

Abstract
Binding of p53 to miR-34a promoter activates the expression of tumor suppressive miR-34a. Oncogenic HPV infection downregulates miR-34a expression through viral E6 degradation of p53. In this report, we found that miR-34a specifically targets p18Ink4c, a CDK4 and CDK6 inhibitor induced by E2F transactivation. HPV18+ HeLa cells with ectopic miR-34a expression or by E6 siRNA knockdown-induced expression of endogenous miR-34a exhibited a substantial reduction of p18Ink4c in a dose-dependent manner, but had no effect on p16Ink4a, another member of CDK4/6 inhibitor family. In contrast, de novo infection by oncogenic HPVs of human keratinocyte-derived raft tissues increased p18Ink4c expression. Suppression of endogenous miR-34a in cell lines with a miR-34a inhibitor also increased p18Ink4c. We found that miR-34a suppresses the expression of p18Ink4c by binding to a specific seed match in the 5' UTR of p18Ink4c. Further investigation found remarkable increase of p18Ink4c in cervical precancer lesions and cervical cancer. Immunohistochemical staining of cervical tissue arrays showed increased expression of p18Ink4c in 68% of cervical cancer, 8.3% of chronic cervical inflammation, and 4.8% of normal cervix. Although p18Ink4c inhibits cell proliferation in general and regulates E2F1 expression in HCT116 cells, it appears not to function as a tumor suppressor in cervical cancer cells lacking an intact G1 checkpoint due to viral E7 degradation of pRB. In summary, this study demonstrates an intimate connection among oncogenic HPV E6, p53, miR-34a, and p18Ink4c and identifies p18Ink4c as a possible biomarker for cervical cancer.
Keywords: Human papillomaviruses, microRNAs, p53, cell cycle control, p18ink4c
Introduction
Cervical cancer induced by persistent infection of oncogenic human papillomaviruses (HPV)1 is the second most common cancer in women worldwide.2 Among 15 oncogenic (also called high-risk) HPV types identified, HPV16 and HPV18 are the two most common types associated with ~70% of all cervical cancer cases.3 Two viral oncoproteins, E6 and E7, of HPV16 and HPV18 are responsible for viral oncogenesis, respectively by destabilizing two major cellular tumor suppressors, p53 and pRB, that are essential for cell cycle control.4
MicroRNAs (miRNAs) are a class of noncoding regulatory RNAs in size of 21–25 nucleotides and regulate gene expression by base-pairing with complementary nucleotide sequences (seed matches or binding sites) in the 5' and 3' untranslated regions (UTRs) of target mRNAs. 5–7 Several hundred genes in human genome have been shown to encode miRNAs.7 Tumor suppressive miR-34a in proliferating cells is a direct transcriptional target of p53 and its expression is transactivated by the binding of p53 to a consensus p53 binding site in the miR-34a promoter region.8–10 However, p53-independent upregulation of miR-34a can be triggered in cells undergoing terminal differentiation 11 or senescence.12 In contrast, cancer cells may inactivate miR-34a expression by aberrant CpG methylation.13 It has been documented that miR-34a exercises remarkable posttranscriptional effects on gene expression of cell cycle regulators, including cyclin E2, cyclin D1, CDK4, CDK6, E2F1, E2F3, E2F5, Bcl-2, and SIRT1.8–10,14–17 By affecting the expression of cell cycle regulators, miR-34a regulates cell cycle progression, cellular senescence, and apoptosis.
Cervical cancer, like many other cancers,18 displays aberrant expression of oncogenic and tumor suppressive miRNAs. 19,20 We found that cervical cancer and their derived cell lines express much reduced levels of miR-34a as a result from viral E6 destabilization of p53.11 Given the evidence that infection by oncogenic HPVs is a necessary factor for the development of cervical cancers,1 we further investigated the consequence of E6-mediated reduction of miR-34a and searched for miR-34a targets that may contribute to cervical carcinogenesis associated with oncogenic HPV infection. In this report, we provide the first compelling evidence that p18Ink4c, a CDK4 and CDK6 inhibitor of INK4 family,21 is a prominent target of miR-34a. Importantly, an increased expression of p18Ink4c in cervical precancer lesions and cervical cancer could serve as a possible biomarker of cervical precancer/cancer as well as oncogenic HPV infections.
Materials and Methods
Cell lines and human tissues
HPV16+ CaSki cells and HPV18+ HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS at 37°C and 5% CO2. HCT116 cells derived from colon cancer were grown in McCoy' 5A medium with 10% FBS at 37°C and 5% CO2. Cervical cancer and normal cervical tissue lysates in RIPA buffer were purchased from Protein Biotechnologies (Ramona, CA).
Human primary keratinocytes and organotypic cultures
HPV16 and HPV18 infected human foreskin keratinocytes (HFKs) and human vaginal keratinocytes (HVKs) were grown in monolayer and raft cultures as described.22 The stratified and differentiated raft culture epidermal tissues were collected free from collagen (no fibroblasts) and frozen on dry ice for total cell RNA preparation or were formalin-fixed and paraffin-embedded for tissue sectioning.
Western blotting
Protein samples in 2X SDS sample buffer containing 5% 2-mercaptoethanol were denatured by heating at 95°C for 5 min and separated in a NuPAGE 4–12% Bis-Tris gel (Invitrogen. Carlsbad, CA) in 1X NuPAGE MES SDS running buffer (Invitrogen). After transfer, the nitrocellulose membrane was blocked with 5% nonfat milk in Tris-buffered saline (TBS) for 1 h at room temperature. After rinsing with TBS, the membrane was incubated overnight at 4°C with a primary antibody and then washed 3 times with TTBS [TBS with the addition of Tween 20 at a final concentration of 0.1% (vol/vol)]. Horseradish peroxidase-labeled secondary antibody (Sigma) at 1:10,000 dilution in TTBS was incubated for 1 h at room temperature. After thorough washing 3 times with TTBS, the immunoreactive proteins were detected with an enhanced chemiluminescence substrate (Pierce, Rockford, IL). The signal was captured on X-ray film. Before reprobing with another primary antibody, the membrane was stripped with Restore Western blot stripping buffer (Pierce) according to the manufacturer's instructions and blocked with 5% nonfat milk in TBS. The primary antibodies used were: monoclonal anti-p53 (Calbiochem, San Diego, CA; Ab-6, 1:100), anti-p18Ink4c (Affinity BioReagents, Golden, CO; DCS118, 1:500), anti-p16Ink4a (BD PharMingen, San Jose, CA; G175-405, 1:500), anti-CDK4 (Cell Signaling Technology, Danvers, MA; DCS156, 1:2000), and anti-β-tubulin (Sigma, Tub 2.1, 1:3000), and polyclonal anti-cyclin E2 (Cell Signaling Technology, 1:1000), anti-E2F1 (Cell Signaling Technology, 1:1000) and anti-Cyclin A (Santa Cruz Biotechnology, H-432, 1:1000). A relative protein level in each sample shown in each bar graph was calculated based on its protein band density on Western blot after normalized to β-tubulin for sample loading.
