

Abstract
Alzheimer's disease (AD) is the most common age related neurodegenerative disease. Currently, there are no disease modifying drugs, existing therapies only offer short-term symptomatic relief. Two of the pathognomonic indicators of AD are the presence of extracellular protein aggregates consisting primarily of the Aβ peptide and oxidative stress. Both of these phenomena can potentially be explained by the interactions of Aβ with metal ions. In addition, metal ions play a pivotal role in synaptic function and their homeostasis is tightly regulated. A breakdown in this metal homeostasis and the generation of toxic Aβ oligomers are likely to be responsible for the synaptic dysfunction associated with AD. Therefore, approaches that are designed to prevent Aβ metal interactions, inhibiting the formation of toxic Aβ species as well as restoring metal homeostasis may have potential as disease modifying strategies for treating AD. This review summarizes the physiological and pathological interactions that metal ions play in synaptic function with particular emphasis placed on interactions with Aβ. A variety of therapeutic strategies designed to address these pathological processes are also described. The most advanced of these strategies is the so-called ‘metal protein attenuating compound’ approach, with the lead molecule PBT2 having successfully completed early phase clinical trials. The success of these various strategies suggests that manipulating metal ion interactions offers multiple opportunities to develop disease modifying therapies for AD.
Keywords: copper, zinc, amyloid, synaptic toxicity, homeostasis, oxidative stress, chelator, bioinorganic chemistry, drug
Introduction
Dementia is characterized by the loss of or decline in memory and other cognitive abilities. Alzheimer's disease (AD) (Alzheimer et al., 1995) is the most common type of dementia and accounts for an estimated 60–80% of cases. AD is an irreversible, progressive neurodegenerative disorder leading invariably to death usually within 7–10 years of diagnosis. Age is the dominant risk factor in AD, and the progressive nature of neurodegeneration ultimately leads to synaptic failure and neuronal damage (Masters and Beyreuther, 1988) in cortical areas of the brain essential for memory and higher mental functions. The increase in the number of new cases of AD is the direct consequence of an improvement in life expectancy. There are currently 18 million people worldwide with AD and this figure is projected to nearly double by 2025 to 34 million (Alzheimer's Association, 2010). In the USA alone the estimated annual cost of caring for over 5 million AD patients is $172 billion (Alzheimer's Association, 2010). Besides the monetary cost, the spouses, relatives and friends of AD patients experience emotional, physical and financial stresses during the years of caregiving that is impossible to quantify. Clearly, AD represents a major socio-economic problem which requires better assessment, management and effective therapies in order to ease the burden of the disease. However, despite an ever increasing knowledge of genetics, epidemiology, risk factors and neuropathological mechanisms, there is still no cure for AD.
The pathognomonic indicators of disease are the presence of senile plaques (Kromer Vogt et al., 1990), neurofibrillary tangles, neurophil threads, amyloid-β peptide (Aβ) deposition and selective loss of neurons and decreased synaptic density in the post-mortem brain. Deposition of amyloid plaques in AD is most obvious pathological feature; the major constituent of these deposits being the Aβ peptide that is proteolytically cleaved from the membrane bound amyloid precursor protein (APP). Genetic evidence from cases of familial AD indicates that Aβ metabolism is linked to the disease. While the precise function of APP is still debated; evidence suggests it has functions in maintaining metal homeostasis. These include roles in modulating Cu efflux from cells (Bellingham et al., 2004) and acting as a ferroxidase (Duce et al., 2010). This ferroxidase activity of APP is inhibited by Zn.
Over the past 20 years, there has been mounting evidence implicating aberrant metal biochemistry in the pathogenesis of AD. As the data have been accumulated there has also been a growing awareness of the critical role that transition metal ions play in modulating neurotransmission at the glutamatergic synapses in the cortex and hippocampus, processes that are pivotal to properly functioning memory. Therefore, the ability to manipulate metal homeostasis and localization offers the potential for the development of new therapeutic strategies for the treatment of AD. It is this metal biology and therapeutic strategies that is the subject of this review.
Metals and the synapse
Synaptic dysfunction has been implicated as the primary cause of the memory deficits associated with AD (Selkoe, 2002). Cu and Zn have been reported to play key roles in regulating synaptic function. Zn is released in high µM concentrations from presynaptic glutamatergic nerve terminals into the synaptic cleft upon neuronal activation. This pool of synaptic Zn interacts with a variety of receptors, ion channels and transporters. Zn is co-released into the synapse with the neurotransmitter glutamate. While glutamate is known to stimulate the N-methyl-D-aspartate receptor (NMDAr), Zn inhibits NMDAr in a voltage dependent and voltage independent manner, implicating the metal in a modulatory role (Smart et al., 2004). The zinc transporter protein ZnT3 is primarily responsible for loading Zn into the synaptic vesicles. A recent study examining the effects of knocking out ZnT3 in mice found that these animals had age dependent deficits in learning and memory that were associated with decreased levels of a range of synaptic proteins including SNAP25, PSD25 and NMDAr subunits 2a and b (Adlard et al., 2010). These results lead the authors to suggest that these knockout animals represented a ‘phenocopy for the synaptic and memory deficits of Alzheimer's disease’. This statement was further supported by evidence that levels of this protein are significantly decreased in human brain tissue as a consequence of ageing and in AD. Cu also plays a role in regulating synaptic function as it is released from the post-synaptic side of the cleft following NMDA activation, this release is facilitated by trafficking of ATP7a, mutations in which can cause Menkes disease (Schlief et al., 2006a,b;).
