

Abstract
The endothelins (ET) are a group of proteins that act through G-protein coupled receptors. Endothelin-1 (ET-1) was initially identified as a potent vasoconstrictor and dysregulation of the ET axis contributes to pathological processes responsible for cardiovascular disease states. More recently, the ET axis, in particular ET-1 acting through the endothelin A receptor (ETA), has been implicated in the development of several cancers through activation of pathways involved in cell proliferation, migration, invasion, epithelial-mesenchymal transition, osteogenesis and angiogenesis. The endothelin B receptor (ETB) may counter tumour progression by promoting apoptosis and clearing ET-1; however, it has recently been implicated in the development of some tumour types including melanomas and oligodendrogliomas. Here, we review emerging preclinical and clinical data outlining the role of the ET axis in cancer, and its antagonism as an attractive and challenging approach to improve clinical cancer management. Clinical data of ETA antagonists in patients with prostate cancer are encouraging and provide promise for new ETA antagonist-based treatment strategies. Given the unexpected opportunities to affect pleiotrophic tumorigenic signals by targeting ETA-mediated pathways in a number of cancers, the evaluation of ET-targeted therapy in cancer warrants further investigation.
Keywords: endothelin, endothelin receptor, endothelin antagonist, ETA receptor, ETB receptor, GPCR, cancer, zibotentan
Introduction
The endothelin (ET) axis consists of three 21-amino acid peptides, ET-1, ET-2 and ET-3, two distinct rhodopsin-like G-protein coupled receptor (GPCR) subtypes, endothelin A and endothelin B (ETA and ETB respectively) and the endothelin-converting enzymes (ECEs), which catalyze the generation of biologically active ET (Rubanyi and Polokoff, 1994). The first major role that was identified for ET-1 was as a potent vasoconstrictor (Rubanyi and Polokoff, 1994). The ET axis has also been shown to play a key role in a number of tissues and systems including somatosensory, respiratory, circulatory, endocrine, urogenital, visual, digestive and the central nervous system (CNS) (Rubanyi and Polokoff, 1994). Dysregulation of the ET axis was initially shown to contribute to the pathological processes responsible for cardiovascular disease states including systemic and pulmonary hypertension, and congestive heart failure (Rubanyi and Polokoff, 1994; Goldie, 1999). More recently, the ET axis has been implicated in a number of other cell signalling pathways such as apoptosis and cell growth (Bagnato and Natali, 2004), which are also common with other GPCR-based signalling pathways such as those induced by angiotensin II (Leung, 2004). The identification of the ET axis in such signalling pathways led to the investigation of its role in the development and progression of cancer. The ET axis was shown to have a relevant role in various cancer cells and stromal cells leading to autocrine/paracrine feedback loops, which promote the development and progression of tumours. Such processes include cell proliferation, escape from apoptosis, angiogenesis, invasion and metastatic dissemination, aberrant osteogenesis and modification of nociceptive stimuli (Nelson et al., 2003). This review will discuss the role of the ET axis in cancer, the pharmacological processes of ET receptor antagonism and recent advances in the development of ET-targeted treatments for patients with cancer.
Endothelin function in normal physiology and pathology
ET-1 is primarily secreted by endothelial cells through both constitutive and regulated (or rapid release) pathways (Russell and Davenport, 1999). In addition, ET-1 is secreted to a lesser extent by a range of other cell types including macrophages, leukocytes, fibroblasts, vascular smooth muscle cells (VSMC), cardiomyocytes, tubular epithelial cells, mesangial cells and podocytes (Nunez et al., 1990; Resink et al., 1990; Firth and Ratcliffe, 1992; Kohan, 1997). ET-2 is secreted in the kidney and intestine, and ET-3 in the brain, intestine, lung and kidney. Two pathways for the clearance of ET-1 have been identified. The first of these is ETB-mediated uptake followed by lysosomal degradation (Bremnes et al., 2000) and the second is through the catabolism of ET-1 by extracellular neutral endopeptidase 24.11 (NEP, neprilysin) (Abassi et al., 1992).
The downstream effects of ET-1 are mediated by two distinct receptor subtypes, ETA and ETB (Rubanyi and Polokoff, 1994). These receptors display different ligand selectivity; however, both receptors bind to ET-1 with equal affinity (Davenport, 2002). The intracytoplasmic C termini of the two ET receptor types differ, which results in their divergent intracellular effects following activation by ET-1 (Nussdorfer et al., 1999). ETA and ETB are differentially expressed in a variety of normal tissues including the vasculature (VSMC, heart, cardiomyocytes, fibroblasts), CNS (trigeminal nerve, brain cells, small diameter sensory neurons), renal epithelial and endothelial cells, prostate, breast and female reproductive tissues (Molenaar et al., 1993; Karet and Davenport, 1996; Kuc and Davenport, 2004; Schinelli, 2006; Smollich and Wülfing, 2008; Khodorova et al., 2009; Chichorro et al., 2010). This differential tissue expression of the ET receptor subtypes is dynamic and contributes to the different actions of the three ETs (Levin, 1995; Nelson et al., 1996).
Extracellular binding of ET-1 to ETA activates a non-linear, highly interconnected signalling network. Many processes activated by this network are involved in normal cell function and in the development and progression of cancer including cell proliferation, apoptosis, cell invasion and metastasis, angiogenesis, osteogenesis and nociception (Figure 1) (Smollich and Wülfing, 2007; Bagnato and Rosanò, 2008; Nelson, 2009). Dysregulation of ETA activation can therefore promote tumour development and progression. Moreover, many cancerous cell types, including prostate, ovarian, renal, pulmonary, colorectal, cervical, breast, bladder and melanoma, have been reported to secrete ET-1, and the ET axis has been implicated in a range of tumours suggesting that it is a very attractive target for cancer therapy (Asham et al., 2001; Nelson, 2003; Bagnato et al., 2005; Bagnato and Rosanò, 2008). Indeed, ET-1 plasma levels were increased in patients with colorectal cancer as well as in a syngeneic rat model of colorectal cancer in which inhibition of the ETA with a selective antagonist (BQ123) significantly reduced tumour weight of metastatic lesions to the liver (Asham et al., 2001). Alterations in the relative ratios of ETA to ETB can incite the progression of cells from a normal phenotype to a more malignant phenotype (Nelson et al., 1996; 1997; Bagnato et al., 1999; Godara et al., 2005). In fact, down-regulation of ETB expression has been reported in many tumour types including prostate cancer (Nelson et al., 1996; Bagnato and Rosanò, 2008). In addition, the ETB gene is frequently hypermethylated leading to reduced or absent receptor expression (Nelson et al., 1997; Pao et al., 2001; Jerónimo et al., 2003). Increased ETA expression was observed with advancing tumour stage and grade in patients with local and metastatic prostate cancer (Nelson, 2003). Furthermore, the ET-1 clearance pathway is disrupted in prostate cancer; ETB expression and NEP protein levels are reduced resulting in increased local ET-1 levels (Nelson, 2003; Bagnato and Rosanò, 2008). ET-1 and ETA are also overexpressed in primary and metastatic ovarian tumours and dysregulation of this autocrine signalling pathway is believed to be a driver of disease progression (Salani et al., 2000; Bagnato et al., 1995; 2005;).
Figure 1.
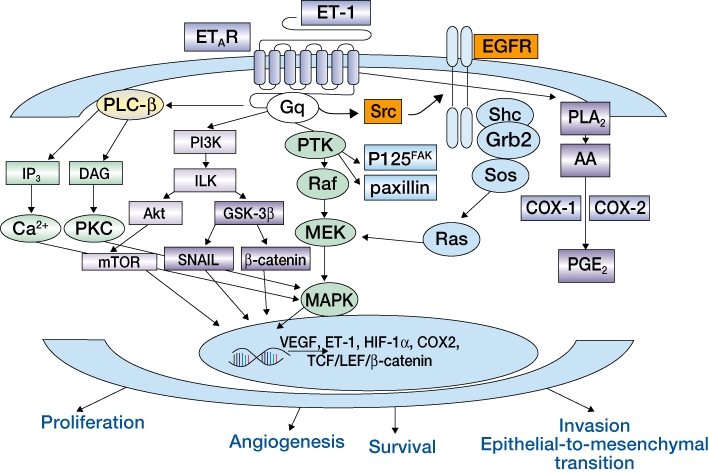
Endothelin-1 (ET-1) signalling through the endothelin A receptor (ETA) in cancer cells (Bagnato and Rosanò, 2008). Reprinted with permission from Elsevier. AA, arachidonic acid; COX-1, cyclo-oxygenase 1; COX-2, cyclo-oxygenase 2; DAG, diacylglycerol; EGFR, epidermal growth factor receptor; ETAR, endothelin A receptor; Gq, G protein q; GSK-3, glycogen synthase kinase-3; HIF-1α, hypoxia-inducible factor 1-α; ILK, integrin-linked kinase; IP3, inositol 1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; MEK, mitogenactivated protein kinase/extracellular signal-regulated kinase kinase; mTOR, mammalian target of rapamycin; PGE2, prostaglandin E2; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PLA2, phospholipase A2; PLC, phospholipase C; PTK, protein-tyrosine kinase; VEGF, vascular endothelial growth factor.
In normal cells and a number of tumour types, activation of ETB by ET-1 has been shown to affect processes involved in the inhibition of cancer; inducing cell death by apoptosis and promoting ET-1 clearance (Okazawa et al., 1998; Dupuis et al., 2000). Recently, ETB activation has been implicated in a range of cancers. ETB is overexpressed in melanomas (Lahav, 2005) and oligodendrogliomas (Anguelova et al., 2005), and has been shown to correlate with malignant melanoma development and progression (Lahav, 2005). In addition, gene expression profiling and immunohistochemical analysis of human melanoma biopsies and cell lines indicated the ETB as a tumour progression marker associated with an aggressive phenotype (Bittner et al., 2000; Demunter et al., 2001). However, reports of the overexpression of ETB in some tumour types, such as lung cancer, have been conflicting which may be reflective of the methodological variations used to detect ETB (Ahmed et al., 2000; Knight et al., 2009). Activation of ETB by ET-1 or ET-3 has been shown to trigger downstream pathways involved in the progression of cutaneous melanoma and blockade of ETB inhibited human melanoma xenograft growth (Bagnato et al., 2004). Furthermore, inhibition of ETB has been shown to block glioma cell proliferation and induce apoptosis (Paolillo et al., 2010).
Increased vasodilation in tumours can occur through ETB, which may aid in the delivery of anticancer treatments to the tumour. Indeed, the selective ETB agonist SPI-1620 (IRL-1620) was shown to selectively and transiently increase tumour blood flow, allowing increased delivery of chemotherapy agents to tumours in a breast cancer model in rats and a solid tumour model in dogs (Rai et al., 2006; Selting et al., 2008).