Immunohistochemical (IHC) staining
IHC staining of HFK and HVK raft tissues or human cervical tissues with high-grade cervical intraepithelial neoplasia (CIN II) or cervical cancer 23 were carried out with formalin-fixed, paraffin-embedded tissue sections using Vectastain ABC kit (Vector Laboratories, Burlingame, CA). For human tissue array analysis, combined array panels were purchased from Cybrdi (Gaithersburg, MD). Each panel contains formalin-fixed tissues from 63 individual patients with cervical carcinomas in combination of normal cervical tissues with or without chronic inflammations. Each dot on the slide represents a diseased or normal tissue spot from one specimen that was pathologically confirmed. The slides were treated with 1X Antigen Retrieval Citra Plus Buffer accordance to manufacturer’s instruction (BioGenex, San Ramon, CA) and then treated with 3% H2O2 in Dulbecco’s PBS (DPBS) for 15 min, washed and blocked with normal horse serum in DPBST [DPBS with the addition of Tween 20 at a final concentration of 0.1% (vol/vol)]. Sections were incubated with anti-p18Ink4c antibody (Santa Cruz Biotechnology, clone 118.2) or anti-p16Ink4a antibody (BD PharMingen G175-405) overnight at 4°C, followed by secondary antibody for 1 h incubation at room temperature, ABC reagent was then added and the specific signal was developed with DAB substrate kit (Vector Laboratories, Burlingame, CA). The relative level of p18Ink4c staining in cervical tissues was measured by signal intensity of a line crossing over the staining cervical area by using an ImageJ software (http://rsb.info.nih.gov/ij/).24
RT-PCR
Total RNA from HeLa cells and HPV18-infected primary HFKs in monolayer cultures or in stratified and differentiated raft tissues were used for RT-PCR with the following primers: oXHW103, 5'-GGGACCTAGAGCAACTTACTAG-3' (forward), and oXHW104, 5' CAAATCACAGGCGGTGTCC-3' (reverse), for p18Ink4c RNA; oZMZ252, 5'-ATCCAACACGGCGACCCTAC-3' (forward), and oZMZ253, 5'-GACCTTCGAGCATTCCAG-3' (reverse), for HPV18 E6E7 RNA; oXHW37, 5'-ACGAGCCGAACCACAACG-3' (forward), and oXHW38, 5'-TTCCAGCACCGTGTCCGT-3' (reverse), for HPV18 E4 RNA; oZMZ269, 5'-GTCATCAATGGAAATCCCATCACC-3' (forward), and oZMZ270, 5'-TGAGTCCTTCCACGATACCAAA-3' (reverse), for GAPDH RNA as a sample loading control.
Northern blotting
Northern blot of miR-34a detection from total cell RNA was performed as described in a previous report.11 For p18Ink4c mRNA detection, HeLa and HCT116 cells were transfected with 30 nM miR-34a or a control miRNA twice at an interval of 48 h. Total RNAs harvested from cells at 96 h were analyzed by Northern blot using a 32P-labeled p18Ink4c cDNA probe prepared from HeLa cell RNA with the primer pair of oXHW103 and oXHW104.
RNAi
Synthetic double-stranded siRNA219 which targets the HPV18 E6 coding region or siRNA209 which targets the HPV16 E6 coding region 25,26 was used for transfection of HeLa cells or CaSki cells as described.11 Synthetic double-stranded p18Ink4c siGenome SMART pool (Dharmacon) was transfected into HeLa or HCT116 cells by using siPORT NeoFX Transfection Agent (Ambion) according to the manufacturer's instructions. After 48 h, the cells were counted, split and transfected again with the same dose of the siRNA. After an additional 48 h, the cells were counted and protein samples were prepared by direct lysis of the cells in 2X SDS protein sample buffer containing 5% of 2-mercaptoethanol.
miRNAs and miRNA inhibitors
A miR-34a precursor and a negative nonspecific control miRNA precursor were purchased from Ambion for transfection of HeLa cells and CaSki cells as described.11 PNAs miR-34a inhibitor and a miRNA inhibitor negative control were purchased from PANAGENE (www.panagene.com, Daejeon, Korea) and transfected into HeLa and CaSki cells based on the protocol of the manufacturer. These inhibitors are peptide nucleic acids (PNAs)-based miRNA inhibitors with high specificity and stability inside cells and not toxic to cells and require no tranfection reagents. Cells were seeded into a 6-well plate one day before transfection in complete growth medium without antibiotics. 200 nM of PNAs miR-34a inhibitor and negative control inhibitor were diluted separately in 150 µl of Opti-MEM I medium, incubated 15 min at room temperature, and then added into the corresponding wells containing cells and medium. The cells were incubated at 37°C in 5% CO2 for 48 h.
Plasmids, plasmid construction and transfection
Eukaryotic p18Ink4c expression vector corresponding to GenBank AF041248 (p18Ink4c clone 40) used in this study was a gift from Alexandre Blais.27,28 pMIR-REPORT-Luciferase vector (Ambion) was used to construct all reporter plasmids by insertion of a putative miR-34a binding site from the p18Ink4c 3' or 5' UTRs or by insertion of the entire p18Ink4c 3' UTR at the Spe1/HindIII sites. The resulting plasmids pXHW5 and pXHW6 contain a putative wt (pXHW5) and mt (pXHW6) miR-34a binding site from the p18Ink4c 3'UTR, respectively. pXHW23 has an insertion of the entire p18Ink4c 3'UTR (nt 1721–2086 according to GenBank accession number, AF041248) amplified from HeLa cell total RNA. pXHW26 and pXHW27 have an insertion of four repeats of wt (pXHW26) or mt (pXHW27) putative miR-34a binding site from the p18Ink4c 5' UTR, respectively. All insertions were confirmed by sequencing.
HeLa cells and HCT116 cells (6 × 105/well) in 6-well plates were transfected with 2 µg of a p18Ink4c-expressing plasmid clone C40 or 2µg of p3XFLAG as a control. The cells at 48 h after transfection were counted, split and transfected again with the plasmids described above and counted again 24 h after the second transfection.
Dual-luciferase assay
The assay was conducted with HeLa cells at 4.5 × 104 cells per well in a 24-well plate, transfected with 30 nM of miR-34a precursor or a negative control miRNA precursor (Ambion) by using siPORT NeoFX Transfection Agent (Ambion) according to the manufacturer's instructions. 24 h later, the cells were cotransfected for another 24 h with 200 ng of the testing firefly luciferase reporter plasmid together with 20 ng of a Renilla luciferase plasmid pRL-TS 29 by using Lipofectamine 2000 (Invitrogen). The supernatant of the cell lysate was examined for dual luciferase activities using Dual-Luciferase Reporter Assay System (Promega). Relative luciferase activity was calculated by dividing the light unit readings obtained from a firefly luciferase reporter construct by the light unit readings obtained from the Renilla luciferase reporter.
Statistical analysis
Chi square (x2) test was used in Figure 4d left panel and Table 1. Student t-test was used in Figure 2b right panel, Figure 4d right panel and Fig. 5b–c.
Figure 4.