While metals play a role in a variety of essential biological functions the transport and utilization of these are generally tightly regulated as the aspects of their chemistry that make them physiologically useful can also give rise to pathological phenomena. The classic example of this is the storage, transport and utilization of oxygen, the same chemistry that allows metal-based enzymes to do this is essentially the same chemistry that facilitates the production of toxic reactive oxygen/nitrogen species that gives rise to oxidative stress. As a result, nature has evolved a complex system of metal transporters and chaperones that tightly regulate metal homeostasis and pools of ‘free’ metal ions are rare, where in a biological context we mean that ‘free’ metals are bound by labile low affinity and therefore readily exchangeable ligands. Interestingly, it appears that both the Zn that is released from the presynaptic terminals and Cu that is released from the post-synaptic terminals following synapse stimulation are predominately in a ‘free’ exchangeable form. In healthy tissue these released metal ions will be retaken up by the cells by metal chaperones such as metallothionein-3.
Aβ interactions with Cu/Zn
Another molecule that is known to be released into the synapse is Aβ; therefore, the synapse is one location where Aβ and ‘free’ Cu and Zn are present in sufficiently high quantities to promote an interaction. Aβ deposition begins within the synapse (Terry, 1996), and the amyloid plaques can be described as metal sinks as remarkably high concentrations of Cu (∼400 µM), Zn (∼1 mM) and Fe (∼1 mM) have been reported in senile plaques isolated from AD subjects (Lovell et al., 1998). The potential role that metals may play in amyloid plaque formation is highlighted by the finding that age- and female-sex-related plaque formation in Tg2576 transgenic mice was reduced by genetic ablation of the ZnT-3 protein (Lee et al., 2002).
Two of the pathognomonic features of AD; Aβ deposition and oxidative stress can potentially be attributed to Aβ's interactions with metal ions. Synthetic Aβ will react with both Zn and Cu to form aggregates while Aβ will react with Cu to generate reactive oxygen species (ROS) (Bush, 2003; Smith et al., 2007). In vitro studies utilizing synthetic Aβ have shown that low µM levels of Zn2+ will induce protease-resistant aggregation and precipitation of Aβ (Bush et al., 1994; Huang et al., 1997; 1999; 2000;). Huang et al. (1997) tested the Zn induced Aβ aggregation for reversibility in vitro and found that low concentration of Zn2+ (20 nmol) induced protease-resistant (β-sheet) aggregation and precipitation of Aβ; this aggregation could be reversed by the use of metal chelators. Similar phenomena were observed with Cu2+ and Fe3+ at physiological conditions (pH = 7) and the aggregation was exaggerated at acidic pH (Atwood et al., 1998).
Aβ coordination of Cu leads to the generation of ROS involving the reduction of the oxidation state of the coordinated Cu2+ to Cu+ and this reduction must be accompanied by the oxidation of another moiety, endogenous molecules such as thiols, ascorbate, lipid or Aβ side-chains (Butterfield et al., 2001; Turnbull et al., 2001; Barnham et al., 2003a,b; Puglielli et al., 2005). Mass spectrometry has shown that Cu2+ ions are able to oxygenate Aβ at a number of different residues including the histidine residues 6, 13, 14 and methionine 35 (Atwood et al., 2000; Schoneich, 2002; Barnham et al., 2003a). Other redox mediated modifications include aldehyde adducts to the lysine residues (Chen et al., 2007) and tyrosine modified to moieties such as DOPA, dopamine, dopamine quinone, dihydroxyindol and isodityrosine (Ali et al., 2005; 2006;). Tyrosine is particularly susceptible to free radical attack due to the conjugated aromatic ring. Elevated dityrosine and 3-nitrotyrosine within the neuronal lesions in AD brain have been reported. In vitro Aβ42 in the presence of Cu2+ and H2O2 forms dityrosine cross-linked oligomers (Barnham et al., 2004; Atwood et al., 2004), a modification that is resistant to proteolysis. The formation of dityrosine linkage in Aβ facilitates further peptide aggregation, leading to the formation of higher order oligomers.
Aβ toxicity
A variety of hypotheses have been proposed for the mechanism by which Aβ induces its neurotoxic effects, the one point of consensus most hypotheses have is the requirement for Aβ to aggregate. Nearly every conceivable aggregate from dimer to fully formed fibrillar structures have been reported as toxic. In general, the various aggregates have been very poorly characterized, though there is a growing interest in soluble oligomers, which appear to be particularly toxic (Lesne et al., 2006). It is possible that covalent cross-linking of Aβ (e.g. dityrosine formation generated by Cu oxidation) contributes to the formation of these toxic soluble species (Barnham et al., 2004). Cu has been shown to potentiate the toxicity of Aβ to neuronal cell cultures (Atwood et al., 1999; Duce et al., 2006; Fodero-Tavoletti et al., 2007). Zn induced Aβ oligomers have been shown to elicit toxic responses in hippocampal brain slices via interactions with the NMDAr (Deshpande et al., 2009). The detailed mechanisms of Aβ toxicity have recently been described elsewhere (Cappai and Barnham, 2008).
Cu/Zn binding sites on Aβ
As the metal ions Cu and Zn are able to modulate Aβ structure and function there has been much interest in delineating the nature of the metal coordination site. Aβ possesses histidine residues at positions 6, 13 and 14, which form a structural element that enables Aβ to coordinate transition metal ions. A variety of spectroscopic studies have confirmed that the histidine residues constitute the principal site(s) of metal coordination. However, many of these studies have given contradictory results and failed to define a definitive structure for the metal binding site(s). Drew et al. (2009a,b;) were able to explain these apparent contradictions in the literature using a combination of multifrequency electron paramagnetic resonance and site specific 15N- and 13C-labelling which indicated that there was not a single metal binding but rather an interconverting ensemble of structures. This concept of the Cu coordination site being pleomorphic was further validated by the results from the Faller group (Dorlet et al., 2009; Hureau et al., 2009).