The role of ET in immune response was initially established in dendritic cells which were shown to produce large amounts of ET-1 and significantly increase the expression of ET receptors upon maturation (Guruli et al., 2004). Selective blockade of ETA significantly reduced expression of the mature dendritic marker CD83, down-regulated dendritic cell ability to stimulate T cells and promoted dendritic cell apoptosis, whereas selective ETB blockade resulted in increased expression of CD83 and improved dendritic cell survival. ETB has also been shown to have a potential role in immune responses associated with cancer (Guruli et al., 2004; Juergens et al., 2008; Kandalaft et al., 2009; Verri et al., 2009; Wang et al., 2010). Overexpression of ETB in tumour endothelium has been observed in ovarian tumours, and activation of these receptors by ET-1 was reported to suppress T-cell adhesion and homing to tumours (Kandalaft et al., 2009). The success of immune therapy depends on the ability of effector T cells to infiltrate tumours. ETB blockade with the selective ETB antagonist BQ788 increased T-cell homing to tumours, which resulted in the tumour responding to otherwise ineffective immunotherapy in vivo without changes to the systemic antitumour immune response (Buckanovich et al., 2008). It has therefore been suggested that ETB antagonists warrant clinical testing in combination with passive or adaptive immunotherapy. This approach may help enable selective up-regulation of immunotherapy directly to the tumour compartment via the ETB (Kandalaft et al., 2009). In addition, this therapy combination may inhibit the significant autoimmune toxicities observed with current immunomodulatory approaches (Kandalaft et al., 2009).
There is a growing body of evidence implicating ET-2 in the progression of cancer. Up-regulation of ET-2 mRNA expression was observed in several human breast cancer tumour cell lines and incubation of these cell lines with ET-2 induced chemotaxis (Grimshaw et al., 2004). More recently, ET-2 mRNA was reported to be overexpressed in basal cell carcinoma compared with normal skin, an effect controlled by the Hedgehog signalling pathway (Tanese et al., 2010).
The first study investigating the role of ET-3 in cancer has recently been reported. In this study, ET-3 mRNA expression and protein levels were attenuated in breast cancer tissues compared with normal tissue, an effect caused by hypermethylation of the ET-3 promoter and subsequent gene silencing (Wiesmann et al., 2009). These preliminary data suggest that unlike ET-1 and ET-2, ET-3 may act as a natural tumour suppressor in breast cancer.
ECE-1 expression is significantly elevated in tumours and ECE-1 has been reported to be increased in primary malignant stromal cells compared with benign cells (Dawson et al., 2004). Inhibition of stromal ECE-1 reduced PC-3 (prostate cancer) cell invasion in co-culture and the inclusion of ET-1 to such cultures only partially recovered this effect, suggesting a role for ECE-1 independent of ET-1 activation. A recent investigation of ECE-1 isoforms demonstrated that ECE-1c overexpression increased PC-3 invasion through Matrigel™, whereas ECE-1a overexpression suppressed invasion (Lambert et al., 2008). The ECE-1 isoforms may therefore be relevant targets for the treatment of cancers including castration-resistant prostate cancer (CRPC); however, in terms of ‘drugability’, the approach of ECE inhibition is something that has not met with much success in the clinic. Theoretical limitations that have potentially had an impact on the success of ECE inhibition have been proposed, including redundancy of the ET-1 generating pathways, non-ECE generating big-ET-1 processing enzymes, potential to reduce beneficial effects of the ligand at respective ET receptors and ‘drug-killing’/development-limiting side effects that this approach has brought (Kirkby et al., 2008).
Targeting the ET axis as an anticancer approach
It is currently unknown whether increased plasma ET-1 levels are the cause or consequence of cancers. However, in some cancers such as CRPC, elevated plasma levels have been reported in patients with advanced metastatic disease relative to those with earlier stage disease (Nelson et al., 1995; 2003;). In colorectal adenomas, increased expression of pre-pro ET-1 and ECE mRNA was observed compared with normal colon (Egidy et al., 2000). In breast carcinoma, ET-1 immunoreactivity and mRNA levels were greater in breast ductal carcinoma in situ specimens compared with normal breast tissue (Alanen et al., 2000). These data suggest that higher ET-1 levels in plasma correlate with disease severity and that the modulation of the ET system is an early event in tumorigenesis. Moreover, there is evidence that in cancers, including CRPC, the gene regulating the ETB (which would normally promote apoptosis and clearance of ET-1) has undergone hypermethylation (Knight et al., 2009) which could lead to increased ET-1 and provide a basis for increased cell survival. Indeed, in a recent study the ETB promoter was significantly hypermethylated in nearly 50% of non-small-cell lung cancer (NSCLC) tumour samples investigated. Furthermore, these samples also had reduced ETB mRNA levels compared with unmethylated samples. These data suggest an involvement of ETB epigenetic deregulation in the development and progression of lung cancer and highlight this gene as a promising biomarker for response to regimens modulating the ET axis (Knight et al., 2009). There is also evidence of a similar hypermethylation and de-activation of the ETB in prostate cancer (Nelson et al., 1997). Somatic methylation of CpG island sequences in the ETB in 5/5 human prostate cancer cell lines, 15/21 primary prostate cancer tissues and 8/14 prostate cancer metastases (70% of samples overall) was observed. Normal tissues contained only unmethylated ETB. Treatment of human prostatic carcinoma cell line cultures with 5-azacytidine induced ETB mRNA expression, suggesting that CpG island methylation changes might accompany the apparent transcriptional silencing of ETBin vivo.
Whether the reduction in ETB functionality per se leads to increased binding of the ligand to the ETA remains to be elucidated. The loss of ETB functionality will permit ET-1 to reside and accumulate which may drive increased receptor expression to accommodate increasing concentrations of ligand. ETA/ET-1 expression analyses in tumour samples from patients tend to support that up-regulation or increase in ligand and receptor expression do occur and these phenomena support increased binding of the ligand to ETA.
Inhibition of the ET axis using specific, selective and dual competitive ET receptor antagonists represents an attractive targeted approach for the treatment of cancer. There are currently over 15 ETA and/or ETB antagonists being evaluated in clinical trials for a variety of indications, including cardiovascular disease and cancer. The selective ETA and ETB antagonists BQ123 and BQ788 have been valuable tools for the assessment of ET receptor antagonism and have been used extensively in preclinical models. However, as these agents are both peptides their utility in the clinical setting has been limited. Although no data in humans with BQ788 have been published, a number of studies have been performed using BQ123, generally in a setting in which access to small molecule non-peptide agents has not been possible (Spratt et al., 2001). To date, the dual ETA/ETB antagonist bosentan, the selective ETA antagonists atrasentan and YM-598, and the specific ETA antagonist zibotentan are the only ET receptor antagonists that have been evaluated in both the preclinical and clinical oncology settings (Table 1).
Table 1.
Clinical evaluation of endothelin antagonists in cancer*
| Compound (target receptor) | Study population and intervention | Trial status | Results and conclusions |
|---|---|---|---|
| Bosentan (Dual competitive ETA/ETB) | Phase II, metastatic melanoma (monotherapy; n = 35) (Kefford et al., 2007) | Completed | •Stable disease seen in six patients at 12 weeks |
| •No treatment responses | |||
| Phase II, metastatic melanoma (in combination with dacarbazine, placebo-controlled; n = 80) (Kefford et al., 2010) | Completed | •No difference in time to progression seen at 12 months | |
| YM598 (Selective ETA) | Phase II, prostate cancer (monotherapy) | Terminated | |
| Phase II, prostate cancer (in combination with mitoxantrone and prednisone) | Terminated | ||
| Atrasentan (Selective ETA) | Phase III, metastatic prostate cancer (placebo-controlled, monotherapy; n = 809) (Carducci et al., 2007) | Completed | •No reduction in disease progression |
| •Study design and prior assumption of progression rates may have limited the ability to define clinical benefit | |||
| Phase III, non-metastatic prostate cancer (placebo-controlled, monotherapy; n = 941) (Nelson et al., 2008) | Completed | •No statistically significant difference in time to progression | |
| •Large regional differences in TTP suggest trial conduct may have influenced results | |||
| Phase III, prostate cancer with bone metastases (in combination with docetaxel and prednisone) | Ongoing | (data not yet available) | |
| Phase II, hormone-refractory prostate cancer (double-blind, randomized monotherapy; n = 288) (Carducci et al., 2003) | Completed | •Trend towards prolongation of disease | |
| •Statistically significant delay in PSA | |||
| Phase II, hormone-naive prostate cancer (monotherapy) | Completed | (data not yet available) | |
| Phase II, prostate cancer with bone metastases (in combination with zoledronic acid; n = 44) (Michaelson et al., 2006) | Completed | •No evidence of additive or synergistic effects of combination therapy | |
| Phase I-II, metastatic prostate cancer (in combination with docetaxel; n = 31) (Armstrong et al., 2008) | Completed | •Survival comparable to that seen with docetaxel and prednisone, but rate of PSA decline lower than expected | |
| Phase I-II, NSCLC (in combination with paclitaxel and carboplatin; n = 44) (Chiappori et al., 2008) | Completed | •Lack of positive response data may reflect deficiencies in clinical trial design and low dose | |
| Phase II, renal carcinoma (monotherapy; n = 94) (Manola et al., 2007) | Completed | •6-month progression-free rates did not support use as first-line monotherapy | |
| Phase I, malignant glioma (monotherapy; n = 25) (Phuphanich et al., 2008) | Completed | •Primarily a safety study, but two partial responses observed | |
| Zibotentan (specific ETA) | Phase III, prostate cancer with bone metastases (monotherapy) | Completed | (data not yet available) |
| Phase III, non-metastatic prostate cancer (monotherapy) | Recruiting | (data not yet available) | |
| Phase III, prostate cancer, metastatic (in combination with docetaxel) | Ongoing | (data not yet available) | |
| Phase II, prostate cancer with bone metastases (placebo-controlled, monotherapy; n = 312) (James et al., 2010) | Complete | •No statistically significant improvement in TTP, but overall survival extended versus placebo | |
| Phase II, prostate cancer, metastatic, patients previously treated with chemotherapy (monotherapy; n = 24) | Ongoing | (data not yet available) | |
| Phase II, NSCLC (in combination with pemetrexed) | Completed | (data not yet available) | |
| Phase I, metastatic prostate cancer (in combination with docetaxel; n = 31) (Trump et al., 2010) | Completed | •Activity observed with the combination, meriting further evaluation | |
| Phase I, advanced solid malignancies in elderly Chinese patients (monotherapy) | Recruiting | (data not yet available) |
Not including planned clinical trials. ETA, endothelin A receptor; ETB, endothelin B receptor; NSCLC, non-small-cell lung cancer; PSA, prostate-specific antigen; TTP, time to tumour progression.