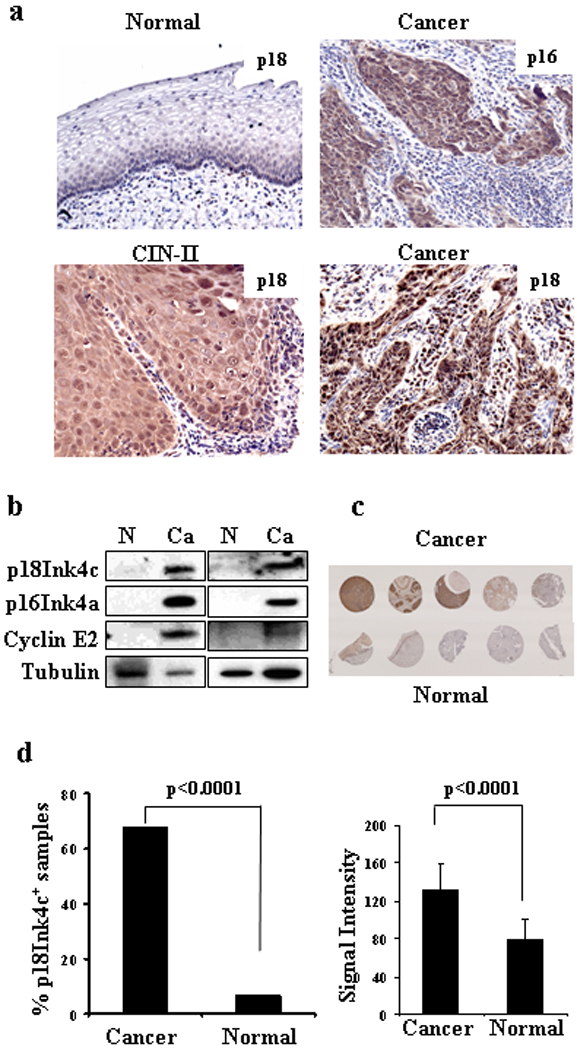
p18Ink4c is highly expressed in cervical CIN lesions and cervical cancer tissues. (a) IHC staining of p18Ink4c in normal cervical tissues and cervical tissues with HPV16+ high-grade CIN or cervical cancer. p16Ink4c staining served as a positive control. Tissue sections with p18Ink4c or p16Ink4a staining are 10× magnification and CIN-II with p18Ink4c staining is 20× magnification. (b) Expression of p18Ink4c, p16Ink4a, and cyclin E2 in paired normal and cervical cancer tissues as measured by Western blot. β-tubulin served as a loading control. (c) p18Ink4c expression in paired normal and cervical cancer tissue arrays by IHC staining. (d) Cervical cancer samples in cervical tissue arrays are frequently positive for p18Ink4c staining (left panel) and express more p18Ink4c proteins (right panel).
Table 1.
Frequency of increased p18Ink4c expression in normal and diseased cervical tissues
| aTissue types | Sample size (n) | bNo. of p18+ | % | P value |
|---|---|---|---|---|
| Cervical cancer | 78 | 53 | 68 | 0.0001c |
| Grade I | 5 | 2 | 40 | |
| Grade II | 39 | 30 | 76.9 | |
| Grade III | 34 | 21 | 61.8 | |
| Chronic cervicitis | 24 | 2 | 8.3 | |
| Normal cervix | 21 | 1 | 4.8 |
Grade I and II were well and moderately differentiated cancers and Grade III poorly differentiated cancer.
A sample was tested as p18Ink4c positive when its staining intensity was above the non-tumor tissue mean value plus two standard deviations.
Compared with chronic cervicitis or normal cervix alone or both in x2 test.
Figure 2.
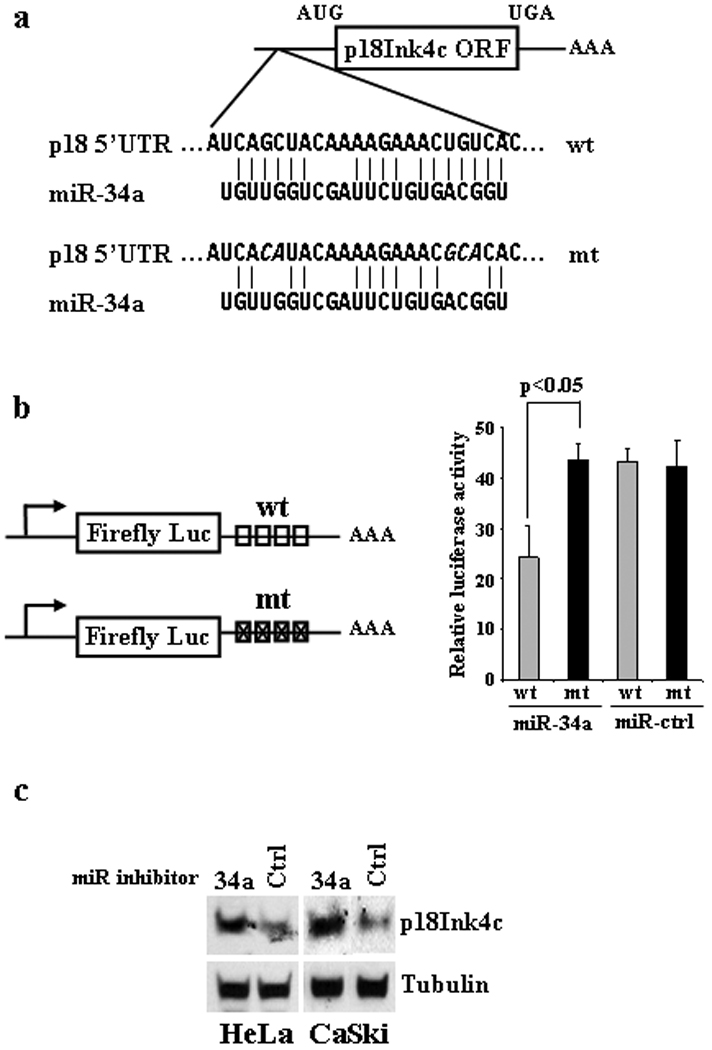
p18Ink4c is a direct downstream target of miR-34a. (a) Schematic depiction of a miR-34a seed match in the 5' UTR of p18Ink4c mRNA and its point mutations (Italics). (b) Response of a luciferase reporter containing a wt or mt miR-34 seed match in its 3' UTR to ectopic miR-34a suppression. The left panel shows diagrams of reporter plasmid 3' UTRs with inserted 4 repeats of wt (boxes) or mt (boxes with an × cross) miR-34a seed match from the p18Ink4c 5' UTR. HeLa cells were cotransfected with 30 nM of miR-34a or control (Ctrl) miRNA in combination with either one of the miR-34a reporter plasmids (200 ng) plus an Renilla luciferase expression plasmid (20 ng). Cell lysates 48 h after transfection were analyzed for dual luciferase assays. Relative luciferase activity was calculated as described in Materials and Methods and is summarized as a mean ± SD for each construct on the right panel. One representative of two separate experiments, each in triplicate, is shown. P<0.05 by one-tailed t-test. (c) Increase of p18Ink4c expression by a miR-34a inhibitor. HPV18+ HeLa cells and HPV16+ CaSki cells 48 h after transfection with 200 nM of a miR-34a specific inhibitor or an inhibitor negative control (Ctrl) were analyzed by Western blot for p18Ink4c expression. β-tubulin served as a loading control.
Figure 5.