The difficulties in defining the metal binding site(s) on Aβ have been mirrored by the various attempts to define the metal binding affinity. As Aβ metal interactions lead to peptide aggregation and precipitation this complicates attempts to define thermodynamic parameters for these interactions. As a result, a variety of binding constants for Aβ/metal interactions have been published, with Kds ranging from high µM to aM. The experimental difficulties associated with obtaining accurate thermodynamic data have recently reviewed (Xiao et al., 2010). The current best estimates for the binding affinity for Aβ with Zn is in low µM range while for Cu it is in the sub nM range (Faller, 2009). A more detailed review of the various spectroscopic characterizations of the Aβ metal binding site and critical appraisal the determination of associated binding affinities has recently been published (Faller, 2009).
Therapeutic strategies
Current medications (United States Food and Drug Administration approved) that have been developed for the treatment of AD include inhibitors of acetylcholinesterase activity (Birks et al., 2006) and antagonists of NMDAr (Areosa et al., 2005). At best these drugs are able to produce modest symptomatic improvements in some patients; they do not cure or stop disease progression (Takeda et al., 2006). As a result, there is a need to search for new therapeutic strategies that target the underlying pathogenic mechanisms in AD.
The majority of attempts to develop disease modifying drugs for AD have centred on reducing levels of Aβ in the brain. This can be achieved by either lowering the levels of Aβ production by inhibiting the processing of APP by either beta-amyloid converting enzyme or γ-secretase or increasing clearance primarily through the use of immunization. To date, both approaches have had toxicity issues hinder development. For example, recently a large scale clinical trial of the Eli Lilly's γ-secretase inhibitor semagacestat had to be halted due to the increased risk of skin cancer and worsening cognitive performance (Extance, 2010). Similarly, the first active immunotherapy trial targeting Aβ had to be halted due to a number of patients developing meningoencephalitis (Orgogozo et al., 2003).
Metal protein attenuating compounds for AD
One approach to developing potential disease modifying drugs for AD is to inhibit the interactions between Aβ and metals that drive the pathological features of AD such as amyloid deposition and oxidative stress. Broadly speaking, there are two potential classes of molecule capable of preventing such interactions. The first involves inhibiting Aβ/metal interactions by selectively occupying the metal binding site on Aβ, thus preventing metal coordination. The initial metal binding site constitutes His-6, His-13 and His-14. However, these residues are natively unstructured in the absence of metal ions, and as a result the design of classic ‘lock and key’ inhibitors is problematic. However, alternative metal based approaches will be discussed later in the review. A second pharmacological approach is to identify a class of molecules that will effectively compete with the peptide for the metal ions. Metal chelators have been used clinically for decades to target metal overload diseases such as haemochromatosis (Fe) and Wilson's disease (Cu). The chelators used to treat these diseases such as desferrioxime, penicillamine and trientine are intended to deplete metal from overloaded tissue and facilitate its excretion. As such these chelators have very high metal binding affinities and are hydrophilic. These types of chelators are inappropriate as potential therapeutics for AD. Firstly, the drug has to cross the blood brain barrier and this generally requires small hydrophobic molecules. Secondly, while there are some reports of increases in metal levels in the AD brain, there are equally as many reports that dispute this. As such AD could not be described as a metal overload disease, but perhaps is better described as one where metal homeostasis is perturbed resulting in miss location of metal ions. As these metals are involved in essential neurological functions just removing them with high affinity chelators is potentially problematic.
One goal is to develop compounds that target this mislocated metal bound to Aβ and restore it to where it came from, that is, within the synapse. To differentiate this approach from the high affinity metal depleting chelators traditionally used in medicine this approach has been labelled metal protein attenuating compounds (MPACs). The prototypic MPAC is clioquinol (CQ, 5-chloro-7-iodo-8-hydroxyquinoline) (Figure 1), a small hydrophobic molecule that has moderate affinity for metal ions and is capable of crossing the blood-brain barrier. In vitro studies (Cherny et al., 2001) showed that CQ was able to inhibit metal mediated aggregation and ROS production by Aβ. CQ was then given orally in a blinded study to Tg2576 transgenic mice, the results showed a 49% decrease in brain Aβ burden compared with non-treated controls after 9 weeks of treatment. Moreover, treatment did not lead to a systematic decrease in metal levels, which is most probably due to the drug's moderate binding affinities (Cherny et al., 2001). The success of CQ in the transgenic mouse trials encouraged the testing of this molecule in clinical trials. The effect of oral CQ treatment in a randomized, double-blind, placebo-controlled pilot Phase II clinical trial (Ritchie et al., 2003) of moderately severe AD patients was evaluated. The results showed a statistically significant prevention of cognitive deterioration during a 36 week period in the more severely affected patients (baseline Alzheimer's Disease Assessment Scale – cognitive subscale ≥25). There was also a significant decline in plasma Aβ42 in the CQ group compared with an increase in the placebo group. Subsequent clinical studies of this compound were not pursued due to manufacturing difficulties associated larger scale chemical synthesis.
Figure 1.