BQ123 and BQ788 (Banyu Pharmaceutical Co; Merck)
BQ123 is a highly soluble, potent, and selective ETA antagonist with IC50 values of 8.3 nM and 61 µM for human ETA and ETB, respectively (Ishikawa et al., 1992), and in isolated porcine coronary arteries, BQ123 had an antivasoconstriction pA2 value of 7.4 (Fukami et al., 1995). BQ123 was the first ETA antagonist to be developed and its use in preclinical models has had great value in demonstrating a role for the ETA in cancer cell growth, proliferation, survival, migration and invasion, and pain (Rosanòet al., 2001; Del Bufalo et al., 2002; Grant et al., 2007; Schmidt et al., 2007; Zhang et al., 2008). BQ123 remains widely used in preclinical studies defining the physiology of the ET axis (Battistini et al., 2006); however, due to the cost of its development and the need for parenteral administration (the peptide is hydrolyzed by peptidases in the systemic circulation and gastrointestinal tract) (Motte et al., 2006), the use of BQ123 has been limited to small-scale clinical trials.
BQ788 is a potent and selective ETB antagonist with IC50 values of 1300 nM and 1.2 nM for human ETA and ETB respectively (Ishikawa et al., 1994). In isolated rabbit pulmonary arteries rich in ETB, BQ788 potently antagonized vasoconstriction produced by an ETB selective agonist with a pA2 value of 8.4 (Ishikawa et al., 1994). Preclinical studies with BQ788 have reported a role for ETB in the survival, growth and metastasis of melanoma and glioma cells (Lahav et al., 1999; Lahav, 2005; Paolillo et al., 2010). Furthermore, BQ788 inhibited several pathways mediated by ET-1, including bronchoconstriction and cell proliferation, and was also shown to inhibit clearance of perfused ET-1 (Okada and Nishikibe, 2002). BQ788 has also been used to identify the role of ETB in control of vascular tone in vessels that supply tumours, implying that ET-1 via ETB dilates vessels supplying breast tumours in the rat (Gulati and Rai, 2004) and that arteries supplying human colorectal tumours were more sensitive to ET-1 due to increased ETB responsiveness (Ferrero et al., 2008). More recently, BQ788 has been used to identify the role of ETB in the growth and invasion of lymphatic endothelial cells and vessels (Spinella et al., 2009) and mediation of the endothelial barrier in T-cell homing to tumours (Buckanovich et al., 2008). As with BQ123, the use of this antagonist in clinical trials has been limited because of the cost of development and the systemic method of its administration (Battistini et al., 2006).
Bosentan (Actelion Pharmaceuticals Ltd)
Bosentan is a dual competitive ETA and ETB antagonist, which competitively inhibited the specific binding of 125I-ET-1 on ETA rich human smooth muscle cells with an inhibitor constant (Ki) of 4.7 nM, and on ETB rich human placenta with a Ki of 95 nM. In addition, ET-1-induced contractions in isolated rat aorta, and contractions induced by the selective ETB agonist sarafotoxin S6C in rat trachea, were competitively antagonized by bosentan with pA2 values of 7.2 and 6.0, respectively (Clozel et al., 1994).
In preclinical studies of human melanoma cell lines, bosentan was observed to inhibit proliferation, decrease cell viability and DNA synthesis and induce apoptosis (Sekulic et al., 2002; Berger et al., 2006). These preclinical studies provided a rationale to investigate this agent in the clinical cancer setting. A single-arm, Phase II uncontrolled study indicated that bosentan monotherapy may be of benefit to patients with stage IV metastatic melanoma, achieving disease stabilization in 19% of patients at week 6, with confirmation at week 12; five patients still had stable disease after 24 weeks and two remained stable after more than 2 years on study treatment (Kefford et al., 2007). Following these positive results, a Phase II randomized, double-blind, placebo-controlled proof-of-concept study in a similar patient population reported no beneficial effect on time to tumour progression (TTP) or other efficacy parameters [progression-free survival (PFS) and overall survival (OS)] when bosentan was combined with first-line dacarbazine chemotherapy (Kefford et al., 2010). The authors of this study suggested that the failure of this trial may be due to the abnormally high TTP observed in the placebo group, the stringent selection criteria of patients in this study and/or the 50% risk reduction rate selected for efficacy (Kefford et al., 2010).
Bosentan has been extensively investigated in the cardiovascular setting and it is licensed for the treatment of pulmonary arterial hypertension and reduction of digital ulcer formation in patients with systemic sclerosis. There are currently no ongoing trials of bosentan in the cancer setting.
YM-598 [Astellas Pharma (formerly Yamanouchi)]
YM-598 is a potent selective ETA antagonist developed through the modification of bosentan. This agent inhibited 125I-ET-1 binding to cloned human ETA and ETB with a Ki of 0.697 nM and 569 nM, respectively, and antagonized ET-1-induced vasoconstriction in isolated rat aorta with a pA2 value of 7.6 (Yuyama et al., 2003).
ETA inhibition with YM-598 significantly reduced tumour growth and liver metastasis in an in vivo model of gastric cancer (Fukui et al., 2007). In addition, YM-598 significantly inhibited ET-1-induced potentiation of nociception in murine models of cancer pain (Yuyama et al., 2004a,b;). The beneficial effect of YM-598 in preclinical models of cancer pain led to the initiation of two randomized Phase II clinical trials to investigate the impact of this agent on pain in patients. The first of these studies evaluated YM-598 monotherapy in patients with localized prostate cancer and the other investigated YM-598 combined with mitoxantrone and prednisone in patients with metastatic prostate cancer. Both trials were terminated due to a lack of pain reduction (Battistini et al., 2006). Previous studies have demonstrated the difficulty in translating preclinical studies of pain perception into the clinical setting, particularly where comparison with opiate analgesia is concerned (Bell et al., 2006), which may, in part explain the failure of these trials. The clinical development of YM-598 has been discontinued.
Atrasentan (Abbott Laboratories)
Atrasentan (ABT-627) is a selective ETA antagonist, which competitively inhibited 125I-ET-1 binding to cloned human ETA and ETB with Ki values of 69 pM and 139 nM, respectively, and antagonized ET-1-induced vasoconstriction in isolated rat aorta with a pA2 value of 9.2 (Opgenorth et al., 1996). Atrasentan decreased the binding affinity of ET-1 without affecting the receptor density, indicating that it is a competitive inhibitor of ET-1 binding, with 800- to 1800-fold selectivity for ETA compared with ETB (Wu-Wong et al., 2002).
A range of preclinical investigations of atrasentan in the cancer setting have demonstrated its potential anticancer activity. In brief, atrasentan dose dependently inhibited ET-1-driven prostate cancer cell line (PPC-1) proliferation (Nelson et al., 1996), inhibited neoangiogenesis in a cervical cancer xenograft model (Bagnato et al., 2002) and reduced osteoblastic bone metastases in mice inoculated with the ZR-75-1 breast cancer line (Guise et al., 2003). When atrasentan was combined with paclitaxel or docetaxel, additive antitumour, pro-apoptotic and antiangiogenic effects were observed in ovarian cancer cells and prostate cancer cells, respectively (Rosanòet al., 2003; Banerjee et al., 2007).
As shown in Table 1, a number of Phase II and Phase III clinical trials of atrasentan have been completed. Several of these were performed in men with CRPC. The largest Phase II trial enrolled 288 men with asymptomatic CRPC and radiographic evidence of metastatic disease. Patients were randomized to receive once-daily atrasentan 2.5 mg, 10 mg or placebo. The primary endpoint of TTP was significantly increased in a subset of evaluable patients [defined before unmasking of the study by excluding patients who did not meet study-defined prostate-specific antigen (PSA) or antiandrogen withdrawal inclusion criteria, were taking excluded medications, received less than 50% of scheduled doses or fewer than 20 total doses, or initiated excluded medications during the study; n = 244], from 129 days in the placebo group to 196 days in the atrasentan 10 mg group (P = 0.021). However, median TTP was not significant in the intent to treat (ITT) population (median 183, 178 and 137 days, for atrasentan 10 mg, 2.5 mg and placebo respectively; P = 0.13 and P = 0.29 for comparisons of atrasentan 10 mg and 2.5 mg with placebo, respectively). The secondary endpoint of time to PSA progression was significant in the ITT population with a median time of 155 days for atrasentan 10 mg, 141 days for atrasentan 2.5 mg and 71 days for placebo (P = 0.002 and P = 0.055 compared with placebo, respectively). In the evaluable population, median time to PSA progression was also significantly longer in the atrasentan 10 mg group compared with placebo (P = 0.002). A favourable tolerability profile of atrasentan was also observed in this patient population (Carducci et al., 2003). This potential to delay the progression of CRPC along with the favourable tolerability profile led to the initiation of two Phase III studies of atrasentan in this disease setting. These Phase III, randomized, double-blind, placebo-controlled trials of once-daily atrasentan 10 mg in the metastatic and non-metastatic CRPC setting failed to meet their primary endpoint of TTP or their secondary endpoint of time to PSA progression (Carducci et al., 2007; Nelson et al., 2008). In the first of these studies in 809 patients with metastatic CRPC, atrasentan did not reduce the risk of disease progression relative to placebo [hazard ratio (HR), 0.89; 95% confidence interval (CI) 0.76, 1.04; P = 0.136; Figure 2A]. Most patients progressed radiographically at the first 12 week bone scan without concomitant clinical progression (Carducci et al., 2007). In the second study of 941 patients with non-metastatic CRPC, there was a 93 day delay in the median TTP (defined as the onset of metastases) following treatment with atrasentan 10 mg; however, this was not statistically significant when compared with placebo (HR, 0.92; 95% CI 0.77, 1.09; P = 0.288; Figure 2B). There were, however, large geographic differences observed between the US and non-US sites in this second study. The difference in TTP was 81 and 180 days longer in the US and non-US sites, respectively, following atrasentan treatment compared with placebo treatment. It is thought that the failure of these two Phase III studies of atrasentan may be due to the design (method of assessment of disease progression in the metastatic CRPC trial) and conduct (geographical differences observed in the non-metastatic CRPC trial) of the trials. Atrasentan was generally well tolerated in these studies. The most common adverse events associated with treatment were headache, rhinitis and peripheral oedema, which reflect the vasodilatory and fluid-retention properties of ETA antagonism (Carducci et al., 2007; Nelson et al., 2008).