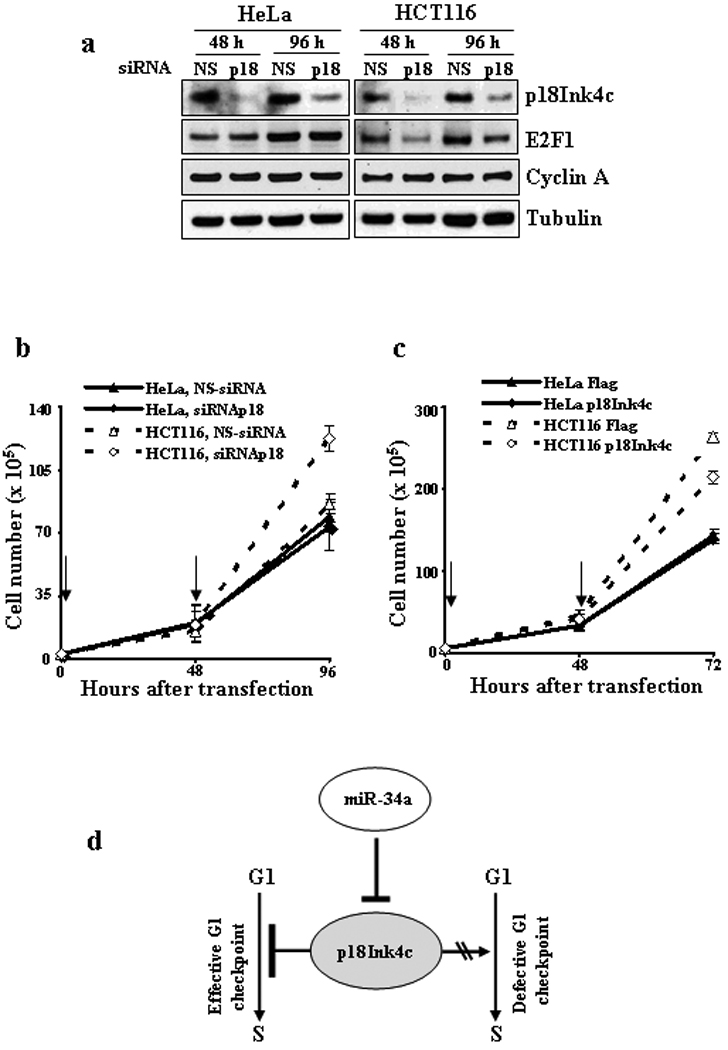
p18Ink4c does not function as a cell cycle inhibitor at G1 phase in HPV18+ HeLa cells. (a–b) siRNA knockdown of p18Ink4c expression in HPV− HCT116 cells represses E2F1 expression and promotes cell proliferation, but reduction of p18Ink4c in HeLa cells does not. Both HeLa cells and HCT116 cells were transfected with 50 nM of p18Ink4c-specific siRNA or non-specific (NS) control siRNA. Protein samples were collected at indicated time points from the transfected cells and were analyzed for the expression of p18Ink4c, E2F1 and cyclin A by Western blot (a). β-tubulin served as a loading control. Arrows in (b) mark transfection time points. Cell numbers at indicated time points after RNAi transfection were counted for each cell line in triplicates and data were averaged from two separate experiments. Only HCT116 cells treated with p18Ink4c siRNA grew faster (P<0.01 at 96 h) than the cells treated with a nonpecific control siRNA (b). (c) Overexpression of p18Ink4c suppresses cell growth of HCT116 cells, but not HeLa cells. See other details in (b). HCT116 cells with overexpressed p18Ink4c by transient transfection of a p18Ink4c expression vector grew slower (P<0.05 at 72 h) than the cells transfected with an empty vector (Flag). (d) A working model for increased p18Ink4c expression in cancers with or without a functional G1 checkpoint.
Results
miR-34a regulates p18Ink4c expression in oncogenic HPV infections
Given the fact that HPV16 and HPV18 infection inhibits the expression of tumor suppressive miR-34a by E6 destabilization of p53,11 we wish to understand mechanistically the role of this inhibition in development of cervical cancer. To search for miR-34a targets in oncogenic HPV infections, we first knocked down the expression of HPV18 E6 by an E6-specific siRNA 25 in HeLa cells. We hypothesized that knockdown of E6 expression would stabilize p53 and increase miR-34a expression, consequently, down-regulating the expression of miR-34a targets. As shown in Fig. 1a and Fig. S1a, the E6 knockdown stabilized p53 and decreased the expression of cyclin E2 and to lesser extent CDK4 and E2F1, three known miR-34a targets.8,17 Four other putative miR-34a targets predicted by TargetScan and Pictar, DLL1, Notch 1, JAG1, and ROCK1, remained unchanged (data not shown). RAD51AP1, DcR3 (TNFRSF6B, TR6), and CDKN2C (p18Ink4c) that have been reported to have altered RNA levels by a miR-34a-expressing retrovirus with RAD51AP and CDKN2C showing an increase and DcR3 showing a decrease. 9 Our examination of RAD51AP1 and DcR3 found no change at the protein level. In the E6 knockdown cells, however, we observed a substantial, dose-dependent reduction of p18Ink4c (Fig. 1a, Fig. S1a), a CDK4 and CDK6 inhibitor. This reduction was much greater than other known miR-34a targets. The level of p16Ink4a, another member of CDK4 and CDK6 inhibitors,21 was not affected, indicating the specificity. The reduction of p18Ink4c expression was further verified by E6 knockdown in an HPV16+ CaSki cell line (Fig. 1b). Together, these results suggest that p18Ink4c is a more prominent target for miR-34a downregulation.
Figure 1.
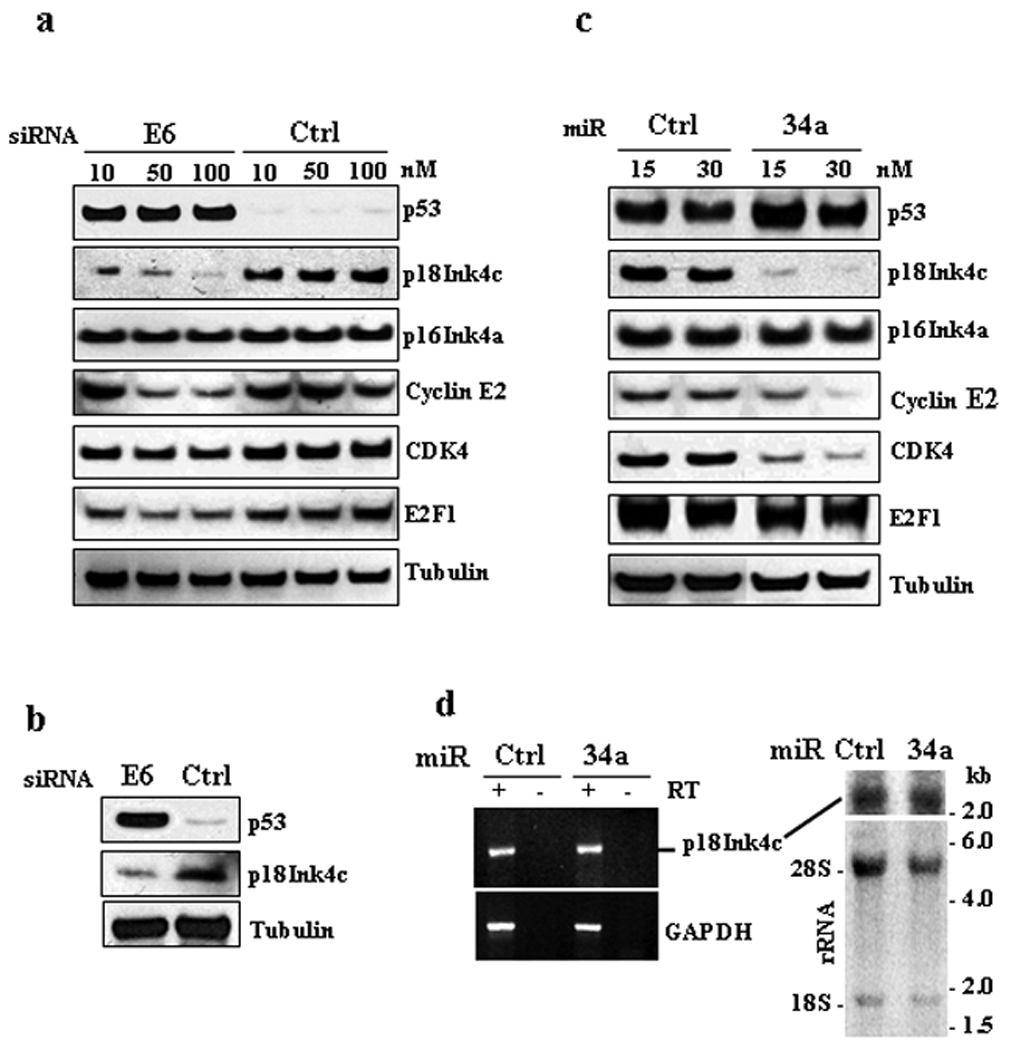
miR-34a regulates p18Ink4c expression. (a) Suppression of E6 expression in HeLa cells by an HPV18 E6-specific siRNA prevents p18Ink4c expression. HPV18+ HeLa cells 48 h after transfection with indicated doses of an HPV18 E6-specific siRNA or a control (Ctrl) siRNA were examined by Western blot for expression of indicated proteins. A relative level of each protein was quantified from each sample after being normalized to β-tubulin and is shown in Supporting Figure S1a. (b) Suppression of E6 expression in CaSki cells by an HPV16 E6-specific siRNA prevents p18Ink4c expression. HPV16+ CaSki cells 48 h after transfection with 40 nM of an HPV16-specific siRNA or a control siRNA were analyzed by Western blot for the expression of p53 and p18Ink4c. β-tubulin served as a loading control. (c), Ectopic miR-34a suppresses the expression of p18Ink4c protein. HeLa cells transfected twice with 15 or 30 nM of miR-34a or a non-specific control (Ctrl) miRNA at an interval of 48 h were examined by Western blot for the expression of indicated proteins 96 h after the first transfection. A relative level of each protein was quantified from each sample after being normalized to β-tubulin and is shown in Supporting Figure S1b. (d) Ectopic miR-34a does not affect p18Ink4c RNA level. Total RNA from HeLa cells 96 h after transfection as described in (c) with 30 nM of miR-34a or a control (Ctrl) miRNA was analyzed by RT-PCR (left) and by Northern blot (right) for p18Ink4c RNA expression. GAPDH RNA detection in RT-PCR served as a loading control. Ribosomal RNA (rRNA) in the agarose gel before blotting were stained with ethidium bromide and served as a loading control for Northern blot (right). An RNA kb marker was used as a size marker of Northern blot.