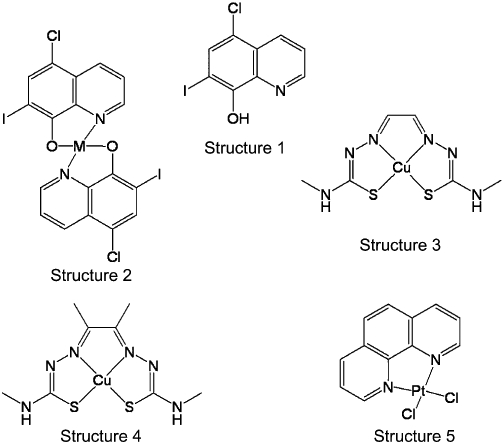
Representative examples of the molecular structures of compounds discussed within this review. Structure 1 is clioquinol (CQ, 5-chloro-7-iodo-8-hydroxyquinoline), the prototypic metal protein attenuating compound (MPAC). Structure 2 is a metal complex of CQ (M = Cu or Zn), which illustrates that the MPAC metal complexes are neutral hydrophobic molecules capable of delivering metals across cellular membranes. Structures 3 and 4 are examples of the M(bis(thiosemicarbazone)) complexes. Structure 3 is glyoxalbis(N (4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(gtsm)). This is a neutral metal complex capable of crossing cellular membranes. In an extracellular environment this complex is very stable; however, intracellularly the oxidation state of the metal is reduced from Cu2+ to Cu+, which destablizes the complex and the metal is made bioavailable. Structure 4 is diacetylbis(N (4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(atsm)). The two additional methyl groups sufficiently alter the redox potential of the metal so there is no intracellular reduction and the complex remains intact (i.e. the metal does not become bioavailable). Structure 5 PtII(1,10-phenanthroline)Cl2 is a representative example of the L-PtCl2 compounds that are designed to coordinate to Aβ and inhibit its toxic interactions.
A novel second generation MPAC PBT2 has been synthesized (Barnham et al., 2003b) that has higher solubility and increased BBB permeability as compared with CQ. When tested in the APP/PS1 transgenic mouse model of AD (Adlard et al., 2008) PBT2 decreased soluble interstitial Aβ within hours, this was accompanied by improved cognitive performance. Moreover, there were significant decreases in insoluble Aβ load and tau phosphorylation. A significant increase in synaptophysin levels was also observed indicating improved synaptic health. Following these successful animal studies PBT2 completed a randomized, double-blind, placebo-controlled Phase II clinical trial in 78 subjects with early AD over a 12 week treatment period. This study demonstrated safety and tolerability at 50 mg and 250 mg daily doses, and reduced CSF levels of Aβ1–42 at the 250 mg dose. Improved cognitive performance in patients taking PBT2 (compared with placebo) was observed when tested on executive function in the Neuropsychological Test Battery (Lannfelt et al., 2008; Faux et al., 2010).
While PBT2 and CQ were originally identified as molecules capable of inhibiting Aβ metal interactions recent results have suggested that these compounds are able to induce neuroprotective signalling cascades (White et al., 2006). When 8-hydroxyquinolines coordinate metals the proton of the phenol group is lost such that a coordination complex of either Cu2+ or Zn2+ with two 8-hydroxyquinoline ligands results in a neutral hydrophobic complex (Figure 1) that is capable of crossing cell membranes (Caragounis et al., 2007; Adlard et al., 2008). Upon entering cells these complexes can induce metal-dependent activation of PI3K and JNK, resulting in JNK-mediated up-regulation of metalloprotease activity (White et al., 2006; Caragounis et al., 2007; Adlard et al., 2008). These proteases are able to degrade Aβ. There is also an increase in phosphorylation of GSK3β. This is an inhibitory phosphorylation of one of the kinases that phosphorylates the tau protein. The ability of the MPACs to inhibit toxic Aβ metal interactions and also promote neuroprotective signalling by transporting these metals into cells suggests that the actions of these compounds rather than acting as simple chelators is much more like that of metal chaperones where they protect against toxic reactions and at the same time promote beneficial interactions.
In addition to Aβ, Tau and GSK3β have also been identified as potential therapeutic targets for AD. To date, the various strategies designed to target these proteins have been very much of a ‘slice and dice’ approach; that is, each protein has been targeted independently of the others. The data that are emerging from the MPAC strategy suggest that this is a more holistic approach to AD capable of having beneficial effects on a number of different protein targets implicated in the pathogenesis of AD.
The encouraging clinical results achieved by CQ and PBT2, has resulted in the metal hypothesis for AD gaining increased scientific support promoting a growing awareness of the therapeutic potential of targeting metal/Aβ-interactions. This in turn has prompted a number of groups to produce a diverse and growing array of metal chelating agents as potential therapeutic agents for AD. We will not attempt a comprehensive review of this literature here as this topic has recently been reviewed in depth (Smith et al., 2007; Scott et al., 2009; Perez et al., 2010). Some of the more interesting compounds synthesized include high affinity metal chelators such as JKL169 (1,1′-xylyl bis-1,4,8,11 tetraaza cyclotetradecane) (Moret et al., 2006)) and chelators with additional functionality built into them. Examples of the additional functionality engineered into chelators include groups that promote blood-brain barrier uptake, for example, moieties such glucose have been added to 3-hydroxy-4-pyridone compounds (Schugar et al., 2007). Other modifications include designing chelators to specifically target Aβ examples include XH1 ([(4-benzothiazol-2-yl-phenylcarbamoyl)-methyl]-{2-[(2-{[(4-benzothiazol-2-yl-henylcarbamoyl)methyl]-carboxymethyl-amino}-ethyl)-carboxymethyl-amino]-ethyl}-amino)-acetic acid) (Dedeoglu et al., 2004) and L2-b (N1,N1-dimethyl-N4-(pyridin-2-ylmethyl)benzene-1,4-diamine) (Choi et al., 2010). Most of these compounds have been shown to be effective at inhibiting Aβ/metal interactions in vitro and some have also shown promise in transgenic animal studies. To date, there is no published clinical data on any of these chelators. Also, it is yet to be ascertained whether this array of compounds are capable of activating the neuroprotective pathways as do the MPACs and whether this will have significant implications for this class of therapeutic agent.