Figure 2.
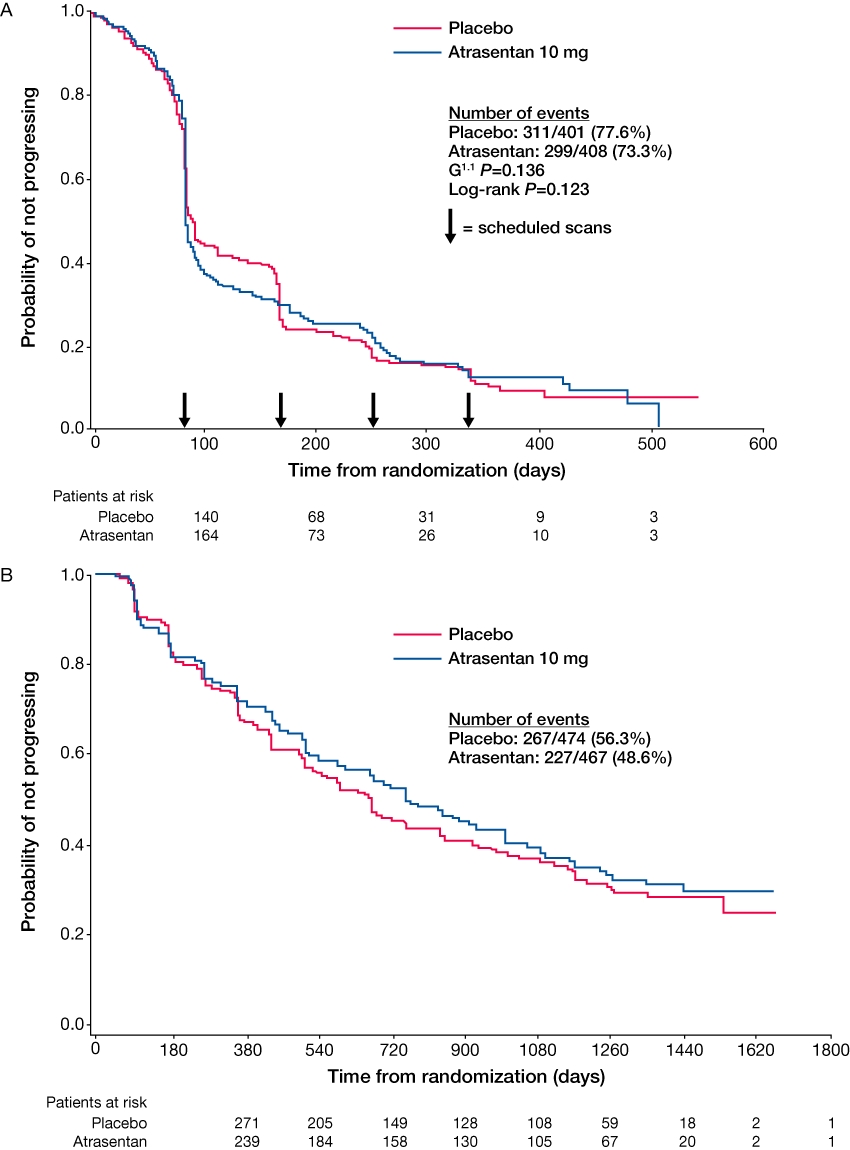
Time to disease progression following treatment with atrasentan in men with (A) metastatic and (B) non-metastatic castration-resistant prostate cancer (Carducci et al., 2007; Nelson et al., 2008). Reprinted with permission from John Wiley and Sons.
Atrasentan has also been investigated in combination with docetaxel in a Phase I-II study of patients with metastatic CRPC to determine the maximum tolerated dose (MTD) of docetaxel in this combination and to investigate preliminary efficacy. The MTD dose of docetaxel once every 3 weeks in combination with once-daily atrasentan 10 mg was 70 to 75 mg·m−2 and PFS and OS were comparable to that seen with docetaxel and prednisone (Armstrong et al., 2008). This combination regimen is currently being investigated in a Phase III trial.
A Phase I-II study of atrasentan in combination with paclitaxel and carboplatin in chemotherapy-naïve patients with stage IIIB and IV NSCLC reported that this combination was well tolerated, and efficacy (Response Evaluation Criteria In Solid Tumors) and median survival were comparable with chemotherapy alone (Chiappori et al., 2008). Atrasentan has also been investigated in a Phase I safety study in patients with progressive or recurrent malignant glioma (Phuphanich et al., 2008), and in a Phase II trial of patients with locally recurrent or metastatic kidney cancer, data from which did not support further investigation as monotherapy in this patient population (Manola et al., 2007).
Zibotentan (AstraZeneca)
Zibotentan (ZD4054) is a specific ETA antagonist, which potently inhibited 125I-ET-1 binding to cloned human ETA expressed in mouse erythroleukaemic cells and membranes with an IC50 value of 21 nM at ETA and an undetectable IC50 value at ETB at concentrations up to 100 µM (Bradbury et al., 1997; Morris et al., 2005b). In vitro competitive binding assays suggest that zibotentan is a less potent inhibitor of ETA activity compared with other selective ETA receptor antagonists (Battistini et al., 2006). However, the relative potency in vitro may not necessarily equate with potency in vivo. In addition, unlike other agents which have activity at both ETA and ETB, the absence of any effect of zibotentan at ETB may be more clinically important than its relative potency versus ETA. Indeed, in clinical trials with zibotentan, population pharmacokinetic data have revealed that steady state plasma concentrations of zibotentan following daily oral administration of 10 mg were well in excess of the non-clinical cellular IC50 values and therefore should achieve sufficient ETA antagonism. In a clinical trial that evaluated the effect of zibotentan on ET-1-induced forearm blood flow in healthy volunteers, it was shown that zibotentan 10 mg and 30 mg antagonized the vasoconstrictor effect of infused ET-1, providing evidence that zibotentan was pharmacologically active at these doses (Morris et al., 2005a). Moreover, no evidence of zibotentan-induced ETB inhibition was detected following administration of zibotentan at doses of 2.5–240 mg, demonstrated by mean plasma levels of ET-1 being within the placebo range at 4 and 24 h post dose (Morris et al., 2005b). In addition, adverse events of headache (a vasodilatory consequence of ETA antagonism) have been reported following treatment with zibotentan 10 mg (James et al., 2010). These data confirmed that zibotentan is the first specific rather than selective ETA antagonist (Bradbury et al., 1997; Morris et al., 2005b). Because of the activities exhibited through ETB, such as induction of apoptosis and clearance of ET-1, specific antagonism of ETA, with no inhibition of ETB, offers the promise of a differentiated anticancer therapy.
A range of preclinical studies have been undertaken with zibotentan, the majority of which were in models of prostate and ovarian cancer. These studies have demonstrated that complete blockade of ETA with zibotentan reversed ET-1-induced inhibition of apoptosis while allowing pro-apoptotic signalling via ETB (Rosanòet al., 2007a). Zibotentan in combination with paclitaxel or docetaxel was also shown to enhance chemotherapy-induced apoptosis compared with either agent alone (Growcott, 2009). A role for zibotentan in the inhibition of cell proliferation and invasion has been reported; zibotentan was observed to inhibit proliferation of human immature pre-osteoblast cells (Growcott, 2009), and dose-dependently inhibit ET-1-mediated changes in cellular invasiveness in human ovarian cancer cells (Rosanòet al., 2007b). Moreover, in human breast cancer cell lines, zibotentan combined with aromatase inhibitors or fulvestrant produced at least an additive inhibition of cell migration and invasion (Growcott, 2009). Zibotentan in combination with pamidronate completely blocked the development of bone metastases and reduced brain and lung metastases in severe combined immune deficient mice inoculated with a human bladder cancer cell line (Growcott, 2009). In addition, inhibition of tumour angiogenesis in prostate, colorectal and ovarian tumour xenografts has been demonstrated following treatment with zibotentan (Curwen et al., 2007; Rosanòet al., 2007b). Furthermore, in a murine xenograft of ovarian carcinoma, tumour growth and metastasis were reduced following treatment with zibotentan. The tumour growth inhibition was enhanced with the addition of a cytotoxic drug (paclitaxel) or a molecular inhibitor (gefitinib) (Rosanòet al., 2007a,b, 2009). These and other preclinical findings provided a rationale for the use of zibotentan in clinical studies.
The efficacy and safety of zibotentan in patients with metastatic CRPC who were pain free or mildly symptomatic for pain was studied in a double-blind, placebo-controlled Phase II clinical trial (James et al., 2009; 2010;). Patients (n = 312) were randomized to receive once-daily zibotentan 10 mg, 15 mg or placebo. The primary endpoint was TTP, defined as time from randomization to clinical progression, objective progression of soft tissue metastasis on CT scan, or death in the absence of progression. Secondary endpoints included OS and time to PSA progression. Three analyses were conducted; at the primary interim analysis there was no difference between the zibotentan groups and placebo for TTP (4.0, 3.8 and 3.6 months for zibotentan 10 mg, 15 mg and placebo respectively; zibotentan 10 mg vs. placebo, HR 0.88; 80% CI 0.71, 1.09; zibotentan 15 mg vs. placebo, HR 0.83; 80% CI, 0.66, 1.03). However, a signal for prolonged OS was observed in the zibotentan treatment groups versus placebo. At the second interim analysis, TTP was not significantly increased following treatment with zibotentan 10 mg or 15 mg when compared with placebo (4.6, 3.8 and 3.7 months respectively; zibotentan 10 mg vs. placebo, HR 1.09; 80% CI 0.91, 1.31; P = 0.553; zibotentan 15 mg vs. placebo, HR 0.94; 80% CI 0.78, 1.14; P = 0.702). However, the secondary endpoint of OS was significantly increased from 17.3 months to 24.5 months in patients receiving zibotentan 10 mg compared with patients receiving placebo (HR 0.55; 80% CI 0.41, 0.73; P = 0.008). Compared with placebo, patients receiving zibotentan 15 mg also had an improvement in OS with a median survival of 23.5 months (HR 0.65; 80% CI 0.49, 0.86; P = 0.052) (James et al., 2009). At the final analysis (Figure 3), the difference in OS was still evident, although it had decreased in patients receiving zibotentan 10 mg and 15 mg when compared with placebo (median OS 23.5, 23.9 and 19.9 months, respectively) (James et al., 2010). However, consistent with previous analyses, HRs of less than one were sustained for both zibotentan 10 mg (HR 0.83, 80% CI 0.67, 1.02, P = 0.254) and 15 mg (HR 0.76, 80% CI 0.61, 0.94, P = 0.103). Plasma ET-1 levels were measured at baseline and at 4 weeks and 8 weeks following randomization. A small increase from baseline was observed in those patients treated with zibotentan whereas little change in plasma ET-1 levels was observed in placebo treated patients. Zibotentan was well tolerated, with the most commonly reported adverse events considered to be related to zibotentan treatment being peripheral oedema, headache and nasal congestion.