As the stabilized p53 in E6 knockdown cells has pleiotropic effects, we investigated whether p18Ink4c is a downstream target of miR-34a or p53. HPV18+ HeLa cells with ectopic expression of miR-34a were examined for the expression of p18Ink4c and other known miR-34a targets. A dose-dependent reduction of p18Ink4c protein was observed by ectopic expression of miR-34a (Fig. 1c and Fig. S1b), but a high dose (30 nM) of ectopic miR-34a showed no effect on p18Ink4c RNA as measured by RT-PCR (Fig. 1d, left panel) and by Northern blot (Fig. 1d, right panel), suggesting that the reduced expression of p18Ink4c protein was at the posttranscriptional level. Ectopic miR-34a in HeLa cells exhibited only a moderate suppression on the expression of two known miR-34a targets, cyclin E2 and CDK4,8 and no effect on E2F117 and p16Ink4a (Fig. 1c and Fig. S1b). Ectopic expression of miR-34a also had no effect on DLL1, Notch 1, JAG1, ROCK1, RAD51AP1, and DcR3 (data not shown). As positive feedback, ectopic miR-34a increased p53 expression 14,17 (Fig. 1c and Fig. S1b). Based on these observations, we conclude that miR-34a targets p18Ink4c for translational suppression.
p18Ink4c mRNA contains a miR-34a binding site in its 5' UTR
To confirm that miR-34a directly targets p18Ink4c mRNA and contributes to translational suppression of p18Ink4c, we searched p18Ink4c mRNA (GenBank accession number, AF041248) for potential miR-34a seed matches and predicted two putative miR-34a binding sites, one each in the 5' UTR and 3' UTR of p18Ink4c mRNA (Fig. S2). As the putative miR-34a seed match in the p18Ink4c 3' UTR has one nucleotide mismatch, subsequent insertion of this seed match or the entire p18Ink4c 3' UTR into the firefly luciferase reporter’s 3' UTR was found no effect on luciferase activity in the presence of ectopic miR-34a (data not shown). We then examined in the reporter plasmid the putative miR-34a binding site in the 5' UTR of p18Ink4c which has a better seed match (Fig. 2a). As shown in Figure 2b, a reduced luciferase activity was found in response to the overexpressed miR-34a, but introduction of point mutations into the seed match prevented this response, demonstrating that the miR-34a seed match in the 5' UTR of p18Ink4c is a functional binding site.
Anti-miR-34 inhibitor was further used to investigate p18Ink4c as a direct target of endogenous miR-34a in vivo. As expected, suppression of endogenous miR-34a in HeLa cells and CaSki cells with a miR-34a inhibitor increased p18Ink4c expression, whereas introduction of a miRNA inhibitor negative control into the cells had no effect (Fig. 2c).
De novo infections by oncogenic HPVs of human keratinocyte-derived raft tissues increase p18Ink4c expression
Our previous study demonstrated that miR-34a expression is decreased in oncogenic HPV infection.11 To determine whether productive infection of oncogenic HPVs increases p18Ink4c expression, we examined HPV18-infected raft tissues (Fig. 3a) derived from HFKs for the expression of virus early gene E6 and viral late gene E4 by RT-PCR. Consistent with our previous report,11 reduced miR-34a expression was observed in the raft tissues with HPV18 infection when compared to the uninfected primary rafts (Fig. 3b). As expected, a significant increase of p18Ink4c expression was found in HFK18, HVK16, and HVK18 rafts by IHC staining (Fig. 3c–d). The uninfected primary HFK and HVK rafts expressing high levels of miR-34a exhibited no or only minimal amount of p18Ink4c (Fig. 3b–d). The characteristic nuclear staining of p18Ink4c in the keratinocytes with oncogenic HPV infections indicates that the increased p18Ink4c was primarily in the nucleus. The cells with nuclear p18Ink4c staining in the raft tissues were found mostly in basal and superbasal layers and much less in granular and cornified layers. Together, our data indicate that de novo infections of oncogenic HPVs reduce miR-34a expression, thus leading to increase p18Ink4c expression.
Figure 3.