M(bis(thiosemicarbazone)) complexes
The mechanistic studies of the MPACs highlighted two potential modes of action, the chelating effects inhibiting Aβ toxicity and metal delivery inducing neuroprotective signalling. To ascertain whether the metal delivery aspects of the mechanism of action could have positive therapeutic benefits in the absence of directly targeting Aβ we identified the metal bis(thiosemicarbazone) (btsc) class of compounds as suitable candidates to evaluate this question (Figure 1). Btsc complexes have been investigated as metallodrugs for a number of years and proven to have a broad range of pharmacological activity (Beraldo et al., 2004). In particular, recent interest has focused on the use of btsc ligands as vehicles for the selective delivery of radioactive copper isotopes to hypoxic tissue and leukocytes in assessment of their potential as radiopharmaceuticals (Green et al., 1988; Lewis et al., 2001). CuII(btsc) complexes are stable (log KA∼ 18) (Green et al., 1988), neutral, low molecular weight compounds capable of crossing cell membranes. In some cases, it has been demonstrated that, once inside cells, Cu2+ is reduced by intracellular reductants to Cu+, which subsequently dissociates from the ligand (Kraker et al., 1985; Adonai et al., 2002). Other CuII(btsc) complexes are more resistant to reduction and dissociation and are trapped only in hypoxic cells. This selectivity is remarkably sensitive to the nature of alkyl groups attached to the diimine backbone of the ligand. For example, diacetylbis(N (4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(atsm); Figure 1) features two methyl substituents on the backbone and copper is not released in normal cellular conditions. On the other hand, glyoxalbis(N (4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(gtsm); Figure 1) releases copper intracellularly (Fujibayashi et al., 1997; Dearling et al., 2002). The selective release of copper has been correlated with the Cu2+/Cu+ reduction potential because CuII(atsm) is more difficult to reduce than CuII(gtsm) (by some 160 mV) (Dearling et al., 2002). However, differences in pKa values, and the stability of the reduced state to dissociation of the metal may also be important.
A range of metal bis(thiosemicarbazonato) complexes (MII(btsc), where M = CuII or ZnII) have been evaluated for their ability to increase intracellular metal levels in Chinese hamster ovary cells overexpressing APP (APP-CHO) and the subsequent effect on extracellular levels of Aβ (Donnelly et al., 2008). The CuII(btsc) complexes were engineered to be either stable to both a change in oxidation state and dissociation of metal or susceptible to intracellular reduction and dissociation of metal. Treatment of APP-CHO cells with stable complexes resulted in elevated levels of intracellular copper with no effect on the detected levels of Aβ. Treatment with complexes susceptible to intracellular reduction increased intracellular copper levels but also resulted in a dose-dependent reduction in the levels of Aβ. Treatment with less stable ZnII(btsc) complexes increased intracellular zinc levels with a subsequent dose-dependent depletion of Aβ levels. As with the MPACs the increased levels of intracellular bioavailable copper and zinc initiated a signalling cascade involving activation of PI3K and c-Jun N-terminal kinase and activation of proteases that degraded Aβ.
As both CuII(gtsm) and CuII(atsm) are capable of crossing the blood brain barrier (Fodero-Tavoletti et al., 2010) the therapeutic potential of intracellular copper delivery was evaluated in the APP/PS1 transgenic AD model mice (Crouch et al., 2009). CuII(gtsm) which delivers bioavailable copper significantly inhibited GSK3β in the brains of APP/PS1 transgenic AD model mice. CuII(gtsm) also decreased the abundance of Aβ trimers and phosphorylated tau, and restored performance of AD mice in the Y-maze test to levels expected for cognitively normal animals. Improvement in the Y-maze correlated directly with decreased Aβ trimer levels. CuII(atsm) does not deliver bioavailable copper and had no effects on cognition or disease relevant biomarkers. This study demonstrates that increasing intracellular copper bioavailability can restore cognitive function by inhibiting the accumulation of neurotoxic Aβ trimers and phosphorylated tau (Crouch et al., 2009).
PtLCl2
It is generally accepted that the Aβ peptide is a valid target for therapeutic development and targeting the metal binding site of Aβ is an alternative to the metal chelating strategy of inhibiting Aβ/metal interactions that have been outlined above. Due to it being unstructured in the native state, the de novo design of effective inhibitors of Aβ is problematic. Therefore, a strategy of using metal compounds that target Aβ specifically by taking advantage of its intrinsic affinity for metal ions has been developed. The three histidine residues His-6, His-13 and His-14 are the principle Aβ metal binding ligands (Drew et al., 2009a,b;). Methylation of the imidazole side chains altered Cu binding and inhibited Aβ toxicity (Tickler et al., 2005). Pt(II) complexes have been designed to target the Aβ–metal binding site. Pt(II) compounds are stable and essentially redox inert when present in biological systems. The slow kinetics associated with substitution reactions at the Pt(II) centre means that, once bound to a target, the Pt(II) metal is difficult to displace. The specificity of the interaction between Pt anticancer drugs and DNA has been attributed largely to the ability of the am(m)ine ligands to form hydrogen-bonds to guanine nucleotides of DNA. To promote specific binding to Aβ by Pt(II) complexes, a range of 1,10-phenanthroline ligands (L) were coordinated to Pt(II) to help target the complex to N-terminal domain of Aβ. This was based on the observation that polyaromatic compounds bind to Aβ and inhibit its aggregation (Porat et al., 2006). Moreover, the classic amyloid-binding fluorescent dyes Congo red and thioflavin T are also polyaromatic compounds. L-PtCl2 complexes are highly stable and the likelihood of the chelating ligand, L, dissociating from the metal is remote. It has previously been demonstrated that the 1,10-phenanthroline ligands in the absence of metal bind weakly to Aβ via interactions with the aromatic residues Phe-4, Tyr-10 and Phe-19 on Aβ (Yao et al., 2004). Importantly for this strategy, these residues span the metal binding residues His-6, His-13 and His-14.