Figure 3.
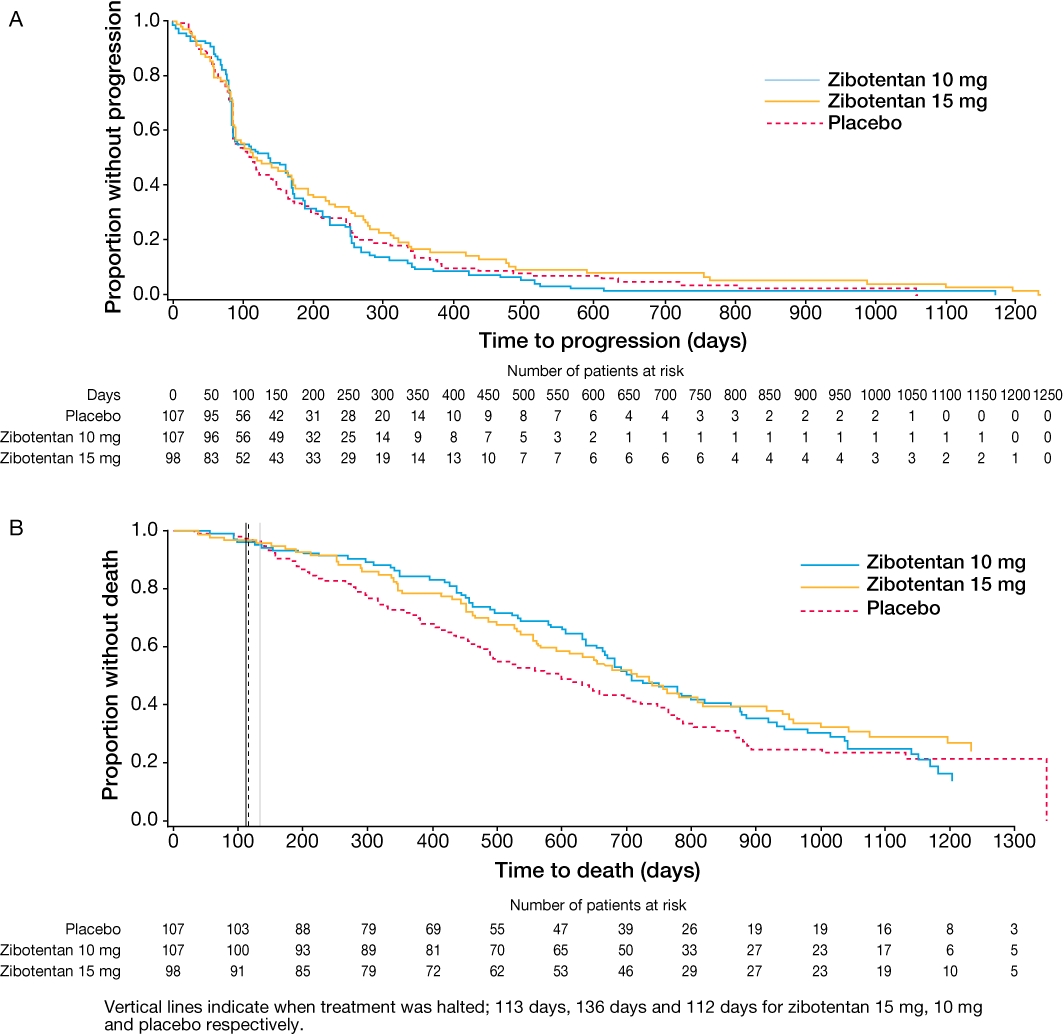
(A) Time to progression and (B) overall survival of patients with castration-resistant prostate cancer treated with zibotentan or placebo (final analysis) (James et al., 2010). Reprinted with permission from John Wiley and Sons.
Zibotentan 10 mg once daily was investigated in combination with docetaxel 75 mg·m−2 once every 3 weeks in a Phase I clinical trial of patients with CRPC to evaluate the tolerability and preliminary efficacy of this combination. This treatment was demonstrated to be well tolerated with no safety concerns and preliminary efficacy was noted (Trump et al., 2010).
A large Phase III clinical trial programme [ENdoTHelin A USE (ENTHUSE)] is further evaluating the therapeutic potential of zibotentan in men with CRPC. Clinical studies to date of zibotentan in prostate cancer, along with preclinical evidence of an anticancer effect of zibotentan in ovarian cancer cells, provide a rationale to investigate this agent in clinical trials of patients with ovarian cancer. As such, a Phase II trial of zibotentan plus carboplatin and paclitaxel, or placebo plus carboplatin and paclitaxel in patients with advanced ovarian cancer sensitive to platinum-based chemotherapy is ongoing. Other zibotentan clinical trials which have either recently been completed, or are ongoing or planned, include a Phase I trial of zibotentan in male, elderly Chinese patients with advanced solid malignancies, and a Phase II trial of zibotentan in combination with pemetrexed in patients with NSCLC.
In general, clinical trials should aim to collect sufficient events and maturity of data to have confidence in the results. There is currently a great deal of discussion regarding the appropriate endpoints for use in clinical trials of patients with cancer. Biomarker-based assessment and PFS are not confounded by subsequent therapy; however, OS is an unambiguous demonstration of clinical benefit. Surrogacy between biomarkers, PFS and OS endpoints has not been formally proved for prostate cancer. The required duration of trials will be dependent on the primary endpoint. In CRPC, trials can be relatively short for agents which are anticipated to have an effect on PSA, circulating tumour cells or PFS which will subsequently translate into OS benefits. However, for targeted agents such as zibotentan which primarily have a cytostatic action and therefore do not affect intermediate endpoints, OS will be the primary endpoint and trials will need to be longer. The zibotentan Phase II study reported a significant survival advantage at the interim analysis which was not present at the final analysis. Treatment was halted at 113 days, 136 days and 112 days for zibotentan 15 mg, 10 mg and placebo, respectively. The Kaplan–Meier curves for each arm were parallel after treatment was stopped which suggests that patients should remain on treatment provided they are deriving benefit and follow-up periods of trials of ETA antagonists should be of sufficient duration to allow the full effect of a treatment to be observed (James et al., 2010). In addition, the survival of placebo-treated patients in prostate cancer trials conducted over the last few years appears to have improved. For example, differences have been observed between the Phase II trial of zibotentan (James et al., 2010) and the recent Sipuleucel-T trial (Kantoff et al., 2010). However, as these studies had different inclusion criteria and were conducted in different geographical areas it is not possible to know whether this reflects true changes in life expectancy for metastatic CRPC over time or if this simply reflects the different inclusion criteria of these prospective randomized studies.
Targeting the ET axis in other diseases
The ETA antagonists sitaxsentan and ambrisentan have been approved for the treatment of pulmonary arterial hypertension; however, these compounds are not currently being evaluated in the oncology setting.
Studies have utilized ETA antagonists, with little improvement in morbidity and mortality, for the treatment of diseases such as chronic heart failure (CHF), where ET-1 and the ETA are substantially elevated and correlate with the severity of disease (Kelland and Webb, 2006). Several reasons for this lack of efficacy have been proposed. It is thought that the benefits of ET blockade in CHF may derive from a truly ETA selective approach. Patients with CHF who had selective blockade of the ETB with BQ-788 experienced systemic vasoconstriction and elevation of plasma ET-1 concentrations. Endothelin dysfunction was improved after ETA blockade, but not combined ETA/ETB blockade in the coronary microcirculation of patients with ischaemic heart disease. Treatment with high doses of selective ETA antagonists can result in ETB blockade, causing increased ET-1 concentrations resulting in chronic systemic vasoconstriction (Kelland and Webb, 2006). However, in trials of patients with CHF, many patients benefit from long-term treatment with ETA antagonists even though there are some poorly understood negative outcomes after initial acute dosing (Kelland and Webb, 2006).
Conclusions
GPCRs have been linked to many processes involved in normal cell function and dysfunction of these receptors is associated with the development and progression of a range of diseases including cancer. Indeed, the ET-1/ETA axis is associated with the development and progression of several types of cancer and is therefore an interesting target for the treatment of tumours. Although many preclinical and clinical studies have suggested that the ET axis contributes towards the development and progression of cancers, the specific mechanisms by which this occurs are still to be elucidated.
A range of specific and selective ETA antagonists and dual ET receptor antagonists have undergone preclinical and clinical studies showing variable efficacy in the cancer setting. Several Phase II clinical trials have reported positive results, such as the promising OS signal observed in the trial of zibotentan in patients with metastatic CRPC. In addition to their potential as monotherapies, experimental and preclinical evidence suggests that ETA antagonists may be able to potentiate the therapeutic efficacy of conventional cytotoxic drugs, offering a rationale for clinical evaluation of this approach. Another novel and promising cancer treatment strategy is the incorporation of ETA blockade with other molecular-targeted drugs to therapeutically overcome compensatory mechanisms of escape. Careful study design and learning from the experience of studies completed to date will be the key to fully evaluating the potential of these approaches. This will be supported by improved preclinical models, including three-dimensional tumour cultures and co-culture models, which more accurately reflect the disease in man. While such models are in development, the use of existing models (largely validated using cytotoxic agents) should take into account the different mechanisms of action between ET antagonists and cytotoxic agents.
A role for ETB in other cancer types such as melanoma and glioma is also emerging. Furthermore, the potential role of ETB in tumour-related immune responses is intriguing and warrants further investigation. Agonism of ETB has also been proposed as an alternative approach to block the effects of ETA. SP-1620 (IRL-1620) has been investigated in this context (Takai et al., 1992) and the first Phase I clinical trial of this agent, currently recruiting, is being undertaken in patients with recurrent or progressive carcinoma.
A more personalized approach to the treatment of cancers will allow targeted agents such as ETA antagonists to be used to treat those patients who will derive the greatest benefit. However, appropriate patient selection will require clinically validated markers of disease and response to treatment. Suitable markers that are reliable and easy to detect are needed to help identify those patients who are most likely to respond to ETA antagonism.
To date, specific inhibition of ETA with antagonists such as zibotentan has shown the most promise as a therapeutic approach for the treatment of cancer providing the rational basis for the design of more focused clinical trials. The results of the clinical trials of zibotentan as a monotherapy and in combination with cytotoxic drugs are awaited to unravel the opportunities to interfere with critical tumorigenic signals by targeting ETA-mediated pathways.