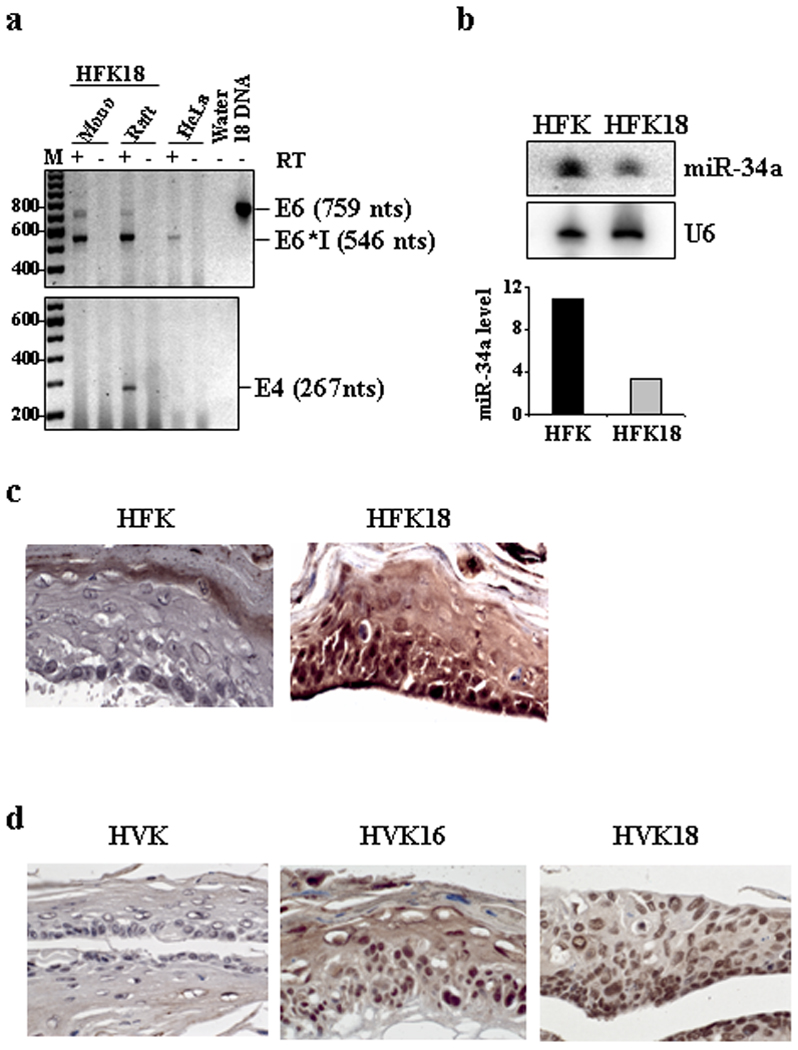
Oncogenic HPV infection reduces miR-34a expression leading to increase p18Ink4c expression. (a) HPV18 infection and gene expression in monolayer and raft cultures derived from HFKs (HFK18). Total RNA from each culture was analyzed by RT-PCR for expression of HPV18 oncogene E6 and late gene E4. Total RNA from HeLa cells served as a positive control of HPV18 E6. HPV18 DNA (18 DNA) served as a size marker for unspliced E6 RNA. (b) Downregulation of miR-34a expression in HFK rafts with HPV18 infection (HFK18). Total RNA from the raft cultures harvested on day 10 was examined by Northern blot for miR-34a expression. U6 snRNA served as a loading control. Bar graph shows relative miR-34a levels in the corresponding sample in the Northern blot after being normalized to U6 snRNA for sample loading. (c) Increased p18Ink4c expression in HFK18 raft cultures as measured by IHC staining. (d) Increased p18Ink4c expression in raft cultures derived from HVKs infected with HPV16 (HVK16) or HPV18 (HVK18) as measured by IHC staining. Tissue sections in (c) and (d) are in 20 × magnification.
Increased expression of p18Ink4c in high-grade cervical intraepithelial neoplasia and cervical cancer tissues
Finding of the increased p18Ink4c expression in productive infection of oncogenic HPVs led us to further analyze the expression of p18Ink4c in cervical tissues obtained from patients with high-grade CIN or cervical cancer. As shown in Figure 4a, increased p18Ink4c expression was also found in HPV16+ CIN II lesions and cervical cancer by IHC staining. High-risk HPV E7-mediated increase of p16Ink4a 30 was used as a positive control. The increased p18Ink4c expression in cervical cancer along with high-risk HPV E7-mediated increase of p16Ink4a 30 and cyclin E2, 31,32 a CDK2 partner protein for G1/S phase transition, was further verified by Western blotting using cell lysates from paired normal and cervical cancer tissues (Fig. 4b). As all cases of cervical cancer (99.9%) are virtually attributable to oncogenic HPV infection,1,3 the increased p18Ink4c expression in cervical cancer could be interpreted as a result from oncogenic HPV infection.
We further examined the possibility of using p18Ink4c as a biomarker for cervical cancer by IHC staining of additional cervical tissue sections. A total of 123 pathologically confirmed tissue samples in cervical tissue arrays, including 78 cancer tissues, 24 inflammation tissues, and 21 normal tissues were stained for p18Ink4c expression (Fig. 4c). We defined a sample positive for p18Ink4c staining based on its staining signal intensity above a mean signal value of all 45 non-cancer tissues plus two standard derivations, measured by using an Image J software. By using this cut-off value, positive p18Ink4c staining was found in 53 (68%) cervical cancer tissues, 2 (8.3%) cervical samples with chronic inflammation, and 1 (4.8%) normal cervical tissue (Table 1). There were no obvious differences among cervical cancer grades. The positive p18Ink4c detection rate in cervical cancer group was significantly higher (P<0.0001) than that in the normal cervical tissue group plus cervical tissues with chronic inflammation (Fig. 4d, left bar graph). In addition, the averaged signal intensity of p18Ink4c-positive staining samples was also significantly higher (P<0.0001) in cervical cancer tissues than that in the cancer-free normal tissues (Fig. 4d, right bar graph).
p18Ink4c in cervical cancer cells does not function as a cell cycle inhibitor
p18Ink4c has been shown to inhibit CDK4 and CDK6 function 21 and many types of human cancer have reduced expression of p18Ink4c.33–39 An increased expression of p18Ink4c in cervical cancer due to viral E6-mediated decrease of p53 and miR-34a would be expected to prevent cancer cell growth and inhibit the development of cervical cancer. We proposed that viral E7 inactivates pRB 4,40 inducing disassociation of E2F from pRB and this results in disruption of normal G1 checkpoint 40. Therefore, the defect of p18Ink4c function in cervical cancer cells may be attributable to viral E7 expression. To test this hypothesis, we knocked down p18Ink4c expression by siRNA in HPV18+ HeLa cells and in HPV− HCT116 cells, a colon cancer cell line that contains wt pRB 41 and retains an intact G1 checkpoint, and examined the effect of p18Ink4c knockdown on the expression of E2F1 and cyclin A, two downstream pRB targets. As shown in Fig. 5a and Fig. S3, E2F1 expression was repressed upon p18Ink4c knockdown in HCT116 cells, but not in HeLa cells. Cyclin A only showed a small increase at 48 h of p18Ink4c knockdown in HCT116 cells. As expected, knocking down p18Ink4c expression promoted the growth of HPV− HCT116 cells, but had no effect on HPV18+ HeLa cells expressing viral E7 25 (Fig. 5b). Consistent with this, overexpression of p18Ink4c inhibited the growth of HCT116 cells, but not HeLa cells (Fig. 5c). Based on these results, we conclude that increased expression of p18Ink4c could not lead to tumor suppression when cancer cells lack a G1 checkpoint, but does so when the G1 checkpoint is intact (Fig. 5c).
