vivo aortic vessels. This may in part explain the negative association we have previously observed between 20-HETE and endothelial function.(6)
Acknowledgements
NC Ward gratefully acknowledges the support of a National Health and Medical Research Council of Australia CJ Martin Postdoctoral Fellowship.
Funding NCW was supported by a National Health & Medical Research Council of Australia CJ Martin Fellowship [404108].
JFK was supported by National Institute of Health grants [AG027081, HL081587 and HL092122]
Reference List
- 1.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol.Chem. 2003;278:30821–30827. doi: 10.1074/jbc.M304471200. [DOI] [PubMed] [Google Scholar]
- 3.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 4.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 5.Wang JS, Singh H, Zhang F, Ishizuka T, Deng H, Kemp R, et al. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ.Res. 2006;98:962–969. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- 6.Ward NC, Rivera J, Hodgson J, Puddey IB, Beilin LJ, Falck JR, et al. Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation. 2004;110:438–443. doi: 10.1161/01.CIR.0000136808.72912.D9. [DOI] [PubMed] [Google Scholar]
- 7.Ward NC, Tsai IJ, Barden A, van Bockxmeer FM, Puddey IB, Hodgson JM, et al. A single nucleotide polymorphism in the CYP4F2 but not CYP4A11 gene is associated with increased 20-HETE excretion and blood pressure. Hypertension. 2008;51:1393–1398. doi: 10.1161/HYPERTENSIONAHA.107.104463. [DOI] [PubMed] [Google Scholar]
- 8.Ward NC, Puddey IB, Hodgson JM, Beilin LJ, Croft KD. Urinary 20-hydroxyeicosatetraenoic acid excretion is associated with oxidative stress in hypertensive subjects. Free Radic.Biol.Med. 2005;38:1032–1036. doi: 10.1016/j.freeradbiomed.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Cheng J, Ou JS, Singh H, Falck JR, Narimhaswamy D, Pritchard KA, et al. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am.J Physiol. 2008;294:H1018–H1026. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 10.Schulz E, Anter E, Zou MH, Keaney JF., Jr. Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation. 2005;111:3473–3480. doi: 10.1161/CIRCULATIONAHA.105.546812. [DOI] [PubMed] [Google Scholar]
- 11.Zou MH, Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin.Exp.Pharmacol.Physiol. 2008;35:535–545. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ.Res. 2009;104:496–505. doi: 10.1161/CIRCRESAHA.108.187567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkis A, Roman RJ. Role of cytochrome p450 metabolites of arachidonic acid in hypertension. Curr.Drug Metab. 2004;5:245–256. doi: 10.2174/1389200043335603. [DOI] [PubMed] [Google Scholar]
- 15.Chen K, Thomas SR, Keaney JF., Jr. Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic.Biol.Med. 2003;35:117–132. doi: 10.1016/s0891-5849(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 16.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, et al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J.Biol.Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]