Acknowledgments
We thank Dr Claire Routley from Mudskipper who provided editorial assistance funded by AstraZeneca.
Glossary
Abbreviations
- CHF
chronic heart failure
- CRPC
castration-resistant prostate cancer
- ECE
endothelin-converting enzyme
- ET
endothelin
- ETA
endothelin A receptor
- ETB
endothelin B receptor
- GPCR
G-protein coupled receptor
- ITT
intent to treat
- MTD
maximum tolerated dose
- NEP
neutral endopeptidase
- NSCLC
non-small-cell lung cancer
- OS
overall survival
- PFS
progression-free survival
- PPC-1
prostate cancer cell line
- PSA
prostate-specific antigen
- TTP
time to tumour progression
Conflicts of interest
Anna Bagnato has no conflicts of interest to declare. Marilena Loizidou received research consumables from AstraZeneca to investigate the efficacy of zibotentan in colorectal cancer models. Beth Pflug has received funding support from AstraZeneca for basic science/translational studies on the ET axis. Jon Curwen and Jim Growcott are employees of AstraZeneca, and Jim Growcott holds AstraZeneca stock options.
Supporting Information
Teaching Materials; Figs 1–3 as PowerPoint slide.
References
- Abassi ZA, Tate JE, Golomb E, Keiser HR. Role of neutral endopeptidase in the metabolism of endothelin. Hypertension. 1992;20:89–95. doi: 10.1161/01.hyp.20.1.89. [DOI] [PubMed] [Google Scholar]
- Ahmed SI, Thompson J, Coulson JM, Woll PJ. Studies on the expression of endothelin, its receptor subtypes, and converting enzymes in lung cancer and in human bronchial epithelium. Am J Respir Cell Mol Biol. 2000;22:422–431. doi: 10.1165/ajrcmb.22.4.3795. [DOI] [PubMed] [Google Scholar]
- Alanen K, Deng DX, Chakrabarti S. Augmented expression of endothelin-1, endothelin-3 and the endothelin-B receptor in breast carcinoma. Histopathology. 2000;36:161–167. doi: 10.1046/j.1365-2559.2000.00795.x. [DOI] [PubMed] [Google Scholar]
- Anguelova E, Beuvon F, Leonard N, Chaverot N, Varlet P, Couraud PO, et al. Functional endothelin ET B receptors are selectively expressed in human oligodendrogliomas. Brain Res Mol Brain Res. 2005;137:77–88. doi: 10.1016/j.molbrainres.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Armstrong AJ, Creel P, Turnbull J, Moore C, Jaffe TA, Haley S, et al. A Phase I-II study of docetaxel and atrasentan in men with castration-resistant metastatic prostate cancer. Clin Cancer Res. 2008;14:6270–6276. doi: 10.1158/1078-0432.CCR-08-1085. [DOI] [PubMed] [Google Scholar]
- Asham E, Shankar A, Loizidou M, Fredericks S, Miller K, Boulos PB, et al. Increased endothelin-1 in colorectal cancer and reduction of tumour growth by ETA receptor antagonism. Br J Cancer. 2001;85:1759–1763. doi: 10.1054/bjoc.2001.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato A, Natali PG. Endothelin receptors as novel targets in tumor therapy. J Transl Med. 2004;2:16. doi: 10.1186/1479-5876-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato A, Rosanò L. The endothelin axis in cancer. Int J Biochem Cell Biol. 2008;40:1443–1451. doi: 10.1016/j.biocel.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Bagnato A, Tecce R, Moretti C, Di Castro V, Spergel D, Catt KJ. Autocrine actions of endothelin-1 as a growth factor in human ovarian carcinoma cells. Clin Cancer Res. 1995;1:1059–1066. [PubMed] [Google Scholar]
- Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra R, et al. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res. 1999;59:720–727. [PubMed] [Google Scholar]
- Bagnato A, Cirilli A, Salani D, Simeone P, Muller A, Nicotra MR, et al. Growth inhibition of cervix carcinoma cells in vivo by endothelin A receptor blockade. Cancer Res. 2002;62:6381–6384. [PubMed] [Google Scholar]
- Bagnato A, Rosanò L, Spinella F, Di Castro V, Tecce R, Natali PG. Endothelin B receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res. 2004;64:1436–1443. doi: 10.1158/0008-5472.can-03-2344. [DOI] [PubMed] [Google Scholar]
- Bagnato A, Spinella F, Rosanò L. Emerging role of the endothelin axis in ovarian tumor progression. Endocr Relat Cancer. 2005;12:761–772. doi: 10.1677/erc.1.01077. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Hussain M, Wang Z, Saliganan A, Che M, Bonfil D, et al. In vitro and in vivo molecular evidence for better therapeutic efficacy of ABT-627 and taxotere combination in prostate cancer. Cancer Res. 2007;67:3818–3826. doi: 10.1158/0008-5472.CAN-06-3879. [DOI] [PubMed] [Google Scholar]
- Battistini B, Berthiaume N, Kelland NF, Webb DJ, Kohan DE. Profile of past and current clinical trials involving endothelin receptor antagonists: the novel ‘-sentan’ class of drug. Exp Biol Med (Maywood) 2006;231:653–695. [PubMed] [Google Scholar]
- Bell RF, Wisløff T, Eccleston C, Kalso E. Controlled clinical trials in cancer pain. How controlled should they be? A qualitative systematic review. Br J Cancer. 2006;94:1559–1567. doi: 10.1038/sj.bjc.6603162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Y, Bernasconi CC, Juillerat-Jeanneret L. Targeting the endothelin axis in human melanoma: combination of endothelin receptor antagonism and alkylating agents. Exp Biol Med (Maywood) 2006;231:1111–1119. [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Bradbury RH, Bath C, Butlin RJ, Dennis M, Heys C, Hunt SJ, et al. New non-peptide endothelin-A receptor antagonists: synthesis, biological properties, and structure-activity relationships of 5-(dimethylamino)-N-pyridyl-,-N-pyrimidinyl-,-N-pyridazinyl-, and -N-pyrazinyl-1-naphthalenesulfonamides. J Med Chem. 1997;40:996–1004. doi: 10.1021/jm9604585. [DOI] [PubMed] [Google Scholar]
- Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000;275:17596–17604. doi: 10.1074/jbc.M000142200. [DOI] [PubMed] [Google Scholar]
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- Carducci MA, Padley RJ, Breul J, Vogelzang NJ, Zonnenberg BA, Daliani DD, et al. Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: a randomized, phase II, placebo-controlled trial. J Clin Oncol. 2003;21:679–689. doi: 10.1200/JCO.2003.04.176. [DOI] [PubMed] [Google Scholar]
- Carducci MA, Saad F, Abrahamsson PA, Dearnaley DP, Schulman CC, North SA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer. 2007;110:1959–1966. doi: 10.1002/cncr.22996. [DOI] [PubMed] [Google Scholar]
- Chiappori AA, Haura E, Rodriguez FA, Boulware D, Kapoor R, Neuger AM, et al. Phase I/II study of atrasentan, an endothelin A receptor antagonist, in combination with paclitaxel and carboplatin as first-line therapy in advanced non-small cell lung cancer. Clin Cancer Res. 2008;14:1464–1469. doi: 10.1158/1078-0432.CCR-07-1508. [DOI] [PubMed] [Google Scholar]
- Chichorro JG, Fiuza CR, Bressan E, Claudino RF, Leite DF, Rae GA. Endothelins as pronociceptive mediators of the rat trigeminal system: Role of ETA and ETB receptors. Brain Res. 2010;1345:73–83. doi: 10.1016/j.brainres.2010.04.075. [DOI] [PubMed] [Google Scholar]
- Clozel M, Breu V, Gray GA, Kalina B, Löffler B-M, Burri K, et al. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J Pharmacol Exp Ther. 1994;270:228–235. [PubMed] [Google Scholar]
- Curwen J, Hughes G, Hickinson M, Curtis N, Growcott JW, Isherwood B, et al. The impact of ZD4054, a specific endothelin A receptor antagonist, on tumor blood supply, invasion, and the bone microenvironment. Mol Cancer Ther. 2007;6:abst A272. [Google Scholar]
- Davenport AP. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol Rev. 2002;54:219–226. doi: 10.1124/pr.54.2.219. [DOI] [PubMed] [Google Scholar]
- Dawson LA, Maitland NJ, Turner AJ, Usmani BA. Stromal-epithelial interactions influence prostate cancer cell invasion by altering the balance of metallopeptidase expression. Br J Cancer. 2004;90:1577–1582. doi: 10.1038/sj.bjc.6601717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bufalo D, Di Castro V, Biroccio A, Salani D, Rosanò L, Spinella F, et al. Endothelin-1 acts as a survival factor in ovarian carcinoma cells. Clin Sci (Lond) 2002;103(Suppl. 48):302S–305S. doi: 10.1042/CS103S302S. [DOI] [PubMed] [Google Scholar]
- Demunter A, De Wolf-Peeters C, Degreef H, Stas M, van den Oord JJ. Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch. 2001;438:485–491. doi: 10.1007/s004280000362. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Jasmin JF, Prié S, Cernacek P. Importance of local production of endothelin-1 and of the ETB receptor in the regulation of pulmonary vascular tone. Pulm Pharmacol Ther. 2000;13:135–140. doi: 10.1006/pupt.2000.0242. [DOI] [PubMed] [Google Scholar]
- Egidy G, Juillerat-Jeanneret L, Jeannin JF, Korth P, Bosman FT, Pinet F. Modulation of human colon tumor-stromal interactions by the endothelin system. Am J Pathol. 2000;157:1863–1874. doi: 10.1016/S0002-9440(10)64825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero E, Labalde M, Fernández N, Monge L, Salcedo A, Narvaez-Sanchez R, et al. Response to endothelin-1 in arteries from human colorectal tumours: role of endothelin receptors. Exp Biol Med. 2008;233:1602–1607. doi: 10.3181/0802-RM-69. [DOI] [PubMed] [Google Scholar]
- Firth JD, Ratcliffe PJ. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J Clin Invest. 1992;90:1023–1031. doi: 10.1172/JCI115915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami T, Nagase T, Fujita K, Hayama T, Niiyama K, Mase T, et al. Structure-activity relationships of cyclic pentapeptide endothelin A receptor antagonists. J Med Chem. 1995;38:4309–4324. doi: 10.1021/jm00021a021. [DOI] [PubMed] [Google Scholar]