Discussion
In this report, we identified p18Ink4c as a possible biomarker for cervical precancer lesions and cervical cancer, which is upregulated by oncogenic HPV E6 via p53-miR-34a pathway. p18Ink4c is an important member of the mammalian INK4 family of CDK4 and CDK6 inhibitors and functions as a tumor suppressor preventing cell cycle progression from G1 to S phase. 42,43 Loss or reduced expression of p18Ink4c has been found in many human cancers 36,38 and was proven experimentally to promote the development of various malignancies.35,43 Regulation of p18Ink4c expression has been described at the transcriptional level. Several cellular transcription factors, E2F, EKLF1, and GATA3, are involved in the positive or negative transactivation of p18Ink4c expression.28,35,44 In this report, we demonstrated that regulation of p18Ink4c expression also takes place at the posttranscriptional level by the cellular miRNA, miR-34a. We found that ectopic miR-34a expression or stabilization of p53 to induce endogenous expression of miR-34a in HPV18+ HeLa cells led to reduction of p18Ink4c. In contrast, suppression of miR-34a activity by an anti-miR-34a inhibitor enhanced p18Ink4c expression.
However, our observation conflicts with a recent report in which miR-34a appeared to increase CDKN2C (p18Ink4c) RNA level in HCT116 cells transduced by a miR-34a-expressing retrovirus.9 HCT116 cell line is an HPV− colon cancer cell line that expresses wt p53 and an increased level of wt pRB.41 To determine that the discrepancy was not a result of using different cell lines, we analyzed HCT116 cells (Fig. S4a–b) along side HeLa cells (Fig. 1c–d) for p18Ink4c expression subsequent to miR-34a transfection. A similar reduction of p18Ink4c protein was observed in HCT116 cells without alteration in p18Ink4c RNA levels as determined by RT-PCR (Fig. S4a–b). These results reaffirm the ability of miR-34a to repress p18Ink4c expression at the posttranscriptional level in both HPV+ and HPV− cells.
Despite the increased expression in cervical cancer and in HPV-containing head and neck cancer,45,46 p18Ink4c exerts no suppressive roles in cancer cell growth and tumor development. We demonstrated that the suppressive activity of p18Ink4c relies on a functional G1 checkpoint in cells with normal expression of wt pRB (Fig. 5d). Oncogenic HPV infection results in viral E7 destabilization of pRB essential for cyclin D and CDK4 or CDK6 to function 40 and thereby creating a dysfunctional G1 checkpoint. In this environment, p18Ink4c has no inhibitory effect on cyclin D/CDK4 or CDK6 activities in HPV-induced cancers. This was supported by the observation that HPV18+ HeLa cells continued proliferating in the presence of high levels of p18Ink4c, but HCT116, a cell line expressing wt pRB, did not. We confirmed that HeLa cells even express naturally a much higher level of p18Ink4c when compared with HCT116 cells (Fig. S4c). In addition, overexpression of p18Ink4c in HCT116 cells suppressed the cell growth. Consistent with this, ectopic expression of p18Ink4c was found to inhibit the cell growth of U2OS cells expressing wt pRB, but not Saos-2 cells lacking pRB due to a deletion in exon 21–27 of the RB1 gene.42 Thus, our findings in this report support the concept that cancer cells with destabilized pRB can continue to proliferate with p18Ink4c overexpression, similar to the upregulation of p16Ink4a by the high-risk E7.30,47 It is worth noting that the expression of both p18Ink4c and p16Ink4a is transactivated by E2F transcription factors.28,48 Viral oncoprotein E7 destabilizes pRB leading to dissociate E2F from pRB in the pRB/E2F complex and thereby enhances transcription of both p18Ink4c and p16Ink4a. We showed in this report that translation of p18Ink4c, but not p16Ink4a, further depends on viral E6 destabilization of p53 to reduce miR-34a expression. Thus, our data provides the compelling evidence distinguishing p18Ink4c from p16Ink4a in its expression by both E6 and E7 regulation.
We have demonstrated that p18Ink4c knockdown had no effect on E2F1 expression in HPV18+ HeLa cells with a disrupted pRB pathway by viral E7, but led to a remarkable reduction of E2F1 in HPV− HCT116 cells with a wt pRB (Fig. 5a). Ectopic expression of miR-34a in HeLa cells also had no effect on E2F1 expression (Fig. 1c). Together, these data provide evidence that E2F1 expression appears pRB-independent in HeLa cells and pRB-dependent in HCT116 cells. Because p18Ink4c knockdown increases CDK4/6 activities, this leads to promote pRB phosphorylation and thereby dissociate pRB with E2F1 in HCT116 cells. Despite that E2F1 transactivates the genes for DNA synthesis in S phase and also functions as a tumor suppressor to prevent cell proliferation, the dissociated E2F1 subjects to cell cycle regulation. E2F1 decreases at the late S phase and is proteolytically degraded in G2/M phase. 49 Although not fully understood, this is perhaps one of the mechanisms by which p18Ink4c knockdown lead to E2F1 reduction in HCT116 cells. In this regard, cyclin A expression appears less sensitive than E2F1 to p18Ink4c knockdown. We observed only a minimal effect on cyclin A expression in HCT116 cells with p18Ink4c knockdown.
The ability of HPV16 and HPV18 infection to suppress miR-34a 11 and to increase p18Ink4c was verified in raft cultures with productive virus infection. Increased expression of p18Ink4c was also observed in about 70% of cervical cancer. As persistent oncogenic HPV infection is a leading cause of cervical cancer, a positive p18Ink4c staining in cancer-free cervical tissues might suggest a transient or early HPV infection. However, the negative p18Ink4c staining in some of cervical cancer in our study remains to be understood and might relate to tissue sample preparation or low levels of high-risk E6 and E7 expression 50. Nevertheless, the finding that p18Ink4c is overexpressed in cervical cancer suggests that p18Ink4c may serve as a possible biomarker, in addition to p16Ink4a, for cervical precancer lesion diagnosis and cervical cancer early detection.
Supplementary Material
Acknowledgments
We thank Alexandre Blais of Ottawa Institute of Systems Biology, University of Ottawa for providing human p18Ink4c expression plasmids and Greg Hannon of Cold Spring Harbor Laboratory for providing miR-34a CDK4-luciferase reporter. We also thank Rick Dreyfuss of NIH Medical Arts and Media Center for his excellent assistance of tissue section photography. This study was supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, and the Center for Cancer Research. This work was also supported partially by a grant (AI057988 to C.M.) from US National Institutes of Health.
Abbreviations
- HPV
human papillomaviruses
- CDK
cyclin-dependent kinase
- miRNA
microRNAs
- UTRs
untranslated regions
- DMEM
Dulbecco’s modified Eagle’s medium
- RIPA buffer
radioimmunoprecipitation assay buffer
- INK4
inhibitor of cyclin-dependent kinase 4
- HFKs
human foreskin keratinocytes
- HVK
human vaginal keratinocytes
- TBS
Tris-buffered saline
- SDS
sodium dodecyl sulfate
- IHC staining
immunohistochemical staining
- CIN
cervical intraepithelial neoplasia
Footnotes
Two brief statements: 1. p18Ink4c is a downstream target of miR-34a; 2. Increased expression of p18Ink4c, like p16Ink4a, in HPV16 and HPV18 infected cells and cervical cancer may be used as a biomarker for diagnosis of HPV infections.