A range of L-PtCl2 complexes were examined in a variety of in vitro assays and shown to bind to Aβ, inhibit neurotoxicity and rescue Aβ-induced synaptotoxicity in mouse hippocampal slices (Barnham et al., 2008). Coordination of the complexes to Aβ altered the chemical properties of the peptide inhibiting amyloid formation and the generation of ROS. In comparison, the classic anticancer drug cisplatin did not affect any of the biochemical and cellular effects of Aβ. This implies that the planar aromatic 1,10-phenanthroline ligands L confer some specificity for Aβ onto the platinum complexes. The potent effect of the L-PtCl2 complexes identifies this class of compounds as potential therapeutic agents for AD. However, the major challenge for the future development of this class of compound is designing compounds that are able to cross the blood brain barrier in sufficient quantity to be an effective agent in vivo. Subsequent to this study it has been demonstrated that ruthenium (Ru) complexes (Valensin et al., 2010) with aromatic ligands were also able to have anti-Aβ effects in vitro.
Concluding remarks
Although more than 100 years have passed since the first reports of AD, development of effective treatments remains challenging as AD pathogenesis is a complex process involving many factors. Metal ions play essential role in a number of essential neurological processes and therefore a breakdown in the homeostatic systems that regulate them can have catastrophic consequences (Figure 2). When Aβ peptide interacts with metal ion not only is there the potential to cause such a breakdown in metal homeostasis but also the generation of toxic Aβ aggregates. Interdicting in these processes has shown great potential in both animal models of AD and in early stage clinical trials.
Figure 2.
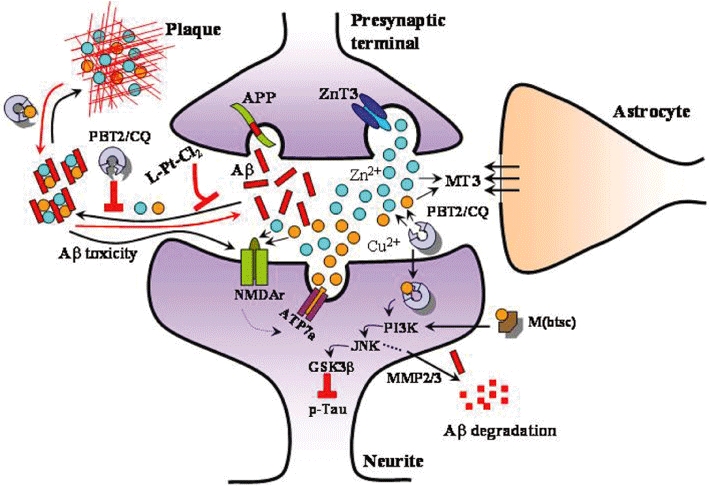
A schematic representation of the synapse and the role that ‘free’ Cu and Zn play in synaptic transmission via modulating N-methyl-D-aspartate receptor (NMDAr) function. When amyloid-β (Aβ) interacts with these metals the peptide aggregates forming toxic oligomers and ultimately amyloid plaques. The toxicity of oligomers is elicited through interactions with the glutamatergic receptors such as the NMDAr. This system offers a number of potential therapeutic strategies. These include the MPACs such as clioquinol (CQ) and PBT2 which inhibit Aβ/metal interactions and facilitate neuroprotective signalling. M(bis(thiosemicarbazone)) (M(btsc)) complexes can also induce neuroprotective signalling. The L-PtCl2 complexes are able to bind to Aβ to inhibit the formation of toxic species.
Acknowledgments
This work was funded by the Australian National Health and Medical Research Council.
Glossary
Abbreviations
- Aβ
amyloid-β peptide
- AD
Alzheimer's disease
- CQ
clioquinol
- MPAC
metal protein attenuating compound
- ROS
reactive oxygen species
Conflict of interest
K. J. B. is a consultant to and share holder of Prana Biotechnology LTD. The company owns and is developing PBT2 as a therapy for AD.
Supporting Information
Teaching Materials; Figs 1–2 as PowerPoint slide.
References
- Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease? J Neurosci. 2010;30:1631–1636. doi: 10.1523/JNEUROSCI.5255-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adonai N, Nguyen KN, Walsh J, Iyer M, Toyokuni T, Phelps ME, et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci U S A. 2002;99:3030–3035. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali FE, Separovic F, Barrow CJ, Cherny RA, Fraser F, Bush AI, et al. Methionine regulates copper/hydrogen peroxide oxidation products of Abeta. J Pept Sci. 2005;11:353–360. doi: 10.1002/psc.626. [DOI] [PubMed] [Google Scholar]
- Ali FE, Leung A, Cherny RA, Mavros C, Barnham KJ, Separovic F, et al. Dimerisation of N-acetyl-L-tyrosine ethyl ester and Abeta peptides via formation of dityrosine. Free Radic Res. 2006;40:1–9. doi: 10.1080/10715760500329721. [DOI] [PubMed] [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, ‘Uber eine eigenartige Erkankung der Hirnrinde’. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. Alzheimer's disease facts and figures. Alzheimers Dement. 2010;6:1–70. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Areosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev. 2005;(3):CD003154. doi: 10.1002/14651858.CD003154.pub3. [DOI] [PubMed] [Google Scholar]
- Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, et al. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- Atwood CS, Huang X, Moir RD, Tanzi RE, Bush AI. Role of free radicals and metal ions in the pathogenesis of Alzheimer's disease. Met Ions Biol Syst. 1999;36:309–364. [PubMed] [Google Scholar]
- Atwood CS, Huang X, Khatri A, Scarpa RC, Kim YS, Moir RD, et al. Copper catalyzed oxidation of Alzheimer Abeta [In Process Citation] Cell Mol Biol (Noisy-Le-Grand) 2000;46:777–783. [PubMed] [Google Scholar]
- Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, et al. Copper mediates dityrosine cross-linking of Alzheimer's amyloid-beta. Biochemistry. 2004;43:560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Ciccotosto GD, Tickler AK, Ali FE, Smith DG, Williamson NA, et al. Neurotoxic, Redox-competent Alzheimer's {beta}-Amyloid Is Released from Lipid Membrane by Methionine Oxidation. J Biol Chem. 2003a;278:42959–42965. doi: 10.1074/jbc.M305494200. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Gautier E, Kok G, Krippner G. 8-Hydroxyquinoline Derivatives. PCT Publication; 2003b. WO2004007461. [Google Scholar]
- Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer's disease beta-amyloid. FASEB J. 2004;18:1427–1429. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Kenche VB, Ciccotosto GD, Smith DP, Tew DJ, Liu X, et al. Platinum-based inhibitors of amyloid-beta as therapeutic agents for Alzheimer's disease. Proc Natl Acad Sci U S A. 2008;105:6813–6818. doi: 10.1073/pnas.0800712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, et al. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- Beraldo H, Gambino D. The wide pharmacological versatility of semicarbazones, thiosemicarba-zones and their metal complexes. Mini Rev Med Chem. 2004;4:31–39. doi: 10.2174/1389557043487484. [DOI] [PubMed] [Google Scholar]
- Birks J, Flicker L. Donepezil for mild cognitive impairment. Cochrane Database Syst Rev. 2006;(3):CD006104. doi: 10.1002/14651858.CD006104. [DOI] [PubMed] [Google Scholar]
- Bush AI. The metallobiology of Alzheimer's disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF, et al. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Cappai R, Barnham KJ. Delineating the mechanism of Alzheimer's disease A beta peptide neurotoxicity. Neurochem Res. 2008;33:526–532. doi: 10.1007/s11064-007-9469-8. [DOI] [PubMed] [Google Scholar]
- Caragounis A, Du T, Filiz G, Laughton KM, Volitakis I, Sharples RA, et al. Differential modulation of Alzheimer's disease amyloid beta-peptide accumulation by diverse classes of metal ligands. Biochem J. 2007;407:435–450. doi: 10.1042/BJ20070579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Kazachkov M, Yu PH. Effect of aldehydes derived from oxidative deamination and oxidative stress on beta-amyloid aggregation; pathological implications to Alzheimer's disease. J Neural Transm. 2007;114:835–839. doi: 10.1007/s00702-007-0697-5. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Choi JS, Braymer JJ, Nanga RP, Ramamoorthy A, Lim MH. Design of small molecules that target metal-A{beta} species and regulate metal-induced A{beta} aggregation and neurotoxicity. Proc Natl Acad Sci U S A. 2010;107:21990–21995. doi: 10.1073/pnas.1006091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci U S A. 2009;106:381–386. doi: 10.1073/pnas.0809057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearling JL, Lewis JS, Mullen GE, Welch MJ, Blower PJ. Copper bis(thiosemicarbazone) complexes as hypoxia imaging agents: structure-activity relationships. J Biol Inorg Chem. 2002;7:249–259. doi: 10.1007/s007750100291. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Cormier K, Payton S, Tseitlin KA, Kremsky JN, Lai L, et al. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer's amyloidogenesis. Exp Gerontol. 2004;39:1641–1649. doi: 10.1016/j.exger.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009;29:4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, et al. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J Biol Chem. 2008;283:4568–4577. doi: 10.1074/jbc.M705957200. [DOI] [PubMed] [Google Scholar]
- Dorlet P, Gambarelli S, Faller P, Hureau C. Pulse EPR spectroscopy reveals the coordination sphere of copper(II) ions in the 1-16 amyloid-beta peptide: a key role of the first two N-terminus residues. Angew Chem Int Ed Engl. 2009;48:9273–9276. doi: 10.1002/anie.200904567. [DOI] [PubMed] [Google Scholar]
- Drew SC, Masters CL, Barnham KJ. Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer's disease amyloid-beta peptide-relevance to N-terminally truncated forms. J Am Chem Soc. 2009a;131:8760–8761. doi: 10.1021/ja903669a. [DOI] [PubMed] [Google Scholar]
- Drew SC, Noble CJ, Masters CL, Hanson GR, Barnham KJ. Pleomorphic copper coordination by Alzheimer's disease amyloid-beta peptide. J Am Chem Soc. 2009b;131:1195–1207. doi: 10.1021/ja808073b. [DOI] [PubMed] [Google Scholar]
- Duce JA, Smith DP, Blake RE, Crouch PJ, Li QX, Masters CL, et al. Linker histone H1 binds to disease associated amyloid-like fibrils. J Mol Biol. 2006;361:493–505. doi: 10.1016/j.jmb.2006.06.038. [DOI] [PubMed] [Google Scholar]
- Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Extance A. Alzheimer's failure raises questions about disease-modifying strategies. Nat Rev Drug Discov. 2010;9:749–751. doi: 10.1038/nrd3288. [DOI] [PubMed] [Google Scholar]
- Faller P. Copper and zinc binding to amyloid-beta: coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem. 2009;10:2837–2845. doi: 10.1002/cbic.200900321. [DOI] [PubMed] [Google Scholar]
- Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, et al. PBT2 rapidly improves cognition in Alzheimer's Disease: additional phase II analyses. J Alzheimers Dis. 2010;20:509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Villemagne VL, Paterson BM, White AR, Li QX, Camakaris J, et al. Bis(thiosemicarbazonato) Cu-64 complexes for positron emission tomography imaging of Alzheimer's disease. J Alzheimers Dis. 2010;20:49–55. doi: 10.3233/JAD-2010-1359. [DOI] [PubMed] [Google Scholar]
- Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A. Copper-62-ATSM: a new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med. 1997;38:1155–1160. [PubMed] [Google Scholar]
- Green MA, Klippenstein DL, Tennison JR. Copper(II) bis(thiosemicarbazone) complexes as potential tracers for evaluation of cerebral and myocardial blood flow with PET. J Nucl Med. 1988;29:1549–1557. [PubMed] [Google Scholar]
- Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel JP, Tanzi RE, et al. Zinc-induced Alzheimer's Abeta1-40 aggregation is mediated by conformational factors. J Biol Chem. 1997;272:26464–26470. doi: 10.1074/jbc.272.42.26464. [DOI] [PubMed] [Google Scholar]
- Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, et al. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- Huang X, Cuajungco MP, Atwood CS, Moir RD, Tanzi RE, Bush AI. Alzheimer's disease, beta-amyloid protein and zinc. J Nutr. 2000;130:1488S–1492S. doi: 10.1093/jn/130.5.1488S. [DOI] [PubMed] [Google Scholar]
- Hureau C, Coppel Y, Dorlet P, Solari PL, Sayen S, Guillon E, et al. Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper(II) environment in the amyloid-beta peptide near physiological pH. Angew Chem Int Ed Engl. 2009;48:9522–9525. doi: 10.1002/anie.200904512. [DOI] [PubMed] [Google Scholar]
- Kraker A, Krezoski S, Schneider J, Minkel D, Petering DH. Reaction of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazonato) Cu(II) with Ehrlich cells. Binding of copper to metallothionein and its relationship to zinc metabolism and cell proliferation. J Biol Chem. 1985;260:13710–13718. [PubMed] [Google Scholar]
- Kromer Vogt LJ, Hyman BT, Van Hoesen GW, Damasio AR. Pathological alterations in the amygdala in Alzheimer's disease. Neuroscience. 1990;37:377–385. doi: 10.1016/0306-4522(90)90408-v. [DOI] [PubMed] [Google Scholar]
- Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lewis J, Laforest R, Buettner T, Song S, Fujibayashi Y, Connett J, et al. Copper-64-diacetyl-bis(N4-methylthiosemicarbazone): an agent for radiotherapy. Proc Natl Acad Sci U S A. 2001;98:1206–1211. doi: 10.1073/pnas.98.3.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Masters CL, Beyreuther K. Neuropathology of unconventional virus infections: molecular pathology of spongiform change and amyloid plaque deposition. Ciba Found Symp. 1988;135:24–36. doi: 10.1002/9780470513613.ch3. [DOI] [PubMed] [Google Scholar]
- Moret V, Laras Y, Pietrancosta N, Garino C, Quelever G, Rolland A, et al. 1,1′-Xylyl bis-1,4,8,11-tetraaza cyclotetradecane: a new potential copper chelator agent for neuroprotection in Alzheimer's disease. Its comparative effects with clioquinol on rat brain copper distribution. Bioorg Med Chem Lett. 2006;16:3298–3301. doi: 10.1016/j.bmcl.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Perez LR, Franz KJ. Minding metals: tailoring multifunctional chelating agents for neurodegenerative disease. Dalton Trans. 2010;39:2177–2187. doi: 10.1039/b919237a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Friedlich AL, Setchell KDR, Nagano S, Opazo C, Cherny RA, et al. Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J Clin Invest. 2005;115:2556–2563. doi: 10.1172/JCI23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- Schlief ML, Gitlin JD. Copper homeostasis in the CNS: a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol Neurobiol. 2006a;33:81–90. doi: 10.1385/MN:33:2:81. [DOI] [PubMed] [Google Scholar]
- Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci U S A. 2006b;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneich C. Redox processes of methionine relevant to beta-amyloid oxidation and Alzheimer's disease. Arch Biochem Biophys. 2002;397:370–376. doi: 10.1006/abbi.2001.2621. [DOI] [PubMed] [Google Scholar]
- Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, et al. Combating Alzheimer's disease with multifunctional molecules designed for metal passivation. Angew Chem Int Ed Engl. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]
- Scott LE, Orvig C. Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem Rev. 2009;109:4885–4910. doi: 10.1021/cr9000176. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer's disease amyloid beta peptide. Biochim Biophys Acta. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Takeda A, Loveman E, Clegg A, Kirby J, Picot J, Payne E, et al. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer's disease. Int J Geriatr Psychiatry. 2006;21:17–28. doi: 10.1002/gps.1402. [DOI] [PubMed] [Google Scholar]
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, et al. Methylation of the imidazole side chains of the Alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutaselike structures and inhibition of neurotoxicity. J Biol Chem. 2005;280:13355–13363. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- Turnbull S, Tabner BJ, El-Agnaf OM, Twyman LJ, Allsop D. New evidence that the Alzheimer beta-amyloid peptide does not spontaneously form free radicals: an ESR study using a series of spin-traps. Free Radic Biol Med. 2001;30:1154–1162. doi: 10.1016/s0891-5849(01)00510-x. [DOI] [PubMed] [Google Scholar]
- Valensin D, Anzini P, Gaggelli E, Gaggelli N, Tamasi G, Cini R, et al. fac-{Ru(CO)(3)}(2+) selectively targets the histidine residues of the beta-amyloid peptide 1-28. Implications for new Alzheimer's disease treatments based on ruthenium complexes. Inorg Chem. 2010;49:4720–4722. doi: 10.1021/ic902593e. [DOI] [PubMed] [Google Scholar]
- White AR, Du T, Laughton KM, Volitakis I, Sharples RA, Xilinas ME, et al. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006;281:17670–17680. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Wedd AG. The challenges of determining metal-protein affinities. Nat Prod Rep. 2010;27:768–789. doi: 10.1039/b906690j. [DOI] [PubMed] [Google Scholar]
- Yao S, Cherny RA, Bush AI, Masters CL, Barnham KJ. Characterizing bathocuproine self-association and subsequent binding to Alzheimer's disease amyloid beta-peptide by NMR. J Pept Sci. 2004;10:210–217. doi: 10.1002/psc.539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.