- Fukui R, Nishimori H, Hata F, Yasoshima T, Ohno K, Yanai Y, et al. Inhibitory effect of endothelin A receptor blockade on tumor growth and liver metastasis of a human gastric cancer cell line. Gastric Cancer. 2007;10:123–128. doi: 10.1007/s10120-007-0421-z. [DOI] [PubMed] [Google Scholar]
- Godara G, Cannon GW, Cannon GM, Jr, Bies RR, Nelson JB, Pflug BR. Role of endothelin axis in progression to aggressive phenotype of prostate adenocarcinoma. Prostate. 2005;65:27–34. doi: 10.1002/pros.20252. [DOI] [PubMed] [Google Scholar]
- Goldie RG. Endothelins in health and disease: an overview. Clin Exp Pharmacol Physiol. 1999;26:145–148. doi: 10.1046/j.1440-1681.1999.03014.x. [DOI] [PubMed] [Google Scholar]
- Grant K, Knowles J, Dawas K, Burnstock G, Taylor I, Loizidou M. Mechanisms of endothelin 1-stimulated proliferation in colorectal cancer cell lines. Br J Surg. 2007;94:106–112. doi: 10.1002/bjs.5536. [DOI] [PubMed] [Google Scholar]
- Grimshaw MJ, Hagemann T, Ayhan A, Gillett CE, Binder C, Balkwill FR. A role for endothelin-2 and its receptors in breast tumor cell invasion. Cancer Res. 2004;64:2461–2468. doi: 10.1158/0008-5472.can-03-1069. [DOI] [PubMed] [Google Scholar]
- Growcott JW. Preclinical anticancer activity of the specific endothelin A receptor antagonist ZD4054. Anticancer Drugs. 2009;20:83–88. doi: 10.1097/CAD.0b013e328320791c. [DOI] [PubMed] [Google Scholar]
- Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–784. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- Gulati A, Rai A. Endothelin-1-induced vasodilatation in rat breast tumor is mediated through endothelin-B receptors. J Cardiovasc Pharmacol. 2004;44(Suppl. 1):S483–S486. doi: 10.1097/01.fjc.0000166308.63808.16. [DOI] [PubMed] [Google Scholar]
- Guruli G, Pflug BR, Pecher S, Makarenkova V, Shurin MR, Nelson JB. Function and survival of dendritic cells depend on endothelin-1 and endothelin receptor autocrine loops. Blood. 2004;104:2107–2115. doi: 10.1182/blood-2003-10-3559. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Fukami T, Nagase T, Fujita K, Hayama T, Niiyama K, et al. Cyclic pentapeptide endothelin antagonists with high ETA selectivity. Potency- and solubility-enhancing modifications. J Med Chem. 1992;35:2139–2142. doi: 10.1021/jm00089a028. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Ihara M, Noguchi K, Mase T, Mino N, Saeki T, et al. Biochemical and pharmacological profile of a potent and selective endothelin B-receptor antagonist, BQ-788. Proc Natl Acad Sci U S A. 1994;91:4892–4896. doi: 10.1073/pnas.91.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James ND, Caty A, Borre M, Zonnenberg BA, Beuzeboc P, Morris T, et al. Safety and efficacy of the specific endothelin-A receptor antagonist ZD4054 in patients with hormone-resistant prostate cancer and bone metastases who were pain free or mildly symptomatic: a double-blind, placebo-controlled, randomized, Phase II trial. Eur Urol. 2009;55:1112–1123. doi: 10.1016/j.eururo.2008.11.002. [DOI] [PubMed] [Google Scholar]
- James ND, Caty A, Payne H, Borre M, Zonnenberg BA, Beuzeboc P, et al. Final safety and efficacy analysis of the specific endothelin A receptor antagonist zibotentan (ZD4054) in patients with metastatic castration-resistant prostate cancer and bone metastases who were pain-free or mildly symptomatic for pain: a double-blind, placebo-controlled, randomized Phase II trial. BJU Int. 2010;106:966–973. doi: 10.1111/j.1464-410X.2010.09638.x. [DOI] [PubMed] [Google Scholar]
- Jerónimo C, Henrique R, Campos PF, Oliveira J, Caballero OL, Lopes C, et al. Endothelin B receptor gene hypermethylation in prostate adenocarcinoma. J Clin Pathol. 2003;56:52–55. doi: 10.1136/jcp.56.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juergens UR, Racké K, Uen S, Haag S, Lamyel F, Stöber M, et al. Inflammatory responses after endothelin B (ETB) receptor activation in human monocytes: new evidence for beneficial anti-inflammatory potency of ETB-receptor antagonism. Pulm Pharmacol Ther. 2008;21:533–539. doi: 10.1016/j.pupt.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Kandalaft LE, Facciabene A, Buckanovich RJ, Coukos G. Endothelin B receptor, a new target in cancer immune therapy. Clin Cancer Res. 2009;15:4521–4528. doi: 10.1158/1078-0432.CCR-08-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- Karet FE, Davenport AP. Localization of endothelin peptides in human kidney. Kidney Int. 1996;49:382–387. doi: 10.1038/ki.1996.56. [DOI] [PubMed] [Google Scholar]
- Kefford R, Beith JM, Van Hazel GA, Millward M, Trotter JM, Wyld DK, et al. A Phase II study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage IV metastatic melanoma. Invest New Drugs. 2007;25:247–252. doi: 10.1007/s10637-006-9014-7. [DOI] [PubMed] [Google Scholar]
- Kefford RF, Clingan PR, Brady B, Ballmer A, Morganti A, Hersey P. A randomized, double-blind, placebo-controlled study of high-dose bosentan in patients with stage IV metastatic melanoma receiving first-line dacarbazine chemotherapy. Mol Cancer. 2010;9:69. doi: 10.1186/1476-4598-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland NF, Webb DJ. Clinical trials of endothelin antagonists in heart failure: a question of dose? Exp Biol Med (Maywood) 2006;231:696–699. [PubMed] [Google Scholar]
- Khodorova A, Montmayeur J-P, Strichartz G. Endothelin receptors and pain. J Pain. 2009;10:4–28. doi: 10.1016/j.jpain.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol. 2008;153:1105–1119. doi: 10.1038/sj.bjp.0707516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight LJ, Burrage J, Bujac SR, Haggerty C, Graham A, Gibson NJ, et al. Epigenetic silencing of the endothelin-B receptor gene in non-small cell lung cancer. Int J Oncol. 2009;34:465–471. [PubMed] [Google Scholar]
- Kohan DE. Endothelins in the normal and diseased kidney. Am J Kidney Dis. 1997;29:2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- Kuc RE, Davenport AP. Comparison of endothelin-A and endothelin-B receptor distribution visualized by radioligand binding versus immunocytochemical localization using subtype selective antisera. J Cardiovasc Pharmacol. 2004;44(Suppl. 1):S224–S226. doi: 10.1097/01.fjc.0000166260.35099.d5. [DOI] [PubMed] [Google Scholar]
- Lahav R. Endothelin receptor B is required for the expansion of melanocyte precursors and malignant melanoma. Int J Dev Biol. 2005;49:173–180. doi: 10.1387/ijdb.041951rl. [DOI] [PubMed] [Google Scholar]
- Lahav R, Heffner G, Patterson PH. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci U S A. 1999;96:11496–11500. doi: 10.1073/pnas.96.20.11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert LA, Whyteside AR, Turner AJ, Usmani BA. Isoforms of endothelin-converting enzyme-1 (ECE-1) have opposing effects on prostate cancer cell invasion. Br J Cancer. 2008;99:1114–1120. doi: 10.1038/sj.bjc.6604631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PS. The peptide hormone angiotensin II: its new functions in tissues and organs. Curr Protein Pept Sci. 2004;5:267–273. doi: 10.2174/1389203043379693. [DOI] [PubMed] [Google Scholar]
- Levin ER. Endothelins. N Engl J Med. 1995;333:356–363. doi: 10.1056/NEJM199508103330607. [DOI] [PubMed] [Google Scholar]
- Manola J, Carducci M, Nair S, Liu G, Rousey S, Wilding G. Phase II ECOG trial of atrasentan in advanced renal cell carcinoma. J Clin Oncol. 2007;25:abst 5102. [Google Scholar]
- Michaelson MD, Kaufman DS, Kantoff P, Oh WK, Smith MR. Randomized phase II study of atrasentan alone or in combination with zoledronic acid in men with metastatic prostate cancer. Cancer. 2006;107:530–535. doi: 10.1002/cncr.22043. [DOI] [PubMed] [Google Scholar]
- Molenaar P, O'Reilly G, Sharkey A, Kuc RE, Harding DP, Plumpton C, et al. Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ Res. 1993;72:526–538. doi: 10.1161/01.res.72.3.526. [DOI] [PubMed] [Google Scholar]
- Morris CD, Hughes A, Rose A, Melville V, Webb DJ. ZD4054 reduces endothelin-1-induced forearm vasoconstriction in healthy male volunteers. Proc Am Assoc Cancer Res. 2005a;46:abst 4187. [Google Scholar]
- Morris CD, Rose A, Curwen J, Hughes AM, Wilson DJ, Webb DJ. Specific inhibition of the endothelin A receptor with ZD4054: clinical and pre-clinical evidence. Br J Cancer. 2005b;92:2148–2152. doi: 10.1038/sj.bjc.6602676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motte S, McEntee K, Naeije R. Endothelin receptor antagonists. Pharmacol Ther. 2006;110:386–414. doi: 10.1016/j.pharmthera.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Nelson JB. Endothelin inhibition: novel therapy for prostate cancer. J Urol. 2003;170:S65–S68. doi: 10.1097/01.ju.0000096372.07687.86. [DOI] [PubMed] [Google Scholar]
- Nelson JB. Endothelin receptors as therapeutic targets in castration-resistant prostate cancer. Eur Urol Suppl. 2009;8:20–28. [Google Scholar]
- Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995;1:944–949. doi: 10.1038/nm0995-944. [DOI] [PubMed] [Google Scholar]
- Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, Opgenorth TJ, Bova GS, et al. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996;56:663–668. [PubMed] [Google Scholar]
- Nelson JB, Lee W-H, Nguyen SH, Jarrard DF, Brooks JD, Magnuson SR, et al. Methylation of the 5′ CpG island of the endothelin B receptor gene is common in human prostate cancer. Cancer Res. 1997;57:35–37. [PubMed] [Google Scholar]
- Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer. 2003;3:110–116. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer. 2008;113:2478–2487. doi: 10.1002/cncr.23864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez DJ, Brown MJ, Davenport AP, Neylon CB, Schofield JP, Wyse RK. Endothelin-1 mRNA is widely expressed in porcine and human tissues. J Clin Invest. 1990;85:1537–1541. doi: 10.1172/JCI114601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussdorfer GG, Rossi GP, Malendowicz LK, Mazzocchi G. Autocrine-paracrine endothelin system in the physiology and pathology of steroid-secreting tissues. Pharmacol Rev. 1999;51:403–438. [PubMed] [Google Scholar]
- Okada M, Nishikibe M. BQ-788, a selective endothelin ETB receptor antagonist. Cardiovasc Drug Rev. 2002;20:53–66. doi: 10.1111/j.1527-3466.2002.tb00082.x. [DOI] [PubMed] [Google Scholar]
- Okazawa M, Shiraki T, Ninomiya H, Kobayashi S, Masaki T. Endothelin-induced apoptosis of A375 human melanoma cells. J Biol Chem. 1998;273:12584–12592. doi: 10.1074/jbc.273.20.12584. [DOI] [PubMed] [Google Scholar]
- Opgenorth TJ, Adler AL, Calzadilla SV, Chiou WJ, Dayton BD, Dixon DB, et al. Pharmacological characterization of A-127722: an orally active and highly potent ETA-selective receptor antagonist. J Pharmacol Exp Ther. 1996;276:473–481. [PubMed] [Google Scholar]
- Pao MM, Tsutsumi M, Liang G, Uzvolgyi E, Gonzales FA, Jones PA. The endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cells. Hum Mol Genet. 2001;10:903–910. doi: 10.1093/hmg/10.9.903. [DOI] [PubMed] [Google Scholar]
- Paolillo M, Russo MA, Curti D, Lanni C, Schinelli S. Endothelin B receptor antagonists block proliferation and induce apoptosis in glioma cells. Pharmacol Res. 2010;61:306–315. doi: 10.1016/j.phrs.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Phuphanich S, Carson KA, Grossman SA, Lesser G, Olson J, Mikkelsen T, et al. Phase I safety study of escalating doses of atrasentan in adults with recurrent malignant glioma. Neuro Oncol. 2008;10:617–623. doi: 10.1215/15228517-2008-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A, Rajeshkumar NV, Gulati A. Effect of the ETB receptor agonist, IRL-1620, on paclitaxel plasma pharmacokinetics of breast tumor rats. Exp Biol Med (Maywood) 2006;231:1120–1122. [PubMed] [Google Scholar]
- Resink TJ, Hahn AW, Scott-Burden T, Powell J, Weber E, Buhler FR. Inducible endothelin mRNA expression and peptide secretion in cultured human vascular smooth muscle cells. Biochem Biophys Res Commun. 1990;168:1303–1310. doi: 10.1016/0006-291x(90)91171-n. [DOI] [PubMed] [Google Scholar]
- Rosanò L, Varmi M, Salani D, Di Castro V, Spinella F, Natali PG, et al. Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian carcinoma cells. Cancer Res. 2001;61:8340–8346. [PubMed] [Google Scholar]
- Rosanò L, Spinella F, Salani D, Di Castro V, Venuti A, Nicotra MR, et al. Therapeutic targeting of the endothelin a receptor in human ovarian carcinoma. Cancer Res. 2003;63:2447–2453. [PubMed] [Google Scholar]
- Rosanò L, Di Castro V, Spinella F, Nicotra MR, Natali PG, Bagnato A. ZD4054, a specific antagonist of the endothelin A receptor, inhibits tumor growth and enhances paclitaxel activity in human ovarian carcinoma in vitro and in vivo. Mol Cancer Ther. 2007a;6:2003–2011. doi: 10.1158/1535-7163.MCT-07-0151. [DOI] [PubMed] [Google Scholar]
- Rosanò L, Di Castro V, Spinella F, Tortora G, Nicotra MR, Natali PG, et al. Combined targeting of endothelin A receptor and epidermal growth factor receptor in ovarian cancer shows enhanced antitumor activity. Cancer Res. 2007b;67:6351–6359. doi: 10.1158/0008-5472.CAN-07-0883. [DOI] [PubMed] [Google Scholar]
- Rosanò L, Cianfrocca R, Masi S, Spinella F, Di Castro V, Biroccio A, et al. Beta-arrestin links endothelin A receptor to betacatenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci U S A. 2009;106:2806–2811. doi: 10.1073/pnas.0807158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- Russell FD, Davenport AP. Secretory pathways in endothelin synthesis. Br J Pharmacol. 1999;126:391–398. doi: 10.1038/sj.bjp.0702315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salani D, Di Castro V, Nicotra MR, Rosano L, Tecce R, Venuti A, et al. Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol. 2000;157:1537–1547. doi: 10.1016/S0002-9440(10)64791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinelli S. Pharmacology and physiopathology of the brain endothelin system: an overview. Curr Med Chem. 2006;13:627–638. doi: 10.2174/092986706776055652. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Pickering V, Liu S, Quang P, Dolan J, Connelly ST, et al. Peripheral endothelin A receptor antagonism attenuates carcinoma-induced pain. Eur J Pain. 2007;11:406–414. doi: 10.1016/j.ejpain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Sekulic A, Suresh P, Pittelkow MR, Markovic S. Bosentan, a novel endothelin-A and -B receptor antagonist inhibits proliferation of malignant melanoma cells. (Abstract) Meeting of the American Association for Cancer Research. Rochester, MN, USA: Mayo Clinic; 2002. [Google Scholar]
- Selting K, Kolli P, Henry C, Owen N, Wipke-Tevis D, Eisen M, et al. Use of a novel endothelin-B agonist (SPI-1620) to alter blood flow to solid tumors for improved chemotherapy delivery in dogs with spontaneously-occurring tumors. Proc Am Assoc Cancer Res. 2008;49:abst 365. [Google Scholar]
- Smollich M, Wülfing P. The endothelin axis: a novel target for pharmacotherapy of female malignancies. Curr Vasc Pharmacol. 2007;5:239–248. doi: 10.2174/157016107781024082. [DOI] [PubMed] [Google Scholar]
- Smollich M, Wülfing P. Targeting the endothelin system: novel therapeutic options in gynecological, urological and breast cancers. Expert Rev Anticancer Ther. 2008;8:1481–1493. doi: 10.1586/14737140.8.9.1481. [DOI] [PubMed] [Google Scholar]
- Spinella F, Garrafa E, Di Castro V, Rosanò L, Nicotra MR, Caruso A, et al. Endothelin-1 stimulates lymphatic endothelial cells and lymphatic vessels to grow and invade. Cancer Res. 2009;69:2669–2676. doi: 10.1158/0008-5472.CAN-08-1879. [DOI] [PubMed] [Google Scholar]
- Spratt JC, Goddard J, Patel N, Strachan FE, Rankin AJ, Webb DJ. Systemic ETA receptor antagonism with BQ-123 blocks ET-1 induced forearm vasoconstriction and decreases peripheral vascular resistance in healthy men. Br J Pharmacol. 2001;134:648–654. doi: 10.1038/sj.bjp.0704304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai M, Umemura I, Yamasaki K, Watakabe T, Fujitani Y, Oda K, et al. A potent and specific agonist, Suc-[Glu9,Ala11,15]-endothelin-1(8-21), IRL 1620, for the ETB receptor. Biochem Biophys Res Commun. 1992;184:953–959. doi: 10.1016/0006-291x(92)90683-c. [DOI] [PubMed] [Google Scholar]
- Tanese K, Fukuma M, Ishiko A, Sakamoto M. Endothelin-2 is upregulated in basal cell carcinoma under control of Hedgehog signaling pathway. Biochem Biophys Res Commun. 2010;391:486–491. doi: 10.1016/j.bbrc.2009.11.085. [DOI] [PubMed] [Google Scholar]
- Trump DL, Payne H, Miller K, de Bono JS, Stephenson J, Burris HA, et al. Safety and efficacy of the specific endothelin A receptor antagonist zibotentan (ZD4054) plus docetaxel in patients with metastatic hormone-resistant prostate cancer: results of a phase I study. 2010. ASCO-GU Symposium San Francisco, CA, USA, 5–7 March abst 193.
- Verri WA, Jr, Cunha TM, Magro DA, Guerrero AT, Vieira SM, Carregaro V, et al. Targeting endothelin ETA and ETB receptors inhibits antigen-induced neutrophil migration and mechanical hypernociception in mice. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:271–279. doi: 10.1007/s00210-008-0360-1. [DOI] [PubMed] [Google Scholar]
- Wang HH, Hsieh HL, Wu CY, Yang CM. Endothelin-1 enhances cell migration via matrix metalloproteinase-9 up-regulation in brain astrocytes. J Neurochem. 2010;113:1133–1149. doi: 10.1111/j.1471-4159.2010.06680.x. [DOI] [PubMed] [Google Scholar]
- Wiesmann F, Veeck J, Galm O, Hartmann A, Esteller M, Knuchel R, et al. Frequent loss of endothelin-3 (EDN3) expression due to epigenetic inactivation in human breast cancer. Breast Cancer Res. 2009;11:R34. doi: 10.1186/bcr2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu-Wong JR, Dixon DB, Chiou WJ, Sorensen BK, Liu G, Jae H-S, et al. Pharmacology of endothelin receptor antagonists ABT-627, ABT-546, A-182086 and A-192621: in vitro studies. Clin Sci (Lond) 2002;103(Suppl. 48):107S–111S. doi: 10.1042/CS103S107S. [DOI] [PubMed] [Google Scholar]
- Yuyama H, Sanagi M, Koakutsu A, Mori M, Fujimori A, Harada H, et al. Pharmacological characterization of YM598, an orally active and highly potent selective endothelin ETA receptor antagonist. Eur J Pharmacol. 2003;478:61–71. doi: 10.1016/j.ejphar.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Yuyama H, Koakutsu A, Fujiyasu N, Fujimori A, Sato S, Shibasaki K, et al. Inhibitory effects of a selective endothelin-A receptor antagonist YM598 on endothelin-1-induced potentiation of nociception in formalin-induced and prostate cancer-induced pain models in mice. J Cardiovasc Pharmacol. 2004a;44(Suppl. 1):S479–S482. doi: 10.1097/01.fjc.0000166309.63808.5f. [DOI] [PubMed] [Google Scholar]
- Yuyama H, Koakutsu A, Fujiyasu N, Tanahashi M, Fujimori A, Sato S, et al. Effects of selective endothelin ET(A) receptor antagonists on endothelin-1-induced potentiation of cancer pain. Eur J Pharmacol. 2004b;492:177–182. doi: 10.1016/j.ejphar.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Zhang W-M, Zhou J, Ye Q-J. Endothelin-1 enhances proliferation of lung cancer cells by increasing intracellular free Ca2+ Life Sci. 2008;82:764–771. doi: 10.1016/j.lfs.2008.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.