References
- 1.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine. 2006;24 Suppl 3 doi: 10.1016/j.vaccine.2006.05.111. S3-11-S3/25. [DOI] [PubMed] [Google Scholar]
- 3.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 4.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 6.Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5' UTR as in the 3' UTR. Proc Natl Acad Sci U S A. 104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Wang H-K, McCoy JP, Banerjee NS, Rader JS, Broker TR, Meyers C, Chow LT, Zheng ZM. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA. 2009;15:637–647. doi: 10.1261/rna.1442309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- 13.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 14.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008;582:1564–1568. doi: 10.1016/j.febslet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 16.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 17.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shenouda SK, Alahari SK. MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009;28:369–378. doi: 10.1007/s10555-009-9188-5. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3:e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–6043. doi: 10.1158/0008-5472.CAN-06-0561. [DOI] [PubMed] [Google Scholar]
- 21.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin-Drubin ME, Meyers C. Propagation of infectious, high-risk HPV in organotypic "raft" culture. Methods Mol Med. 2005;119:171–186. doi: 10.1385/1-59259-982-6:171. [DOI] [PubMed] [Google Scholar]
- 23.Guo M, Gong Y, Deavers M, Silva EG, Jan YJ, Cogdell DE, Luthra R, Lin E, Lai HC, Zhang W, Sneige N. Evaluation of a commercialized in situ hybridization assay for detecting human papillomavirus DNA in tissue specimens from patients with cervical intraepithelial neoplasia and cervical carcinoma. J Clin Microbiol. 2008;46:274–280. doi: 10.1128/JCM.01299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majerciak V, Kruhlak M, Dagur PK, McCoy JP, Jr, Zheng ZM. Caspase-7 cleavage of Kaposi sarcoma-associated herpesvirus ORF57 confers a cellular function against viral lytic gene expression. J Biol Chem. 2010;285:11297–11307. doi: 10.1074/jbc.M109.068221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang S, Tao M, McCoy JP, Jr, Zheng ZM. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol. 2006;80:4249–4263. doi: 10.1128/JVI.80.9.4249-4263.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang S, Tao M, McCoy JP, Zheng ZM. Short-term induction and long-term suppression of HPV16 oncogene silencing by RNA interference in cervical cancer cells. Oncogene. 2006;25:2094–2104. doi: 10.1038/sj.onc.1209244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blais A, Labrie Y, Pouliot F, Lachance Y, Labrie C. Structure of the gene encoding the human cyclin-dependent kinase inhibitor p18 and mutational analysis in breast cancer. Biochem Biophys Res Commun. 1998;247:146–153. doi: 10.1006/bbrc.1998.8497. [DOI] [PubMed] [Google Scholar]
- 28.Blais A, Monte D, Pouliot F, Labrie C. Regulation of the human cyclin-dependent kinase inhibitor p18INK4c by the transcription factors E2F1 and Sp1. J Biol Chem. 2002;277:31679–31693. doi: 10.1074/jbc.M204554200. [DOI] [PubMed] [Google Scholar]
- 29.Tang S, Yamanegi K, Zheng ZM. Requirement of a 12-base-pair TATT-containing sequence and viral lytic DNA replication in activation of the Kaposi's sarcoma-associated herpesvirus K8.1 late promoter. J Virol. 2004;78:2609–2614. doi: 10.1128/JVI.78.5.2609-2614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, Dallenbach-Hellweg G, Schmidt D, von Knebel DM. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001;92:276–284. doi: 10.1002/ijc.1174. [DOI] [PubMed] [Google Scholar]
- 31.Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B, Jansen-Durr P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J Virol. 1995;69:6389–6399. doi: 10.1128/jvi.69.10.6389-6399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zariwala M, Liu J, Xiong Y. Cyclin E2, a novel human G1 cyclin and activating partner of CDK2 and CDK3, is induced by viral oncoproteins. Oncogene. 1998;17:2787–2798. doi: 10.1038/sj.onc.1202505. [DOI] [PubMed] [Google Scholar]
- 33.Uziel T, Zindy F, Sherr CJ, Roussel MF. The CDK inhibitor p18Ink4c is a tumor suppressor in medulloblastoma. Cell Cycle. 2006;5:363–365. doi: 10.4161/cc.5.4.2475. [DOI] [PubMed] [Google Scholar]
- 34.Wiedemeyer R, Brennan C, Heffernan TP, Xiao Y, Mahoney J, Protopopov A, Zheng H, Bignell G, Furnari F, Cavenee WK, Hahn WC, Ichimura K, et al. Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell. 2008;13:355–364. doi: 10.1016/j.ccr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY, Ho IC, Perou CM, Xiong Y. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell. 2009;15:389–401. doi: 10.1016/j.ccr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hossain MG, Iwata T, Mizusawa N, Qian ZR, Shima SW, Okutsu T, Yamada S, Sano T, Yoshimoto K. Expression of p18(INK4C) is down-regulated in human pituitary adenomas. Endocr Pathol. 2009;20:114–121. doi: 10.1007/s12022-009-9076-0. [DOI] [PubMed] [Google Scholar]
- 37.Pei XH, Bai F, Smith MD, Xiong Y. p18Ink4c collaborates with Men1 to constrain lung stem cell expansion and suppress non-small-cell lung cancers. Cancer Res. 2007;67:3162–3170. doi: 10.1158/0008-5472.CAN-06-4517. [DOI] [PubMed] [Google Scholar]
- 38.Saab R, Rodriguez-Galindo C, Matmati K, Rehg JE, Baumer SH, Khoury JD, Billups C, Neale G, Helton KJ, Skapek SX. p18Ink4c and p53 Act as tumor suppressors in cyclin D1-driven primitive neuroectodermal tumor. Cancer Res. 2009;69:440–448. doi: 10.1158/0008-5472.CAN-08-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leone PE, Walker BA, Jenner MW, Chiecchio L, Dagrada G, Protheroe RK, Johnson DC, Dickens NJ, Brito JL, Else M, Gonzalez D, Ross FM, et al. Deletions of CDKN2C in multiple myeloma: biological and clinical implications. Clin Cancer Res. 2008;14:6033–6041. doi: 10.1158/1078-0432.CCR-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto H, Soh JW, Monden T, Klein MG, Zhang LM, Shirin H, Arber N, Tomita N, Schieren I, Stein CA, Weinstein IB. Paradoxical increase in retinoblastoma protein in colorectal carcinomas may protect cells from apoptosis. Clin Cancer Res. 1999;5:1805–1815. [PubMed] [Google Scholar]
- 42.Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O'Keefe CL, Matera AG, Xiong Y. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939–2952. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 43.Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tallack MR, Keys JR, Perkins AC. Erythroid Kruppel-like factor regulates the G1 cyclin dependent kinase inhibitor p18INK4c. J Mol Biol. 2007;369:313–321. doi: 10.1016/j.jmb.2007.02.109. [DOI] [PubMed] [Google Scholar]
- 45.Slebos RJ, Yi Y, Ely K, Carter J, Evjen A, Zhang X, Shyr Y, Murphy BM, Cmelak AJ, Burkey BB, Netterville JL, Levy S, et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:701–709. doi: 10.1158/1078-0432.CCR-05-2017. [DOI] [PubMed] [Google Scholar]
- 46.Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, Chen Q, Brandwein-Gensler M, Prystowsky MB, Childs G, Smith RV, Belbin TJ. Gene expression profiles in HPV-infected head and neck cancer. J Pathol. 2007;213:283–293. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 47.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:2221–2226. doi: 10.1073/pnas.94.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ressler S, Scheiden R, Dreier K, Laich A, Muller-Holzner E, Pircher H, Morandell D, Stein I, Viertler HP, Santer FR, Widschwendter A, Even J, et al. High-risk human papillomavirus E7 oncoprotein detection in cervical squamous cell carcinoma. Clin Cancer Res. 2007;13:7067–7072. doi: 10.1158/1078-0432.CCR-07-1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.